

Abstract
Heparin accelerates inhibition of factor XIa (fXIa) by the serpins antithrombin (AT) and C1-inhibitor (C1-INH) by more than two orders of magnitude. The mechanism of the heparin-mediated acceleration of fXIa inhibition by these serpins is incompletely understood, as heparin appears to interact with both the catalytic and non-catalytic domains of the protease. We replaced the basic residues of the fXIa 170-loop (Lys-170, Arg-171, Arg-173, Lys-175 and Lys-179 - chymotrypsin numbering) with Ala, using an expression system that allows separation of the fXIa catalytic domain (CD) from non-catalytic domains. Heparin-mediated inhibition of 170-loop CD variants with AT was impaired 3 to 10-fold relative to the wild type (CD-WT). In reactions with C1-INH, Arg-171 was the most critical residue contributing ∼2-3-fold to heparin-mediated inhibition of CD-WT. A template mechanism did not fully account for the effect of heparin with either serpin, as the second-order inhibition rate constants did not exhibit a characteristic bell-shaped dependence on heparin concentrations. Further studies revealed that the C1-INH inhibition of full length fXIa containing Ala substitutions for basic residues of the 148-loop is not enhanced by heparin. Inhibition by AT of a full-length fXIa variant containing an Ala substitution for Arg-37 in the fXIa CD was ∼5-fold greater than for wild type fXIa in the absence of heparin. These results suggest that basic residues of the fXIa 170-loop form a heparin-binding site, and that the accelerating effect of heparin on inhibition of fXIa by AT or C1-INH may be mediated by charge neutralization and/or allosteric mechanisms that overcome the repulsive inhibitory interactions of serpins with basic residues on the fXIa 148 and 37 loops.
Factor XIa (fXIa)1 is a plasma serine protease that catalyzes the conversion of factor IX (fIX) to fIXa in the intrinsic pathway of blood coagulation (1-4). Hereditary deficiency of the fXIa precursor factor XI (fXI) is associated with a mild to moderate bleeding disorder, suggesting that the protease plays a role in maintenance of normal blood clots (5). FXIa is a disulphide-linked homodimer with a molecular mass of ∼160 kDa (6). The N-terminal heavy chain of each fXIa monomer contains four 90-91 amino acid repeats called apple domains, which facilitate interactions with natural ligands such as fIX, high molecular weight kininogen, glycosaminoglycans, and platelet glycoproteins (6-9). The C-terminal light chain of each monomer contains a trypsin-like catalytic domain (3). The proteolytic activity of fXIa is regulated by several serpin inhibitors. Based on second-order association rate constants, protein Z-dependent protease inhibitor (∼3 × 105 M-1 s-1), protease nexin I (∼8 × 104 M-1 s-1), C1 Inhibitor (C1-INH, ∼2 × 103 M-1 s-1) and antithrombin (AT, ∼3 × 102 M-1 s-1) may be physiologic inhibitors of fXIa in plasma (10-15). With the exception of ZPI, inhibition of fXIa by these serpins is dramatically enhanced by heparin and other glycosaminoglycans (11,16).
The mechanism by which heparin accelerates fXIa inhibition by serpins is not well understood. Based on the observation that fXIa inhibition by C1-INH and AT exhibits a bell-shaped dependence on the concentration of the high molecular weight fraction of heparin, it has been hypothesized that heparin functions as a template facilitating non-covalent complex formation between the protease and serpin (14). Such a mechanism is possible, as both serpins (17,18) and fXIa (14,19,20) have heparin binding sites. Previous work indicated that fXIa has two heparin-binding sites located on the apple-3 domain of the heavy chain (14) and the catalytic domain (19). The basic residues of the apple-3 domain that support the interaction with heparin have been mapped by a mutagenesis approach (14), while the evidence for heparin interacting with the catalytic domain of fXIa is derived from a competitive binding study which showed that a cysteine-constrained α-helical peptide spanning fXIa residues 527-542 (168-182 in chymotrypsin numbering [21]) competes with heparin for interaction with the protease (19).
The relative contribution of the two heparin-binding sites to fXIa interactions with C1-INH and AT is not known, and the mechanism by which heparin enhances the reactivity of fXIa with serpins is poorly understood. To address this, we used an expression system that allowed us to isolate monomeric fXIa catalytic domains (CDs) containing alanine substitutions for the basic residues of the 170-helix (Lys-170, Arg-171, Arg-173, Lys-175 or Lys-179) individually or in combination. FXIa CDs were characterized with respect to their ability to hydrolyze the chromogenic substrate S2366 and to undergo inhibition by AT and C1-INH in the absence and presence of high molecular weight heparin or a heparin pentasaccharide fragment incapable of functioning by a template mechanism.
MATERIALS AND METHODS
Proteins and reagents
Human plasma fXIa and AT were from Haematologic Technologies Inc. (Essex Junction, VT). C1-INH was from Sigma (St. Louis, MO). Human factor XIIa (fXIIa) was from Enzyme Research Laboratories (South Bend, IN). Unfractionated heparin (average MW ∼15 kDa) and the AT-binding pentasaccharide fondaparinux sodium (Organon Sanofi-Synthelabo) were from Quintiles Clinical Supplies (Mt. Laurel, NJ). Fractionated high affinity heparin fragments of ∼35 and ∼64 saccharides were generous gifts from Dr. Steven Olson (University of Illinois-Chicago). S2366 (L-pyroglutamyl-L-prolyl-L-arginine-p-nitroanilide) was from Diapharma (West Chester, OH).
Mutagenesis and expression of recombinant proteins
Mutations in the fXIa 170 helix were introduced into a modified human fXI cDNA (fXI-Ser-362,482), which contains serine substitutions for Cys-362 and Cys-482 (fXI numbering [22,23]). A disulfide bond between these residues connects the heavy chains and catalytic domains after cleavage at the activation site, and eliminating the bond allows the catalytic domain (CD) to separate from the heavy chain (22). The basic residues of the fXIa 170-helix, Lys-529, Arg-530, Arg-532, Lys-536 and Lys-540 in the fXI numbering system (23), were changed to alanine using a Quick Change kit (Stratagene, La Jolla, CA [8]). These residues correspond to residues 170, 171, 173, 175 and 179 in the chymotrypsinogen numbering system (21), which will be used hereafter. Recombinant wild type fXIa catalytic domain is designated CD-WT, and mutants are CD-K170A, CD-R171A, CD-R173A, CD-K175A, and CD-K179A. A CD with residues 170, 171, and 173 changed to alanine is designated CD-KRR/A. cDNAs in expression vector pJVCMV were used to transfect HEK-293 cells as described (8). Expression and characterization of full-length wild type fXI (fXI-WT); fXI-R37Q, which contains a Gln substitution for Arg-37 (Arg-395 in fXI numbering); and the autolysis loop variant fXI-144-149A (10), which contains Ala substitutions for Arg-144, Lys-145, Arg-147 and Arg-149 (504, 505, 507 and 509 in fXI numbering), were prepared as described (8). Stably expressing clones were expanded in 175 cm2 flasks, and serum free media (Cellgro Complete, Mediatech, Herndon, VA) was collected every 48 hours, supplemented with benzamidine (5 mM) and stored at -20°C pending purification (22).
