

Abstract
Almost half of adult acute myelogenous leukemia (AML) is normal cytogenetically, and this subgroup shows a remarkable heterogeneity of genetic mutations at the molecular level and an intermediate response to therapy. The finding of recurrent cytogenetic abnormalities has influenced, in a primary way, the understanding and treatment of leukemias. Yet “normal karyotype AML” lacks such obvious abnormalities, but has a variety of prognostically important genetic abnormalities. Thus, the presence of a FLT3-ITD (internal tandem duplication), MLL-PTD (partial tandem duplication), or the increased expression of ERG or EVI1 mRNAs confer a poor prognosis, and an increased risk of relapse. In contrast, the presence of cytoplasmic nucleophosmin or C/EBPA mutations is associated with lower relapse rates and improved survival. Although resistance to treatment is associated with specific mutations, it has also been suggested that the degree to which the leukemia resembles a stem cell in its functional properties provides greater protection from the effects of treatment. Although usually all of the circulating leukemia cells are cleared following treatment, a small residual population of leukemic cells in the bone marrow persists, making this disease hard to eradicate. Increased understanding of the biological consequences of at least some of these mutations in “normal karyotype AML” is leading to more targeted approaches to develop more effective treatments for this disease.
Keywords: AML, FLT3-ITD, MLL-PTD, NPM, BAALC, ERG, C/EBP-α, histone deacetylase, methyltransferase, cytogenetics, stem cell
INTRODUCTION
The evolution of cytogenetics in the latter half of the 20th century has had a tremendous impact on the understanding and treatment of acute myelogenous leukemia (AML). The first cytogenetic abnormalities were identified in the 1960s and the first leukemia cell lines were established during the same period.1–4 The isolation of reverse transcriptase,5–7 and the development of DNA sequencing,8 polymerase chain reaction technology,9 and more recently, fluorescence in situ hybridization (FISH)10 have provided both profound insight and powerful tools for further research. These techniques have already elucidated more than a dozen mutations relevant to our detailed understanding of AML, such as KIT, FLT3 and NRAS mutations.11
We are now in the process of trying to understand the biological and clinical implications of this genetic or molecular heterogeneity. The French-American-British (FAB) classification, proposed by Bennett et al in 197611 (Table 1), is based on morphology and cytochemistry. A few morphologically identifiable acute leukemias are associated with specific cytogenetic abnormalities (eg t(15;17) in acute promyelocytic leukemia; AML, M3), but there is no characteristic morphologic abnormality for the 40% to 45% of AML patients that have a normal karyotype. The prognosis of individuals with normal karyotype AML is intermediate, with a 5-year survival rate reported from 24% to 42%.12–16 Clearly, the interplay between cytogenetics and the biological and clinical expression of the disease is more complex than it appears on the surface.
Table 1.
The FAB classification of AML
| FAB subclassification of AML | ||
|---|---|---|
| M0 | Undifferentiated | ? |
| M1 | Early Myeloid | ? |
| M2 | Late Myeloid | t(8;21) |
| M3 | Promyelocytic | t(15;17) |
| M4 | Myelomonocytic | Inv (16) |
| M5 | Monocytic | t(9;11) |
| M6 | Erythroleukemia | ? |
| M7 | Megakaryocytic | t(1;22), +21 |
Approximately 45% of AML patients have a normal karyotype. For intermediate risk patients, the 5-year-survival rate is 24% to 42%.