All recombinant fXI was purified from conditioned media on an anti-fXI IgG 1G5.12 affinity column (8). After loading, the column was washed with 25 mM Tris-HCl, pH 7.4, 100 mM NaCl (TBS), and eluted with 2M NaSCN in TBS. Protein containing fractions were pooled, concentrated and dialyzed against TBS, and protein concentrations were determined by dye-binding assay (Bio-Rad). FXI (∼200-300 μg/ml) was activated with 5 μg/ml fXIIa at 37°C, and complete activation was confirmed by SDS-PAGE. Activated preparations were passed over a 1G5.12 column to separate the protease from fXIIa. In the case of proteins prepared in fXI-Ser-362,482, the catalytic domains bind to the column, while the heavy chain passes through (22).
FXIa hydrolysis of S2366
The steady-state kinetics of S2366 hydrolysis by fXIa and fXIa CDs (6 nM active sites) was measured in TBS containing 0.1 mg/ml bovine serum albumin (TBSA) and 50-2000 μM S2366. Rates of generation of free p-nitroaniline (pNA) in 100 μL reaction volumes (3 mm path length) were measured by continuous monitoring of absorbance at 405 nm on a SpectraMax 340 microtiter plate reader (Molecular Devices Corp., Sunnyvale, CA) (22). Km and kcat for S2366 hydrolysis were obtained by initial rate analysis of pNA generation as a function of S2366 concentration. Non-linear regression was performed with Scientist Software (MicroMath Scientific Software, Salt Lake City, UT). Estimates of error are ± 2 SD.
Inhibition of fXIa and fXIa catalytic domains by serpins
Rates of fXIa inhibition by AT or C1-INH were measured under pseudo-first-order conditions by a discontinuous assay in the absence and presence of heparins (10). In the absence of heparin, each protease (0.5-1 nM) was incubated at RT (∼25°C) with 100-400 nM AT or C1-INH in 50 μL reactions in TBSA containing 0.1 % PEG 8000. After 80 min incubation, 50 μL S2366 was added (final concentration 0.5 mM), and residual fXIa activity was determined from the rate of S2366 hydrolysis at 405 nm. The observed pseudo-first-order, and second-order rate constants (k2) were calculated as described (10). The same procedure was used to determine k2 values in the presence of fondaparinux except that serpin concentrations were 50-200 nM with incubation times of 30-80 min.
Heparin concentration dependence was determined by incubating each protease with 25-100 nM serpin and 0-50 μM unfractionated heparin or 0-10 μM fractionated heparin in 50-μL reactions in TBSA containing 0.1% PEG 8000. Following 1-10 min incubations at RT, 50 μL S2366 in TBS containing 1 mg/ml Polybrene (to immediately neutralize heparin) was added to a final concentration of 0.5 mM. The catalyzed kobs values at each heparin concentration were determined from a first-order rate equation, and the k2 values were calculated by dividing kobs by the serpin concentration. Plots of k2 as a function of heparin concentration yielded maximal k2 values and optimal concentrations of heparin for each reaction.
RESULTS
Expression, purification and activation of recombinant proteins
Isolated fXIa CDs were prepared using fXI lacking the 362-482 disulphide bond that connects the two fXIa CDs to the non-catalytic heavy chain dimer (HC-D) after proteolysis by fXIIa at the activation cleavage site (22) (Fig. 1A). The CD of fXIa-Ser362-482 (CD-WT) binds to monoclonal antibody 1G5.12 (Fig. 1B, first lane) while the HC-D does not (Fig. 1B, second lane). An SDS-PAGE of fXIa-Ser362-482 170-loop mutants is shown in Fig. 1C (top panel). The gel was run under reducing conditions to demonstrate complete conversion of zymogen (∼75 kDa under reducing conditions) to the heavy chain (HC) and CD of fXIa. The non-reducing gel in the bottom panel of Fig. 1C shows purified CDs, along with the position of residual heavy chain dimer (HC-D) and traces of albumin from the media. A reducing SDS-PAGE of zymogen (left panel) and activated forms (right panel) of full length fXI-WT and fIX-R37Q is shown in Fig. 1D. Both proteins migrate with the expected molecular mass.
Figure 1.
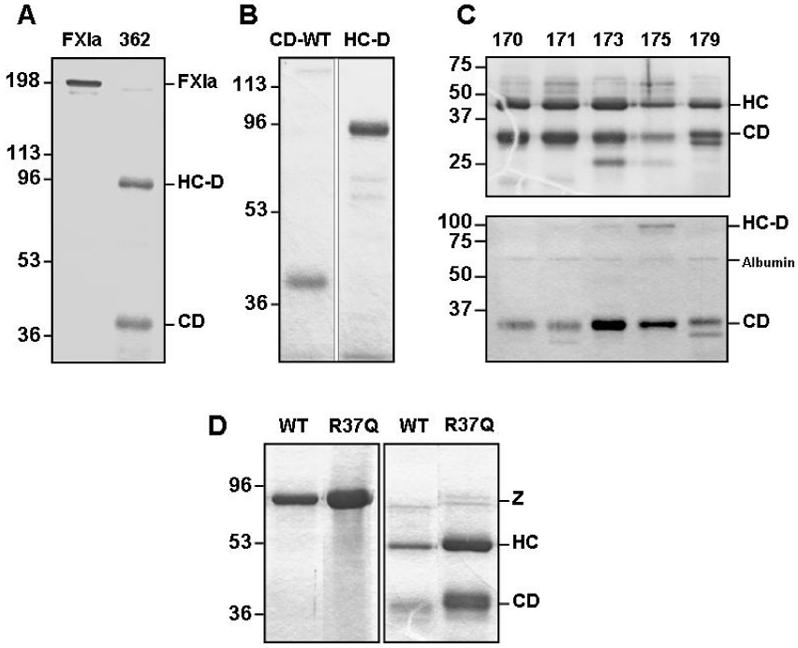
(A) Non-reducing SDS-polyacrylamide gel of full length fXIa and fXIa-Ser362-482 (labeled 362) lacking the disulfide bond. (B) The first lane is the 1G5.12 affinity purified catalytic domain (CD-WT) of fXIa-Ser362-482 and the second lane is the flow-through heavy chain dimer (HC-D). (C) The top panel is a reducing gel of the Ala substitution mutants of fXIa-Ser362-482 and the bottom panel is a non-reducing gel showing purified catalytic domains (CD) of the same mutants, along with the position of residual heavy chain dimer (HC-D) and traces of albumin from conditioned media. (D) Reducing gel of full-length fXI (left panel) and fXIa (right panel). Shown are wild type fXI/fXIa (WT) and fXI/fXIa-R37Q. Abbreviations to the right of the panel are: (Z) - zymogen fXI, (HC) - fXIa heavy chain, and (CD) - fXIa catalytic domain. Positions of molecular mass standards are shown to the left of each panel.
Amidolytic Activity
Kinetic parameters for hydrolysis of S2366 by fXIa CDs are shown in Table 1. Mutants exhibited similar Km and kcat to CD-WT for hydrolysis of this tripeptidyl chromogenic substrate, indicating substitution for the basic residues in the 170-loop does not adversely affect conformation of the active-site pocket, and specifically the P1-P3 residues. This also holds true for full-length fXIa-R37Q (Table 1). FXIa-R37Q, which was prepared prior to development of the system for generating isolated CDs, was included because it provides insight into the mechanism by which AT inhibits fXIa in the presence of heparin (see below).
Table 1. Amidolytic activities of recombinant factor XIa and factor XIa CDs.
The kinetics of S2366 cleavage by fXIa derivatives were determined in TBSA at room temperature, using 6 nM fXIa active sites as described in “Materials and Methods”. All proteases w ere tested in triplicate, and values are ±2 SD.
| Protein | S2366 Cleavage | |
|---|---|---|
| Km (μM) | kcat (s-1) | |
| FXIa-CD-WT | 757 ± 55 | 119 ± 4 |
| FXIa-CD-K170A | 655 ± 46 | 98 ± 3 |
| FXIa-CD-R171A | 812 ± 71 | 115 ± 4 |
| FXIa-CD-R173A | 810 ± 69 | 117 ± 4 |
| FXIa-CD-K175A | 829 ± 77 | 117 ± 5 |
| FXIa-CD-K179A | 764 ± 63 | 116 ± 4 |
| FXIa-CD-KRR/A | 552 ± 36 | 115 ± 3 |
| FXIa-WT | 943 ± 40 | 148 ± 3 |
| FXIa-R37Q | 993 ± 51 | 148 ± 4 |
Reaction of fXIa CDs with AT
The k2 values for inhibition of fXIa CDs by AT in the absence and presence of heparins are shown in Table 2. All CDs exhibited similar inhibition to CD-WT by AT in the absence and presence of fondaparinux (H5). In contrast, with the exception of CD-K179A, the unfractionated heparin-catalyzed AT inhibition was impaired relative to CD-WT for all CD mutants (Table 2). Heparin enhanced AT inhibition of CD-WT 212-fold, but only 37-94-fold for CD mutants. AT inhibition of the triple mutant CD-KRR/A was reduced >10-fold in the presence of heparin compared to CD-WT. The allosteric effect of heparin contributed 2-to 3-fold to the enhancement in AT-mediated inhibition by heparin as demonstrated by k2 values in the presence of fondaparinux (Table 2). The bell-shaped dependence of k2 values on heparin concentrations suggested a template effect may partly account for the heparin cofactor activity. However, the bell-shaped distribution was relatively shallow in amplitude (Fig. 2A), with inhibition of CD-WT showing only a 2-3-fold decline in k2 at an unfractionated heparin concentration of 100 μM (data not shown). This observation raised the possibility that a template effect may not represent the primary mechanism for heparin-mediated inhibition of fXIa CDs.
Table 2. Second-order rate constants for inhibition of full-length fXIa and fXIa CDs by AT in the absence and presence of heparins.
| - cofactor 103 (M-1 s-1) | +H5 103 (M-1 s-1) | +Hep 103 (M-1 s-1) | + Hep/- cofactor (fold) | |
|---|---|---|---|---|
| FXIa-WT | 0.26 ± 0.02 | 0.62 ± 0.04 | 223 ± 18 | 858 |
| FXIa-Q37A | 1.55 ± 0.06 | 2.62 ± 0.04 | 180 ± 10 | 116 |
| FXIa-CD-WT | 0.25 ± 0.02 | 0.60 ± 0.03 | 53 ± 6 | 212 |
| FXIa-CD-K170A | 0.23 ± 0.01 | 0.61 ± 0.02 | 12 ± 1 | 52 |
| FXIa-CD-R171A | 0.49 ± 0.03 | 1.20 ± 0.05 | 18 ± 1 | 37 |
| FXIa-CD-R173A | 0.21 ± 0.01 | 0.56 ± 0.03 | 8 ± 0.5 | 38 |
| FXIa-CD-K175A | 0.18 ± 0.01 | 0.55 ± 0.02 | 17 ± 1 | 94 |
| FXIa-CD-K179A | 0.25 ± 0.02 | 0.68 ± 0.03 | 50 ± 3 | 200 |
| FXIa-CD-KRR/A | 0.22 ± 0.01 | 0.56 ± 0.02 | 4 ± 0.3 | 18 |
All second-order inhibition rate constants (k2) were determined by incubation of fXIa or fXIa CD (0.5-1 nM) with AT (50-1000 nM AT) in TBSA containing 0.1% PEG 8000 for 3-140 min. In AT reactions in the presence of fondaparinux pentasaccharide (H5), 2 μM pentasaccharide was included in each reaction. k2 values in the presence of heparin are derived from Fig. 2A at optimal heparin concentrations. Values for k2 were determined by measuring residual enzyme activity in an amidolytic activity assay as described in “Materials and Methods”. All values are the average of 2-3 measurements ± SD.
Figure 2.

Heparin concentration dependence of fXIa catalytic domain inhibition by AT. (A) The heparin dependence of k2 for AT inhibition of CD-WT (○), K170A (●), R171A (□), R173A (■), K175A (△), K179A (▲) and the triple mutant KRR/A (▽) are shown for unfractionated heparin. k2 values (Table 2) were determined from the remaining activities of CDs as described under “Materials and Methods”. (B) The same as panel A except that k2 values for AT inhibition of fXIa CDs were determined in the presence of increasing concentrations of a high-affinity fractionated heparin composed of ∼64 saccharides. (C) The same as panel A except that k2 values for AT inhibition of fXIa CDs were determined in the presence of increasing concentrations of a high-affinity fractionated heparin composed of ∼35 saccharides.