The heterogeneity of AML at the molecular level can be demonstrated biologically, providing insight into prognosis. As an example, ERG appears to function as an oncogene17,18; it has been associated with prostate cancer,19–21 22 leukemia in Down’s syndrome/trisomy 21,23,24 and Ewing’s sarcoma.25–27 In AML, the increased expression of ERG is associated with a poor prognosis.28
Response to treatment of AML is definitely associated with cytogenetic subtypes. For example, good prognosis is associated with t(15;17), inv(16), t(16;16) and t(8;21); intermediate prognosis is associated with normal karyotype, -Y; and poor prognosis is associated with monosomy 7 or 5, complex cytogenetics, most 11q23 abnormalities and loss of 17p.29 However, these classifications are not fully predictive of response. One has to ask: what underlies these prognoses? For the last few years, our laboratory and others have focused on defining the stem cell “gene signature” and on examining how the presence of stem-cell-like properties of the leukemia cell can serve as a potential determinant of response to treatment. The question remains as to whether we are curing core binding factor (CBF) leukemias [inv(16), t(16;16), t(8,21)] because they are caused by CBF mutations or because the cellular origins of these leukemias are not stem-cell-like? A similar argument can be made for acute promyelocytic leukemia, which is usually CD34-. Morphologically, the M2, M3 and M4eo AMLs are more differentiated than other AMLs, and the target cell in these leukemias is presumably different than that of the FAB M0 and M1 leukemias. This line of reasoning (that poor prognosis AML is stem-cell-like and good prognosis AML is more progenitor-cell-like) does not hold absolutely, because there are well differentiated leukemias, erythroleukemia (M6) and megakaryocytic leukemia (M7), that are extremely difficult to treat with currently available therapies. Even so, several questions remain: does some fundamental aspect of stem cells determine the success of our treatments, and could that critical aspect be their quiescent nature? Are quiescent cells really the reason why we are not curing more patients with this disease? In solid tumors, the presence of quiescent cells may be more important than whether the cancer arose in a stem cell. One in every 10,000 cells in a solid tumor is said to be a stem cell.30,31 However, chemotherapy for most solid tumors does not eliminate all but the 1 stem cell in 104 cancer cells. Factors other than the presence of cancer stem cells clearly affect our ability to achieve complete remissions in patients with solid tumors.
REGULATION OF HEMATOPOETIC STEM CELL QUIESCENCE
We now know that stem cells reside in niches within the endosteal surface of bone, where they are protected from oxidative damage. From there, stem cells can move towards the vascular niche, enter the circulation, and later return to their niche. Several mechanisms keep the stem cells quiescent as long as they are in their niche. Intrinsic cell cycle regulators like p57 and p21 modulate the quiescence of stem cells, but there are also signals that are received by hematopoietic stem cells residing within the stem cell niche that lead to their quiescence. For example, CD56 is a known marker for poor prognosis in almost all AMLs.32–34 A possible biologic explanation for this is that CD56 may be important for the adherence of some of the leukemic stem cells to the niche, so that the cells can receive pro-survival signals that keep them alive. Perhaps certain leukemias, which begin in the earliest stem cells, and others, which occur in slightly more committed cells but have acquired the ability to behave like a stem cell, are able to evade chemotherapy by residing in the highly protective stem cell niche.
Our laboratory has spent the last decade studying myeloid ELF-1-like factor (MEF), an ETS family member that, like AML1B, can strongly transactivate several promoters. This led us to examine whether MEF interacts functionally or physically with AML1 proteins.35 MEF is located on Xq26, where it binds both AML1 and PML. It has now been shown to be involved in the t(X;21) translocation, where it fuses with ERG.36 MEF null murine hematopoietic stem cells showed a marked increase in quiescence. This was seen in vivo, using bromodeoxyuradine (BrdU) uptake studies. Normally, over a 72 hour period, about 60% of the lineage -, Sca-1+, c-kit+, (LSK) cells incorporate BrdU, whereas MEF-null LSK cells incorporate about one-third as much as is BrdU (22%) indicating that they divide at a much slower rate. Similarly, G0/G1 analyses demonstrated that about twice as many MEF null LSK cells are in G0, compared to wild-type LSK cells.37
This enhanced quiescence is associated with chemotherapy resistance. Thus, dosing wild type mice with 5-fluorouracil (5-FU), results in severe neutropenia, with survival of only 2 of 6 wild type mice. However, MEF knockout mice given the same dose of 5-FU have less neutropenia and all 6 survive 5-FU treatment. Bone marrow examination on day 8 after a high dose of 5-FU demonstrates an empty bone marrow in wild-type, “chemosensitive normal” mice, and a relatively hypercellular bone marrow in the MEF-null mouse. Few stem cells are observed in the marrow of the “chemosensitive normal mouse,” whereas, in the chemoresistant MEF-null mouse, the stem cell niche appears relatively full of hematopoietic stem cells. This suggests that the pathways controlled by MEF (and probably other cell cycle regulators) maintain the leukemic stem cells in the niche, keeping them quiescent and protecting them from chemotherapeutic agents. Activation of these pathways may partially explain the reason why we are not particularly effective in treating most cancers.
MUTATIONS AND PROGNOSIS: C/EBP ALPHA AND FLT3, CYTOPLASMIC NUCLEOPLASMIN, AND BAALC
Mutations that are associated with improved prognosis have been identified. For example, mutations resulting in either dominant negative forms of (C/EBP alpha) or to haplo insufficiency of CCAAT/enhancer-binding protein (C/EBP alpha) function are associated with better than average prognosis in AML.14,38–41 While the complete response rates to chemotherapy are not different, fewer patients relapse, and overall survival is better. In addition to C/EBP-alpha mutations, there are a variety of FLT3 mutations detectable in 30% to 40% of AML patients.40 FLT3 can phosphorylate and inactivate C/EBP alpha (leading to a block in differentiation),42,43 and FLT3 mutations are associated with worse outcome in AML.44–46 FLT3 mutations combined with partial tandem duplication (PTD) of the mixed lineage leukemia gene (MLL) are rare but convey an even worse prognosis.47–49
Why should C/EBP-alpha mutations confer a better than average prognosis in normal karyotype AML? Work on a C/EBP-alpha knockout mouse model, published by Zhang and Tenen et al50 demonstrated profound neutropenia that was unresponsive to G-CSF treatment, and the C/EBP-alpha null cells appear to be blocked (ie a maturation arrest) at the promyelocyte stage. Perhaps leukemias that have C/EBP-alpha mutations are arrested at a late stage in myeloid differentiation, and the better-than-average prognosis is due to the type of cell that harbors this mutation or to effects of the loss of C/EBP-alpha function arresting the cell at this stage of differentiation. Either way, the cell itself would be more susceptible to chemotherapy.
In 2005, Falini and colleagues with the GIMEMA Acute Leukemia Working Party published their work demonstrating a high frequency of nucleophosmin (NPM) mutations in patients with normal-karyotype AML.51 Almost 40% of these patients have NPM1 mutations, which lead to frame shifts that cause expression of a nuclear export signal, resulting in mislocalization of NPM to the cytoplasm. Using immunostaining techniques and monoclonal antibodies against anaplastic lymphoma kinase (ALK) and NPM, these researchers identified a large subgroup of patients with normal karyotype AML and cytoplasmic NPM. These mutations have been associated with female sex, higher white blood count (WBC), increased blast percentage, and low or absent CD34 expression. This suggests that cytoplasmic NPM may also be found in a cell that is at a later stage of differentiation than the more difficult-to-treat leukemias, hence its better prognosis. Among the patients with NPMc, who did not also have a FLT3 mutation, prognosis was generally more favorable; that is, they were more likely to respond to induction therapy and stay in remission.
The brain and acute leukemia cytoplasmic (BAALC) gene encodes a protein with still unidentified homology to any known proteins or functional domains. It is expressed in neuroectoderm-derived tissues and hematopoietic cells. When BAALC is overexpressed in AML with normal cytogenetics, it confers a poor prognosis, especially in the presence of FLT3 mutations. In a study reported by Baldus et al,52 pretreatment blood samples were used to measure BAALC expression in 307 adults with AML and normal cytogenetics. Patients were divided into low- and high- BAALC expression groups. This study showed that high BAALC expression was associated with a higher incidence of relapse and a lower overall survival. BAALC is over-expressed in AML, ALL, and in CML in blast crisis, but not in CLL or CML in chronic phase. As yet, we have no clear explanation for this other than the possibility that BAALC expression is characteristic of stem cells with increased potentiality, ie, a cell in an earlier stage. With respect to FLT3 mutations and BAALC expression, 4 subgroups were identified: BAALC low/FLT3 low (n = 125), BAALC high/FLT3 low (n = 110), BAALC low/FLT3 high (n = 12), and BAALC high/FLT3 high (n = 21). Patients with low-risk FLT3 mutation and low BAALC expression showed the best outcome, whereas those with high-risk FLT3 and high BAALC expression had the worst outcome (Figure 1).52
Figure 1.