To further investigate this possibility, AT inhibition of fXIa CDs was monitored in the presence of increasing concentrations of a high-affinity fractionated heparin composed of ∼64-saccharides. Consistent with a small role for the template mechanism in inhibition of fXIa-CD, the k2 values for inhibition of CD by AT exhibited a saturable dependence on heparin concentration up to 20 μM (Fig. 2B, shown up to 10 μM for all CD derivatives). k2 values demonstrated a similar saturable dependence on heparin concentration with a high-affinity fractionated heparin composed of ∼35-saccharides, except that the rates were ∼2-3-fold slower (Fig. 2C). It should be noted that, with the exception of CD-K179A, the optimal concentration of heparin for maximal cofactor effect was shifted from ∼200 nM for CD-WT to ∼1000 nM for the mutants, suggesting a decreased affinity for heparin (Fig. 2A).
The magnitude of the effect of heparin on AT-mediated inhibition was decreased from >800-fold for full-length fXIa-WT to ∼200-fold for CD-WT (Table 2). This suggests an ∼4-fold contribution from the heparin binding site on the apple-3 domain in the HC (14,20), and is consistent with data showing a similar extent of impairment in heparin-enhanced AT inhibition of fXIa with Ala substitutions for basic residues in the apple-3 domain (14). The results are also consistent with the observation that the optimal heparin concentration for AT inhibition of fXIa-WT is 4-fold lower (∼50 nM - data not shown) compared to CD-WT (200 nM - Fig. 2). At the highest heparin concentration tested (100 μM), there was still ∼40-fold acceleration of AT inhibition of CD-WT, indicating a significant allosteric effect on the serpin, the protease, or both. The allosteric effect on AT may account for a 2-3-fold enhancement of fXIa inhibition, if one compares k2 values in the absence and presence of fondaparinux, a pentasaccharide that conformationally activates AT (Table 2) (24).
Previously, it was reported that heparin causes a conformational change in the catalytic pocket of fXIa (25). To determine whether the heparin interaction with CD-WT changes the active-site pocket, the amidolytic activity of CD-WT was monitored in the presence of different heparin concentrations. In contrast to the published results for full-length fXIa (25), heparin or fondaparinux had minimal effect on CD-WT cleavage of S-2366, suggesting the S3-S1 substrate binding sites are unaffected (data not shown). The results are consistent with a report that did not observe an effect of heparin on S2366 cleavage by fXIa (14). It has been observed that residues of the 37-loop of coagulation proteases contribute to the interaction with AT at the C-termini of the serpin reactive center loop (26,27). In fXIa, this loop contains Arg at position 37 (Arg-395 in fXI numbering [23]). Inhibition of full-length fXIa-R37Q by AT (k2 = 1.6 × 103 M-1 s-1) was enhanced ∼7-fold in the absence of heparin (Table 2), raising the possibility that heparin modulates the 37-loop of fXIa to improve its reactivity with AT.
Inhibition of fXIa and fXIa CDs by C1-INH
k2 values for C1-INH-mediated inhibition of fXIa-WT and fXIa CDs are shown in Table 3. All fXIa-CDs were inhibited similarly in the absence of a heparin. Interestingly, the rate constant for C1-INH inhibition of CD-WT was enhanced by an order of magnitude in the presence of fondaparinux (Table 3), and a similar enhancement was observed for all 170-helix CD mutants including the triple mutant CD-KRR/A. This suggests that binding of fondaparinux to the serpin, but not the protease, is responsible for the rate enhancement. Similar to reactions with AT, the concentration dependence of unfractionated heparin-mediated rate enhancement, (Fig. 3A) exhibited a bell-shaped distribution with shallow amplitude that largely disappeared with CD-R171A and CD-RRK/A. The same studies with ∼64-and ∼35-saccharides fractionated heparins did not result in a bell-shaped curve, however, at 10 μM heparin (the highest concentration used in the assay), the accelerating effect of heparin on C1-INH-mediated inhibition of CD-RRK/A was decreased ∼4-fold with both heparin fractions (Fig. 3B shown for ∼64-saccharides only), suggesting either a direct effect or a template-mediated role for the basic residues of the 170-helix interaction with C1-INH.
Table 3. Second-order rate constants for inhibition of full-length fXIa and fXIa CDs by C1-INH.
| - cofactor 103 (M-1 s-1) | +H5 103 (M-1 s-1) | +Hep 103 (M-1 s-1) | +Hep/- cofactor (fold) | |
|---|---|---|---|---|
| FXIa-WT | 1.9 ± 0.2 | 9.1 ± 0.8 | 536 ± 32 | 282 |
| FXIa-CD-WT | 1.0 ± 0.1 | 9.0 ± 0.7 | 750 ± 54 | 750 |
| FXIa-CD-K170A | 1.2 ± 0.1 | 10.0 ± 0.7 | 734 ± 58 | 612 |
| FXIa-CD-R171A | 0.6 ± 0.1 | 5.0 ± 0.3 | 261 ± 18 | 435 |
| FXIa-CD-R173A | 1.3 ± 0.1 | 9.1 ± 0.5 | 667 ± 35 | 513 |
| FXIa-CD-K175A | 1.0 ± 0.1 | 7.3 ± 0.4 | 730 ± 49 | 730 |
| FXIa-CD-K179A | 1.1 ± 0.1 | 8.8 ± 0.5 | 778 ± 53 | 707 |
| FXIa-CD-KRR/A | 0.8 ± 0.1 | 6.3 ± 0.3 | 430 ± 23 | 525 |
| FXIa-144-149A | 19.2 ± 1.8 | 19.5 ± 2.1 | 20.6 ± 1.9 | 1 |
All second-order inhibition rate constants (k2) were determined by incubation of fXIa or fXIa CDs (0.5-1 nM ) with C1-INH (25-200 nM) in TBSA containing 0.1% PEG 8000 for 2-30 min. In reactions in the presence of fondaparinux pentasaccharide (H5), 2 μM pentasaccharide was included in each reaction. k2 values in the presence of heparin are derived from Fig. 3A at optimal heparin concentrations. Values for k2 were determined by measuring residual enzyme activity in an amidolytic activity assay as described in “Materials and Methods”. All values are the average of 2-3 measurements ± SD .
Figure 3.

Heparin concentration dependence of fXIa catalytic domain inhibition by C1-INH. (A) The heparin dependence of k2 for C1-INH inhibition of CD-WT (○), K170A (●), R171A (□), R173A (■), K175A (△), K179A (▲) and the triple mutant KRR/A (▽) are shown for unfractionated heparin. k2 values (Table 3) were determined from the remaining activities of CDs as described under “Materials and Methods”. (B) The same as panel A except that k2 values for C1-INH inhibition of CD-WT (○) and CD-KRR/A (▽) were determined in the presence of increasing concentrations of a high-affinity fractionated heparin composed of ∼64 saccharides.
The observation that inhibition of CD-K170A and CD-R173A by C1-INH was slightly enhanced compared to CD-WT, and that inhibition of CD-R171A was impaired ∼2-fold, suggested an interaction between the reactive center loop of C1-INH and the protease 170-helix. Analysis of maximum k2 values derived from Fig. 3A (shown in Table 3), indicated that these mutations have minimal effect on heparin-mediated inhibition. In the presence of heparin, C1-INH inhibition of CD-WT and CD-RRK/A was enhanced 780- and 530-fold, respectively. These results, and the order of magnitude enhancement of C1-INH-mediated inhibition of CD-WT by fondaparinux, raises the possibility that an allosteric effect of heparin on C1-INH and/or the protease is involved. Alternatively, heparin could enhance the productive interaction of CD-WT with C1-INH by charge neutralization, as has been hypothesized based on crystal structure of an N-terminal deletion mutant of C1-INH (28).
To further investigate this issue, we analyzed the reactivity of a full-length fXIa variant, fXIa-144-149A, in which the four basic residues of the protease autolysis loop have been replaced with alanine (10). The putative heparin-binding sites on the apple-3 domain and 170-loop of the protease domain should be intact in this mutant, and we showed that inhibition of this mutant by C1-INH is enhanced >15-fold compared to fXIa-WT in the absence of heparin (10). Heparin accelerated AT inhibition of fXIa-WT and fXIa-144-149A (Fig. 4) to a similar extent, with a maximal k2 of ∼2 × 105 M-1 s-1 at an optimal heparin concentration for both proteases. This indicates that fXIa-144-149A binds normally to heparin. However, neither heparin nor fondaparinux enhanced inhibition of fXIa-144-149A by C1-INH (Table 3), indicating that heparin enhances C1-INH inhibition of fXIa primarily through an interaction with the serpin. Heparin binding to basic residues on C1-INH may overcome the inhibitory interaction between the serpin and basic residues of the fXIa autolysis loop by charge neutralization. Alternatively, heparin may optimize docking of the C1-INH reactive center loop with the fXIa catalytic pocket by an allosteric effect on the serpin, reminiscent of the mechanism by which heparin enhances AT inhibition of factor Xa (24,27). Similar to fXIa, the autolysis loop of factor Xa contains several basic residues, and heparin binding to AT allosterically exposes a cryptic exosite on the serpin that is recognized by the residues on factor Xa (29). Alanine substitutions for the basic residues in the factor Xa autolysis loop (particularly Arg-150) largely eliminates the interaction of this loop with the heparin activated conformation of AT (29).
Figure 4.

Heparin concentration dependence of AT inhibition of fXIa. Shown is the dependence of k2 for AT inhibition of full-length fXIa-WT (○) and the autolysis loop mutant fXIa-144-149A (●) on heparin concentrations using unfractionated heparin. k2 values were determined from the remaining fXIa activity as described under “Materials and Methods”.
To test the hypothesis that basic residues on C1-INH and the fXIa autolysis loop are involved in repulsive interactions, we studied inhibition of fXIa-144-149A (10) by C1-INH. C1-INH inhibited fXIa-144-149A with 3-fold higher k2 than wild-type fXIa (0.6 × 103 vs. 0.2 × 103 M-1 s-1, respectively) in the absence of heparin. This is consistent with results for autolysis loop mutants of factor Xa, where the effect of heparin on C1-INH-mediated inhibition was decreased from ∼35-fold for wild type factor Xa to ∼4-fold for the factor Xa mutants with substitutions for basic residues in the autolysis loop (data not presented). These results strongly suggest that heparin enhances C1-INH inhibition of fXIa and factor Xa through a charge neutralization mechanism that overcomes an inhibitory interaction between basic residues on the protease autolysis loops and a complementary site on C1-INH. In contrast to the results with AT, inhibition of the fXIa-R37Q by C-INH was impaired ∼2-fold in the absence and presence of fondaparinux. This suggests that, unlike the inhibitory role of basic residues of the autolysis loop, Arg-37 contributes to the fXIa-C1-INH interaction (data not presented).
DISCUSSION
Heparin-like glycosaminoglycans regulate the catalytic activity of fXIa during interactions with macromolecular substrates and inhibitors (4,11,16,20). Previous work indicates that both the catalytic and non-catalytic domains of fXIa interact with heparin. The heparin-binding site on the non-catalytic fXIa HC is located in the apple-3 domain (14,20). Competitive binding studies with peptides containing sequence from the fXIa 170-helix indicate that the basic residues of this region also interact with heparin (19). To understand the contributions of each of these sites to the mechanism of regulation of fXIa by plasma serpins, we expressed and isolated the wild type fXIa catalytic domain (CD) and several CD variants with alanine substitutions for basic residues in the 170-helix. The relative orientations of the side chains of these residues in the three-dimensional fXI CD structure (30) are shown in Fig. 5.
Figure 5.

Structure of the catalytic domain of fXIa. Shown is the peptide backbone for the crystal structure of factor XIa catalytic domain in complex with p-aminobenzamidine. The side chains of basic residues in the 170-helix, 220-loop and Arg-37 are shown in blue. The active site serine residue (Ser-195) is in green. The backbone of autolysis loop residues 144-149 are colored in red. The coordinates (Protein Data Bank code 1ZHM) of the catalytic domain of fXIa were used to prepare the figure (30).