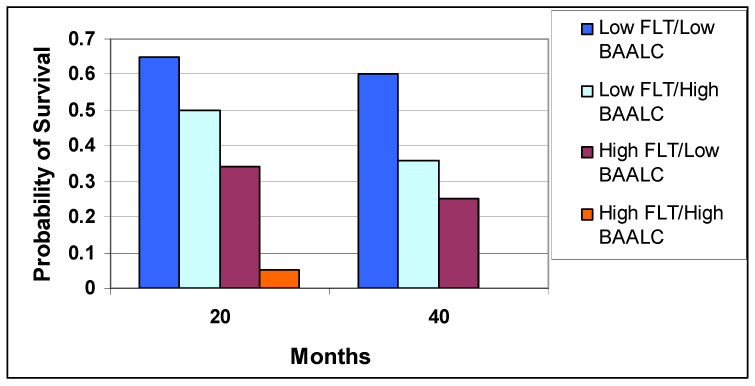
Outcome based on BAALC expression and FLT3 mutation status. The probability of outcome based low- or high-risk FLT3 mutation status and BAALC expression was measured in this cohort of patients. Those with FLT3 low/BAALC low (n-125) had the best outcome, followed by low FLT3/high BAALC (n=110), then high FLT3/low BAALC (n=12), and those with high FLT3/high BAALC (n=21) had the worst outcome.52
Molecular epidemiology is important, but what can it tell the clinician and the patient about therapeutic options? Perhaps poor prognosis mutations portend a poor prognosis despite varying the intensity of treatment. However, the study of BAALC expression suggested that allogenic stem cell transplantation consolidation therapy may be associated with improved long-term outcome for patients with high BAALC expression. Indeed, it is our hope that intensifying the treatment of certain subtypes of normal karyotype AML can lead to improved outcome. However, we not only need to identify these subgroups but also to sufficiently understand the biology involved to target it meaningfully. MLL-PTD is an interesting example of this subtype. Several groups have recently published studies linking MLL-PTD to worse outcomes.53–55 The MLL protein has histone methyltransferase activity, which may be modulated or targeted in the future.56
NEW THERAPIES
Currently, the most promising avenues of research, which take advantage of our increasing understanding of the biologic consequences of mutations in malignant myeloid cells, have focused on inhibiting enzymatic activities, especially tyrosine kinases. However, epigenetic based therapies are showing some success, and combination therapies are being widely explored. Gore and colleagues at Johns Hopkins and our group at MSKCC have reported encouraging phase 1 results using a combination of DNA methyltransferase inhibition followed by histone deacetylase (HDAC) inhibition.57,58 These studies are beginning to address the hypothesis that reversal of aberrant gene silencing can improve remission rates in some patients with normal karyotype AML.57 In vitro work on histone demethylase inhibitors is exploring these same pathways with the idea that the right sequence of inhibitors may be able to restore gene expression, leading to a more differentiated phenotype and better disease control.59 Kinase inhibitors active against FLT3, the mTOR pathway, AKT, and PI3K, among others, are all under investigation.60–64 HDAC inhibitors may overcome the consequences of haploinsufficiency for C/EBP alpha.65,66 Other agents may affect hematopoietic stem cell quiescence, but, so far, our attempts to activate quiescent cells to resume cycling using agents such as GM-CSF or G-CSF have been largely unimpressive.67–69
Immunotherapy in AML goes back to studies of Bacillus Calmette-Guérin (BCG) as an adjuvant to chemotherapy.70 However, this “shotgun” approach failed over time to show clear survival benefit.71 More recently, defining how leukemic cells present antigens, and learning how to maximize that presentation, has led to various approaches intended to allow the individual’s immune system to engage and eliminate leukemic cells.72–74 Additionally, blockade of the export and import of RNA or protein species from the nucleus appears to be a critical factor in translocations in leukemias, through the function of the nuclear pore complex proteins, NUP98, NUP96, and NUP214. Restoring the normal functions of these nuclear pore proteins may be another path to modulating the response to treatment in AML.75,76
SUMMARY AND CONCLUSIONS