For AT, k2 for inhibition of CD-WT was enhanced 212-fold by heparin, and all mutants exhibited some defect in inhibition with the exception of CD-K179A. Specifically, the effect of heparin was reduced 4-fold for CD-K170A, 6-fold for both CD-R171A and CD-R173A, and 2-fold for CD-K175A. Inhibition of the triple mutant CD-KRR/A was reduced 10-fold in the presence of heparin, suggesting that these 170-helix residues make the greatest contribution to the affinity of the CD for heparin. A comparison of the effects of heparin on inhibition of full length fXIa-CD and CD-WT, and consideration of published results (14), indicates the fXIa apple-3 domain contributes ∼4-fold to acceleration of the protease inhibition by AT, probably through a template mechanism. It is interesting to note that unlike factors IXa, Xa and α-thrombin, which have basic C-terminal helices that interact with heparin (31-33), the C-terminal helix of fXIa is acidic and not expected to interact with heparin (34). However, fXIa CD has two basic residues on the 220-loop (Arg-222 and Arg-224) that, together with basic residues on the 170-helix, form a patch capable of interaction with heparin (34). The observation that heparin still enhances AT inhibition of the CD-KRR/A 18-fold suggests that heparin interacts with other residues on the CD, such as those of the 220-loop. The 220-loop of thrombin and vitamin K-dependent coagulation proteases harbor a functionally critical Na+-binding site, with the basic residues of this loop contributing to the coordination of Na+ (35). The corresponding loop in factor XIa does not bind Na+ (35), but may interact with heparin (34). If true, this makes the heparin interaction with fXIa CD quite different from interactions with the protease domains of other coagulation factors, and may account for the inability of heparin to enhance AT inhibition of CD-WT by a template mechanism.
At first glance, the bell-shaped dependence for unfractionated heparin-mediated AT inhibition of CD-WT may suggest a template effect, even though the contribution appears to be relatively small. However, as full-length fXIa-WT and CD-WT exhibit similar affinities for heparin (25), one might expect the heparin cofactor effect to be greatly reduced or eliminated at high heparin concentrations if a bridging mechanism was important (15). Our studies with full-length fractionated heparin indicate that a template mechanism contributes minimally to heparin-enhancement of AT inhibition of CD-WT. The shallow bell-shaped dependence on heparin could be due to the heterogeneity of unfractionated heparin, where less active small fragments may compete with larger molecules at high heparin concentrations for binding to serpin and/or protease. Given this, the reported bell-shaped distribution of rate constants for AT inhibition of fXIa in the presence of heparin should be interpreted with caution. Heparin allosterically enhances AT interactions with factors IXa and Xa (24,36). The analysis of AT inhibition of fXIa with the AT-binding pentasaccharide fondaparinux suggests this mechanism contributes 2-3-fold to the cofactor effect of heparin on fXIa, consistent with data reported in the literature (15).
We did not observe, in contrast to a previous study (25), an effect of heparin on CD-WT cleavage of S2366, indicating that heparin does not allosterically modulate the fXIa S1-S3 substrate binding subsites. Nevertheless, further studies are required, as the crystal structure of fXIa CD in complex with ecotin (34) indicates the heparin binding residues on the 170-helix are part of the S4 loop which may be involved in determining the specificity of the S4 subsite. Since tripeptidyl substrates such as S2366 do not possess a P4 residue, our amidolytic activity assays may not be sensitive to heparin-mediated allosteric changes in the fXIa catalytic pocket.
Residues of the 37-loop of coagulation proteases can influence interactions with AT in the absence and presence of heparin (26,27). Interestingly, inhibition of full-length fXIa-R37Q by AT was increased 7-fold relative to fXIa-WT in the absence of heparin. Furthermore, the enhancing effect of heparin was decreased to the same extent, resulting in similar rates of inhibition for heparin-catalyzed AT inhibition of fXIa-R37Q and fXIa-WT. Heparin may overcome an inhibitory interaction between Arg-37 and AT by an allosteric effect on Arg-37, which is located on a surface loop surrounding the protease active-site pocket (Fig. 5). Therefore, unlike the case of factor Xa, which has a negatively charged 37-loop, Arg-37 in fXIa may contribute to poor inhibition by AT in the absence of heparin. Previous studies indicated that residues in the 37-loop influence the protease specificity for the primed sites (in particular the P3′ site) on substrates and inhibitors (26,37), and the amidolytic activity of fXIa toward substrates that lack a P’ residue (such as S2366) may not accurately report a modulating effect of heparin through the 37-loop on the protease catalytic-site.
Taken as a whole, the results suggest that full-length heparin accelerates AT inhibition of fXIa ∼800-fold, with heparin binding to a specific site on the apple-3 domain contributing ∼4-fold to acceleration of AT inhibition of fXIa (14), and an allosteric effect on the AT reactive center loop contributing ∼3-fold. The remaining 50-fold accelerating effect of heparin in enhancing the AT inhibition of fXIa may be mediated primarily through an allosteric mechanism that appears to involve binding of heparin to basic residues on the catalytic domain including those of the 170-helix. Such an allosteric effect may partly involve overcoming an inhibitory interaction of the basic 37-loop of fXIa with the AT reactive center loop.
The studies with C1-INH suggest that, with the exception of a small decrease for CD-R171A, substitutions for basic residues of the fXIa CD 170-loop do not significantly affect heparin-mediated inhibition (Table 3). These results, in combination with the observation that inhibition by CI-INH was increased by nearly an order of magnitude in the presence of fondaparinux, suggest that heparin does not enhance C1-INH inhibition of fXIa CD by a template mechanism. Recently, we showed that replacing basic residues in the fXIa autolysis loop (148-loop) with Ala (fXIa-144-149A) enhances inhibition by C1-INH 15-20-fold (10). To determine if the cofactor function of heparin involves overcoming an inhibitory interaction between C1-INH and the fXIa autolysis loop, we studied inhibition of fXIa-144-149A by C1-INH in the presence of heparin. Heparin did not affect inhibition at concentrations up to 100 μM, suggesting that heparin interacts with a basic site on the serpin to overcome a repulsive interaction with charged residues on the autolysis loop. This hypothesis is consistent with structural data for the latent form of a recombinant N-terminal deletion mutant of C1-INH (28), where basic residues on strands s2C, S3c and s4C form a large contiguous electro-positive area of 35 Å diameter at the top of the serpin (28). Based on structural homology with other serpins, this patch may overlap an area on the reactive center loop that interacts with the protease to form a non-covalent Michaelis-type complex (27,38). Thus, heparin binding to this region may enhance fXIa inhibition through charge neutralization. The observations that heparin does not enhance C1-INH inhibition of fXIa-144-149A, and that replacement of basic residues in the autolysis loop does not mimic the cofactor effect of heparin, suggests that other mechanisms such as an allosteric effect of heparin on either the serpin or the protease may also contribute to accelerated inhibition of fXIa by C1-INH.