Our therapies generally fail to kill all of the malignant cells, and a small population of cells that is unaffected by therapy is responsible for persistence or recurrence of the disease. There is a need to understand both cancer cell quiescence and the cancer initiating cell, which in some cases may be a stem cell. Although much is known about the cytogenetic and genetic mutations in AML and the prognosis related to these mutations, little is known about how to better treat patients with poor prognosis, other than with allogeneic stem cell transplantation. Given that as many as 25% of AML patients with normal cytogenetics do not have any of the mutations discussed above, a better understanding of the molecular mechanisms underlying these subtypes of the disease will hopefully lead to the discovery of more effective ways to target them.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Nowell PC, Hungerford DA. Chromosome studies in human leukemia. II. Chronic granulocytic leukemia. J Natl Cancer Inst. 1961;27:1013–1035. [PubMed] [Google Scholar]
- 2.Hall B. Mongolism and other abnormalities in a family with trisomy 21–22 tendency. Acta Paediatr Suppl. 1963:SUPPL146–SUPPL191. doi: 10.1111/j.1651-2227.1963.tb05520.x. [DOI] [PubMed] [Google Scholar]
- 3.Moore AE, Southam CM, Sternberg SS. Neoplastic changes developing in epithelial cell lines derived from normal persons. Science. 1956;124:127–129. doi: 10.1126/science.124.3212.127-a. [DOI] [PubMed] [Google Scholar]
- 4.Eagle H. The growth requirements of two mammalian cell lines in tissue culture. Trans Assoc Am Physicians. 1955;68:78–81. [PubMed] [Google Scholar]
- 5.Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970;226:1209–1211. doi: 10.1038/2261209a0. [DOI] [PubMed] [Google Scholar]
- 6.Dulbecco R. Oncogenic viruses: the last twelve years. Cold Spring Harb Symp Quant Biol. 1975;39(Pt 1):1–7. doi: 10.1101/sqb.1974.039.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Temin HM, Mizutani S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 1970;226:1211–1213. doi: 10.1038/2261211a0. [DOI] [PubMed] [Google Scholar]
- 8.Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci U S A. 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullis K, Faloona F, Scharf S, et al. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol. 1986;51(Pt 1):263–273. doi: 10.1101/sqb.1986.051.01.032. [DOI] [PubMed] [Google Scholar]
- 10.Pinkel D, Straume T, Gray JW. Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci U S A. 1986;83:2934–2938. doi: 10.1073/pnas.83.9.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 12.Arthur DC, Berger R, Golomb HM, et al. The clinical significance of karyotype in acute myelogenous leukemia. Cancer Genet Cytogenet. 1989;40:203–216. doi: 10.1016/0165-4608(89)90025-3. [DOI] [PubMed] [Google Scholar]
- 13.Machnicki JL, Bloomfield CD. Chromosomal abnormalities in myelodysplastic syndromes and acute myeloid leukemia. Clin Lab Med. 1990;10:755–767. [PubMed] [Google Scholar]
- 14.Marcucci G, Mrozek K, Bloomfield CD. Molecular heterogeneity and prognostic biomarkers in adults with acute myeloid leukemia and normal cytogenetics. Curr Opin Hematol. 2005;12:68–75. doi: 10.1097/01.moh.0000149608.29685.d1. [DOI] [PubMed] [Google Scholar]
- 15.Mrozek K, Heinonen K, de la CA, Bloomfield CD. Clinical significance of cytogenetics in acute myeloid leukemia. Semin Oncol. 1997;24:17–31. [PubMed] [Google Scholar]
- 16.Swansbury GJ, Lawler SD, Alimena G, et al. Long-term survival in acute myelogenous leukemia: a second follow-up of the Fourth International Workshop on Chromosomes in Leukemia. Cancer Genet Cytogenet. 1994;73:1–7. doi: 10.1016/0165-4608(94)90174-0. [DOI] [PubMed] [Google Scholar]
- 17.Reddy ES, Rao VN, Papas TS. The erg gene: a human gene related to the ets oncogene. Proc Natl Acad Sci U S A. 1987;84:6131–6135. doi: 10.1073/pnas.84.17.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hart AH, Corrick CM, Tymms MJ, Hertzog PJ, Kola I. Human ERG is a proto-oncogene with mitogenic and transforming activity. Oncogene. 1995;10:1423–1430. [PubMed] [Google Scholar]
- 19.Petrovics G, Liu A, Shaheduzzaman S, et al. Frequent overexpression of ETS-related gene-1 (ERG1) in prostate cancer transcriptome. Oncogene. 2005;24:3847–3852. doi: 10.1038/sj.onc.1208518. [DOI] [PubMed] [Google Scholar]