A template effect is not likely to contribute significantly to acceleration of fXIa inhibition by C1-INH, based on the observation that heparin interacts with fXIa-144-149A normally, enhancing the AT inhibition of the mutant similarly to fXIa-WT (Fig. 4). Furthermore, while the optimal heparin concentration for inhibition of CD-KRR/A by C1-INH is shifted from 2 μM to 10 μM, the maximal k2 value for this mutant decreases ∼2-fold and is not affected by increasing heparin concentrations. An allosteric effect for heparin on C1-INH that enhances fXIa inhibition has not been previously supported (28). Hypothetically, the cofactor effect of heparin in C1-INH inhibition of fXIa may be mediated through i) heparin interacting with the basic patch near the serpin reactive center loop, resulting in charge neutralization, ii) heparin binding to fXIa allosterically optimizing the active-site pocket for interaction with C1-INH, and iii) a “sandwich” mechanism (28) where the heparin-bound serpin makes productive interactions with basic residues on the protease. The observation that mutations in the fXIa 170-helix have a minimal effect on heparin-mediated enhancement of inhibition by C1-INH would not support the second mechanism unless heparin binds to another site on the fXIa CD; however, the structural data (28), together with the heparin-independent enhancement of C1-INH inhibition of fXIa-144-149A provide strong support for the first mechanism. The inability of heparin to accelerate C1-INH inhibition of fXIa-144-149A also is consistent with the third option, where the basic autolysis loop of fXIa could provide an interactive site specifically for the heparin-bound serpin. In such a ternary complex, the response to different concentrations of heparin would not assume a bell-shaped distribution, as the basic residues of the autolysis loop do not constitute an independent heparin-binding site. In this mechanism, the cofactor effect of heparin is primarily directed at relieving a steric interaction that down-regulates factor XIa inhibition, rather than enhancing the rate of an otherwise efficient reaction. If the heparin-bound serpin actually makes a productive interaction with basic residues on the autolysis loop, then replacing these residues with acidic ones may produce an effect similar to the cofactor activity of heparin, and result in a dramatic enhancement (greater than that of fXIa-144-149A) in the reactivity of the protease with the serpin that is independent of the polysaccharide. Additional mutagenesis-based studies will also be required to explore the possibility that distinct binding sites on fXIa are utilized in a serpin-dependent manner.
Acknowledgments
This work was supported by grants HL-62565 to ARR and HL-58837 to DG from the National Heart, Lung, and Blood Institute.
Footnotes
- FXI
- factor XI
- fXIa
- activated factor XI
- fXIIa
- activated factor XII
- fXIa-WT
- wild type recombinant fXIa
- CD
- catalytic domain of fXIa
- CD-K170A, CD-R171A, CD-R173A, CD-K175A and CD-K179A
- catalytic domain mutants of factor XIa with residues in the chymotrypsin system (21) substituted with Ala
- C1-INH
- C1-inhibitor
- AT
- antithrombin
- BSA
- bovine serum albumin
- PEG
- polyethylene glycol
REFERENCES
- 1.Wiggins RC, Bouma BN, Cochrane CG, Griffin JH. Role of high molecular-weight kininogen in surface-binding and activation of coagulation factor XI and prekallikrein. Proc. Natl. Acad. Sci. (USA) 1977;74:4636–4640. doi: 10.1073/pnas.74.10.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh PN, Griffin JH. Contributions of human platelets to the proteolytic activation of blood coagulation factors XII and XI. Blood. 1981;57:106–118. [PubMed] [Google Scholar]
- 3.Kurachi K, Davie EW. Activation of human factor XI (plasma thromboplastin antecedent) by factor XIIa (activated Hageman factor) Biochemistry. 1977;16:5831–5839. doi: 10.1021/bi00645a030. [DOI] [PubMed] [Google Scholar]
- 4.Gailani D, Broze GJ., Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 5.Ragni MV, Sinha D, Seaman F, Lewis JH, Spero JA, Walsh PN. Comparison of bleeding tendency, factor XI coagulant activity, and factor XI antigen in 25 factor XI-deficient kindreds. Blood. 1985;65:719–724. [PubMed] [Google Scholar]
- 6.Walsh PN. Platelets and factor XI bypass the contact system of blood coagulation. Thromb. Haemostas. 1999;82:234–242. [PubMed] [Google Scholar]
- 7.Walsh PN, Baglia FA, Jameson BA. Factor XI and platelets: Activation and regulation. Thromb. Haemostas. 1993;70:75–79. [PubMed] [Google Scholar]
- 8.Sun MF, Zhao M, Gailani D. Identification of amino acids in the factor XI apple 3 domain required for activation of factor IX. J. Biol. Chem. 1999;274:36373–36378. doi: 10.1074/jbc.274.51.36373. [DOI] [PubMed] [Google Scholar]
- 9.Sun MF, Gailani D. Identification of a factor IX binding site on the third apple domain of activated factor XI. J. Biol. Chem. 1996;271:29023–29028. doi: 10.1074/jbc.271.46.29023. [DOI] [PubMed] [Google Scholar]
- 10.Rezaie AR, Sun MF, Gailani D. Contribution of basic amino acids in the autolysis loop of factor XIa to serpin specificity. Biochemistry. 2006;45:9427–9433. doi: 10.1021/bi060820+. [DOI] [PubMed] [Google Scholar]
- 11.Knauer DJ, Majumdar D, Fong P-C, Knauer MF. Serpin regulation of factor XIa. J. Biol. Chem. 2000;275:37340–37346. doi: 10.1074/jbc.M003909200. [DOI] [PubMed] [Google Scholar]
- 12.Meijers JCM, Vlooswikj RAA, Bouma BN. Inhibition of human blood coagulation factor XIa by C1 Inhibitor. Biochemistry. 1988;27:959–963. doi: 10.1021/bi00403a018. [DOI] [PubMed] [Google Scholar]
- 13.Patston PA, Gettins P, Beechem J, Schapira M. Mechanism of serpin action: Evidence that C1 inhibitor functions as a suicide substrate. Biochemistry. 1991;30:8876–8882. doi: 10.1021/bi00100a022. [DOI] [PubMed] [Google Scholar]
- 14.Zhao M, Abdel-Razek T, Sun MF, Gailani D. Characterization of a heparin binding site on the heavy chain of factor XI. J. Biol. Chem. 1998;273:31153–31159. doi: 10.1074/jbc.273.47.31153. [DOI] [PubMed] [Google Scholar]