- 20.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 21.Rajput AB, Miller MA, De LA, et al. Frequency of the TMPRSS2:ERG gene fusion is increased in moderate to poorly differentiated prostate cancers. J Clin Pathol. 2007 doi: 10.1136/jcp.2006.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demichelis F, Fall K, Perner S, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–4599. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 23.Drabkin HA, Erickson P. Down syndrome and leukemia, an update. Prog Clin Biol Res. 1995;393:169–176. [PubMed] [Google Scholar]
- 24.Papas TS, Watson DK, Sacchi N, et al. ETS family of genes in leukemia and Down syndrome. Am J Med Genet Suppl. 1990;7:251–261. doi: 10.1002/ajmg.1320370751. [DOI] [PubMed] [Google Scholar]
- 25.Sorensen PH, Lessnick SL, Lopez-Terrada D, et al. A second Ewing's sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet. 1994;6:146–151. doi: 10.1038/ng0294-146. [DOI] [PubMed] [Google Scholar]
- 26.Zucman J, Melot T, Desmaze C, et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumours. EMBO J. 1993;12:4481–4487. doi: 10.1002/j.1460-2075.1993.tb06137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giovannini M, Biegel JA, Serra M, et al. EWS-erg and EWS-Fli1 fusion transcripts in Ewing's sarcoma and primitive neuroectodermal tumors with variant translocations. J Clin Invest. 1994;94:489–496. doi: 10.1172/JCI117360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcucci G, Baldus CD, Ruppert AS, et al. Overexpression of the ETS-related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype: a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23:9234–9242. doi: 10.1200/JCO.2005.03.6137. [DOI] [PubMed] [Google Scholar]
- 29.Bloomfield CD, Lawrence D, Byrd JC, et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998;58:4173–4179. [PubMed] [Google Scholar]
- 30.Steel G. Growth kinetics of tumors. Oxford: Clarendon Press; 1977. [Google Scholar]
- 31.Guo W, Lasky JL, III, Wu H. Cancer stem cells. Pediatr Res. 2006;59:59R–64R. doi: 10.1203/01.pdr.0000203592.04530.06. [DOI] [PubMed] [Google Scholar]
- 32.Chang H, Salma F, Yi QL, et al. Prognostic relevance of immunophenotyping in 379 patients with acute myeloid leukemia. Leuk Res. 2004;28:43–48. doi: 10.1016/s0145-2126(03)00180-2. [DOI] [PubMed] [Google Scholar]
- 33.Raspadori D, Damiani D, Lenoci M, et al. CD56 antigenic expression in acute myeloid leukemia identifies patients with poor clinical prognosis. Leukemia. 2001;15:1161–1164. doi: 10.1038/sj.leu.2402174. [DOI] [PubMed] [Google Scholar]
- 34.Mann KP, DeCastro CM, Liu J, et al. Neural cell adhesion molecule (CD56)-positive acute myelogenous leukemia and myelodysplastic and myeloproliferative syndromes. Am J Clin Pathol. 1997;107:653–660. doi: 10.1093/ajcp/107.6.653. [DOI] [PubMed] [Google Scholar]
- 35.Mao S, Frank RC, Zhang J, Miyazaki Y, Nimer SD. Functional and physical interactions between AML1 proteins and an ETS protein, MEF: implications for the pathogenesis of t(8;21)-positive leukemias. Mol Cell Biol. 1999;19:3635–3644. doi: 10.1128/mcb.19.5.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore SD, Offor O, Ferry JA, et al. ELF4 is fused to ERG in a case of acute myeloid leukemia with a t(X;21)(q25–26;q22) Leuk Res. 2006;30:1037–1042. doi: 10.1016/j.leukres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 37.Lacorazza HD, Yamada T, Liu Y, et al. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell. 2006;9:175–187. doi: 10.1016/j.ccr.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 38.Barjesteh van Waalwijk van Doorn-Khosrovani, Erpelinck C, Meijer J, et al. Biallelic mutations in the CEBPA gene and low CEBPA expression levels as prognostic markers in intermediate-risk AML. Hematol J. 2003;4:31–40. doi: 10.1038/sj.thj.6200216. [DOI] [PubMed] [Google Scholar]
- 39.Preudhomme C, Sagot C, Boissel N, et al. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA) Blood. 2002;100:2717–2723. doi: 10.1182/blood-2002-03-0990. [DOI] [PubMed] [Google Scholar]
- 40.Frohling S, Schlenk RF, Stolze I, et al. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol. 2004;22:624–633. doi: 10.1200/JCO.2004.06.060. [DOI] [PubMed] [Google Scholar]
- 41.Bienz M, Ludwig M, Leibundgut EO, et al. Risk assessment in patients with acute myeloid leukemia and a normal karyotype. Clin Cancer Res. 2005;11:1416–1424. doi: 10.1158/1078-0432.CCR-04-1552. Erratum in: Clin Cancer Res. 2005; 11(15):5659) [DOI] [PubMed] [Google Scholar]
- 42.Zheng R, Friedman AD, Levis M, et al. Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPalpha expression. Blood. 2004;103:1883–1890. doi: 10.1182/blood-2003-06-1978. [DOI] [PubMed] [Google Scholar]
- 43.Radomska HS, Basseres DS, Zheng R, et al. Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J Exp Med. 2006;203:371–381. doi: 10.1084/jem.20052242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–1759. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- 45.Meshinchi S, Woods WG, Stirewalt DL, et al. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001;97:89–94. doi: 10.1182/blood.v97.1.89. [DOI] [PubMed] [Google Scholar]
- 46.Abu-Duhier FM, Goodeve AC, Wilson GA, et al. FLT3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group. Br J Haematol. 2000;111:190–195. doi: 10.1046/j.1365-2141.2000.02317.x. [DOI] [PubMed] [Google Scholar]
- 47.Steudel C, Wermke M, Schaich M, et al. Comparative analysis of MLL partial tandem duplication and FLT3 internal tandem duplication mutations in 956 adult patients with acute myeloid leukemia. Genes Chromosomes Cancer. 2003;37:237–251. doi: 10.1002/gcc.10219. [DOI] [PubMed] [Google Scholar]
- 48.Kuchenbauer F, Schnittger S, Look T, et al. Identification of additional cytogenetic and molecular genetic abnormalities in acute myeloid leukaemia with t(8;21)/AML1-ETO. Br J Haematol. 2006;134:616–619. doi: 10.1111/j.1365-2141.2006.06229.x. [DOI] [PubMed] [Google Scholar]
- 49.Kuchenbauer F, Kern W, Schoch C, et al. Detailed analysis of FLT3 expression levels in acute myeloid leukemia. Haematologica. 2005;90:1617–1625. [PubMed] [Google Scholar]
- 50.Zhang DE, Zhang P, Wang ND, et al. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci U S A. 1997;94:569–574. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(7):254–266. doi: 10.1056/NEJMoa041974. Erratum in: N Engl J Med. 2005; 352(7):740. [DOI] [PubMed] [Google Scholar]
- 52.Baldus CD, Thiede C, Soucek S, et al. BAALC expression and FLT3 internal tandem duplication mutations in acute myeloid leukemia patients with normal cytogenetics: prognostic implications. J Clin Oncol. 2006;24:790–797. doi: 10.1200/JCO.2005.01.6253. [DOI] [PubMed] [Google Scholar]
- 53.Dohner K, Tobis K, Ulrich R, et al. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol. 2002;20:3254–3261. doi: 10.1200/JCO.2002.09.088. [DOI] [PubMed] [Google Scholar]
- 54.Basecke J, Whelan JT, Griesinger F, Bertrand FE. The MLL partial tandem duplication in acute myeloid leukaemia. Br J Haematol. 2006;135:438–449. doi: 10.1111/j.1365-2141.2006.06301.x. [DOI] [PubMed] [Google Scholar]
- 55.Whitman SP, Ruppert AS, Marcucci G, et al. Long-term disease-free survivors with cytogenetically normal acute myeloid leukemia and MLL partial tandem duplication: a Cancer and Leukemia Group B study. Blood. 2007;109:5164–5167. doi: 10.1182/blood-2007-01-069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitman SP, Liu S, Vukosavljevic T, et al. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood. 2005;106:345–352. doi: 10.1182/blood-2005-01-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gore SD, Baylin S, Sugar E, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66:6361–6369. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 58.Maslak P, Chanel S, Camacho LH, et al. Pilot study of combination transcriptional modulation therapy with sodium phenylbutyrate and 5-azacytidine in patients with acute myeloid leukemia or myelodysplastic syndrome. Leukemia. 2006;20:212–217. doi: 10.1038/sj.leu.2404050. [DOI] [PubMed] [Google Scholar]
- 59.Lee MG, Wynder C, Bochar DA, et al. Functional interplay between histone demethylase and deacetylase enzymes. Mol Cell Biol. 2006;26:6395–6402. doi: 10.1128/MCB.00723-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tse KF, Novelli E, Civin CI, Bohmer FD, Small D. Inhibition of FLT3-mediated transformation by use of a tyrosine kinase inhibitor. Leukemia. 2001;15:1001–1010. doi: 10.1038/sj.leu.2402199. [DOI] [PubMed] [Google Scholar]