- 15.Olson ST, Swanson R, Raub-Segall E, Bedsted T, Sadri M, Petitou M, Herault JP, Herbert JM, Björk I. Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Comparison with heparin and low-molecular-weight heparin. Thromb. Haemostas. 2004;92:929–939. doi: 10.1160/TH04-06-0384. [DOI] [PubMed] [Google Scholar]
- 16.Wuillemin WA, Eldering E, Citarella F, de Ruig CP, ten Cate H, Hack CE. Modulation of contact system proteases by glycosaminoglycans. J. Biol. Chem. 1996;271:12913–12918. doi: 10.1074/jbc.271.22.12913. [DOI] [PubMed] [Google Scholar]
- 17.Bos IGA, Hack CE, Abrahams JP. Structural and functional aspects of C1-inhibitor. Immunobiol. 2002;205:518–533. doi: 10.1078/0171-2985-00151. [DOI] [PubMed] [Google Scholar]
- 18.Huntington JA, Olson ST, Fan B, Gettins PGW. Mechanism of heparin activation of antithrombin. Evidence for reactive center loop preinsertion with expulsion upon heparin binding. Biochemistry. 1996;35:8495–8503. doi: 10.1021/bi9604643. [DOI] [PubMed] [Google Scholar]
- 19.Badellino KO, Walsh PN. Localization of a heparin binding site in the catalytic domain of factor XIa. Biochemistry. 2001;40:7569–7580. doi: 10.1021/bi0027433. [DOI] [PubMed] [Google Scholar]
- 20.Ho DH, Badellino K, Baglia FA, Walsh PN. A binding site for heparin in the apple 3 domain of factor XI. J. Biol. Chem. 1998;273:16382–16390. doi: 10.1074/jbc.273.26.16382. [DOI] [PubMed] [Google Scholar]
- 21.Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J. The refined 1.9 Å crystal structure of human α-thrombin: interaction with D-Phe-Pro-Arg chlorometheylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogawa T, Verhamme IM, Sun MF, Bock PE, Gailani D. Exosite-mediated substrate recognition of factor IX by factor XIa: The factor XIa heavy chain is required for initial recognition of factor IX. J. Biol. Chem. 2005;280:23523–23530. doi: 10.1074/jbc.M500894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujikawa K, Chung DW, Hendrickson LE, Davie EW. Amino acid sequence of human factor XI, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry. 1986;25:2417–2424. doi: 10.1021/bi00357a018. [DOI] [PubMed] [Google Scholar]
- 24.Olson ST, Björk I, Sheffer R, Craig PA, Shore JD, Choay J. Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J. Biol. Chem. 1992;267:12528–12538. [PubMed] [Google Scholar]
- 25.Sinha D, Badellino KO, Marcinkiewicz M, Walsh PN. Allosteric modification of factor XIa functional activity upon binding to polyanions. Biochemistry. 2004;43:7593–7600. doi: 10.1021/bi049808c. [DOI] [PubMed] [Google Scholar]
- 26.Rezaie AR. Partial activation of antithrombin without heparin through deletion of a unique sequence on the reactive site loop of the serpin. J. Biol. Chem. 2002;277:1235–1239. doi: 10.1074/jbc.M108544200. [DOI] [PubMed] [Google Scholar]
- 27.Johnson DJD, Li W, Adams TE, Huntington JA. Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 2006;25:2029–2037. doi: 10.1038/sj.emboj.7601089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beinrohr L, Harmat V, Dobo J, Lorincz Z, Gal P, Zavodszky P. C1 inhibitor serpin domain structure reveals the likely mechanism of heparin potentiation and conformational disease. J. Biol. Chem. 2007;282:21100–21109. doi: 10.1074/jbc.M700841200. [DOI] [PubMed] [Google Scholar]
- 29.Yang L, Manithody C, Rezaie AR. Heparin-activated antithrombin interacts with the autolysis of target coagulation proteases. Blood. 2004;104:1753–1759. doi: 10.1182/blood-2004-03-1092. [DOI] [PubMed] [Google Scholar]
- 30.Jin L, Pandey P, Babine RE, Weaver DT, Abdel-Meguid SS, Strickler JE. Mutations of surface residues to promote crystallization of active factor XI as a complex with benzamidine an essential step for the iterative structure-based design factor XI inhibitors. Acta Crystallogr. D. Biol. Crystallogr. 2005;61:1418–1425. doi: 10.1107/S0907444905024340. [DOI] [PubMed] [Google Scholar]
- 31.Yang L, Manithody C, Rezaie AR. Localization of the heparin binding exosite of factor IXa. J. Biol. Chem. 2002;277:50756–50760. doi: 10.1074/jbc.M208485200. [DOI] [PubMed] [Google Scholar]
- 32.Rezaie AR. Identification of basic residues in the heparin-binding exosite of factor Xa critical for heparin and factor Va binding. J. Biol. Chem. 2000;275:3320–3327. doi: 10.1074/jbc.275.5.3320. [DOI] [PubMed] [Google Scholar]
- 33.Sheehan JP, Sadler JE. Molecular mapping of the heparin-binding exosite of thrombin. Proc. Natl. Acad. Sci. (USA) 1994;91:5518–5522. doi: 10.1073/pnas.91.12.5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin J, Pandey P, Babine RE, Gorga JC, Seidl KJ, Gelfand E, Weaver DT, Abdel-Meguid SS, Strickler JE. Crystal structures of the factor XIa catalytic domain in complex with ecotin mutants reveal substrate-like interactions. J. Biol. Chem. 2005;280:4704–4712. doi: 10.1074/jbc.M411309200. [DOI] [PubMed] [Google Scholar]
- 35.Dang QD, Di Cera E. Residue 225 determines the Na+-induced allosteric regulation of catalytic activity in serine proteases. Proc. Natl. Acad. Sci. (USA) 1996;93:10653–10656. doi: 10.1073/pnas.93.20.10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiebe EM, Stafford AR, Fredenburgh JC, Weitz JI. Mechanism of catalysis of inhibition of factor IXa by antithrombin in the presence of heparin or pentasaccharide. J. Biol. Chem. 2003;278:35767–35774. doi: 10.1074/jbc.M304803200. [DOI] [PubMed] [Google Scholar]
- 37.Madison EL, Goldsmith EJ, Gerard RD, Gething M-JH, Sambrook JF, Bassel-Duby RS. Amino acid residues that affect interaction of tissue-type plasminogen activator with plasminogen activator inhibitor 1. Proc. Natl. Acad. Sci. (USA) 1990;87:3530–3533. doi: 10.1073/pnas.87.9.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dementiev A, Petitou M, Herbert JM, Gettins PG. The ternary complex of antithrombin-anhydrothrombin-heparin reveals the basis of inhibitor specificity. Nat. Struct. Mol. Biol. 2004;11:863–867. doi: 10.1038/nsmb810. [DOI] [PubMed] [Google Scholar]