- 61.Zeng Z, Sarbassov dD, Samudio IJ, et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109:3509–3512. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeng Z, Samudio IJ, Zhang W, et al. Simultaneous inhibition of PDK1/AKT and Fms-like tyrosine kinase 3 signaling by a small-molecule KP372-1 induces mitochondrial dysfunction and apoptosis in acute myelogenous leukemia. Cancer Res. 2006;66:3737–3746. doi: 10.1158/0008-5472.CAN-05-1278. [DOI] [PubMed] [Google Scholar]
- 63.Cammenga J, Horn S, Bergholz U, et al. Extracellular KIT receptor mutants, commonly found in core binding factor AML, are constitutively active and respond to imatinib mesylate. Blood. 2005;106:3958–3961. doi: 10.1182/blood-2005-02-0583. [DOI] [PubMed] [Google Scholar]
- 64.Mesa RA, Loegering D, Powell HL, et al. Heat shock protein 90 inhibition sensitizes acute myelogenous leukemia cells to cytarabine. Blood. 2005;106:318–327. doi: 10.1182/blood-2004-09-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang WH, Srihari R, Day RN, Schaufele F. CCAAT/enhancer-binding protein alpha alters histone H3 acetylation at large subnuclear domains. J Biol Chem. 2001;276:40373–40376. doi: 10.1074/jbc.C100505200. [DOI] [PubMed] [Google Scholar]
- 66.Desilets A, Gheorghiu I, Yu SJ, Seidman EG, Asselin C. Inhibition by deacetylase inhibitors of IL-1-dependent induction of haptoglobin involves CCAAT/Enhancer-binding protein isoforms in intestinal epithelial cells. Biochem Biophys Res Commun. 2000;276:673–679. doi: 10.1006/bbrc.2000.3531. [DOI] [PubMed] [Google Scholar]
- 67.Matsui WH, Gladstone DE, Vala MS, et al. The role of growth factors in the activity of pharmacological differentiation agents. Cell Growth Differ. 2002;13:275–283. [PubMed] [Google Scholar]
- 68.Laver J, Shearer P, Krance R, et al. A pilot study of continuous infusion Ara-C in combination with rhG-CSF in relapsed childhood acute myeloid leukemia. Leuk Lymphoma. 1997;26:589–593. doi: 10.3109/10428199709050894. [DOI] [PubMed] [Google Scholar]
- 69.Budel LM, Touw IP, Delwel R, Lowenberg B. Granulocyte colony-stimulating factor receptors in human acute myelocytic leukemia. Blood. 1989;74:2668–2673. [PubMed] [Google Scholar]
- 70.Powles RL, Russell J, Lister TA, et al. Immunotherapy for acute myelogenous leukaemia: a controlled clinical study 2 1/2 years after entry of the last patient. Br J Cancer. 1977;35:265–272. doi: 10.1038/bjc.1977.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Foon KA, Smalley RV, Riggs CW, Gale RP. The role of immunotherapy in acute myelogenous leukemia. Arch Intern Med. 1983;143:1726–1731. [PubMed] [Google Scholar]
- 72.Greiner J, Dohner H, Schmitt M. Cancer vaccines for patients with acute myeloid leukemia--definition of leukemia-associated antigens and current clinical protocols targeting these antigens. Haematologica. 2006;91:1653–1661. [PubMed] [Google Scholar]
- 73.Stripecke R, Levine AM, Pullarkat V, Cardoso AA. Immunotherapy with acute leukemia cells modified into antigen-presenting cells: ex vivo culture and gene transfer methods. Leukemia. 2002;16:1974–1983. doi: 10.1038/sj.leu.2402701. [DOI] [PubMed] [Google Scholar]
- 74.Galea-Lauri J, Darling D, Mufti G, Harrison P, Farzaneh F. Eliciting cytotoxic T lymphocytes against acute myeloid leukemia-derived antigens: evaluation of dendritic cellleukemia cell hybrids and other antigen-loading strategies for dendritic cell-based vaccination. Cancer Immunol Immunother. 2002;51:299–310. doi: 10.1007/s00262-002-0284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Enninga J, Levy DE, Blobel G, Fontoura BM. Role of nucleoporin induction in releasing an mRNA nuclear export block. Science. 2002;295:1523–1525. doi: 10.1126/science.1067861. [DOI] [PubMed] [Google Scholar]
- 76.Rosenblum JS, Blobel G. Autoproteolysis in nucleoporin biogenesis. Proc Natl Acad Sci U S A. 1999;96:11370–11375. doi: 10.1073/pnas.96.20.11370. [DOI] [PMC free article] [PubMed] [Google Scholar]