

Abstract
Pancreatic cancer is one of the deadliest diseases largely due to difficulty in early diagnosis and the lack of effective treatments. KRAS is mutated in more than 90% of pancreatic cancer patients, and oncogenic KRAS contributes to pancreatic cancer tumorigenesis and progression. In this report, using an oncogenic KRASV12-based pancreatic cancer cell model, we developed a chemical genetic screen to identify small chemical inhibitors that selectively target pancreatic cancer cells with gain-of-function KRAS mutation. After screening ∼3,200 compounds, we identified one compound that showed selective synthetic lethality against the KRASV12 transformed human pancreatic ductal epithelial cell over its isogenic parental cell line. These selective KRASV12-synthetic lethal compounds may serve as leads for subsequent development of clinically-effective treatments for pancreatic cancer.
Keywords: High-through-put screening, HTS, Pancreatic Cancer, Pancreatic Ductal Adenocarcinoma, Transformation, RAS, Chemical Genetic Screening, Synthetic Lethal Screening, Cancer Inhibitor
2. Introduction
Pancreatic cancer (PC) is the fourth-leading cause of cancer-related death in the US, with a 5-year survival rate of less than 4% (16;17). PC is resistant to most forms of treatments such as chemotherapy, radiation, and combination therapies that are effective in other tumors. The refractory nature of this disease clearly underscores the need for novel drug discovery research that specifically targets the genetic makeup of pancreatic ductal adenocarcinoma (PDA).
Extensive histopathological and genetic studies have led to a PDA tumor progression model in which the pancreatic ductal epithelium progresses from normal to increased grades of pancreatic intraepithelial neoplasias (PanINs), to invasive adenocarcinoma (14). In parallel with progressive stages of morphological changes is sequential accumulation of genetic alterations in KRAS and tumor suppressors INK4A, TP53 and SMAD4/DPC4. Activating KRAS mutations represent one of the earliest genetic changes associated with the transformation of normal ductal epithelium. KRAS mutations have been detected in pancreatic duct lesions with minimal cytological and architectural atypia, and, occasionally, in the histologically normal pancreas (4;21;24;25;27). The frequency of KRAS mutations correlates with disease progression, reaching almost 100% in PDA. Targeted endogenous expression of an oncogenic KRAS allele in the mouse pancreas is sufficient to drive the development of PanINs, and subsequently, at low frequency, the progression to both locally invasive adenocarcinoma and metastatic disease with sites of spread exactly as found in human pancreatic cancer (1;12;13). These observations suggest that KRAS plays an essential role in the initiation, progression and maintenance of PDA.
Because of its critical importance in PDA development, oncogenic KRAS represents a good target for therapeutic intervention. Consistent with this notion, siRNA specifically targeting oncogenic KRAS allele induces growth inhibition and apoptosis in pancreatic cancer cell lines harboring KRAS mutation. However, small molecules that inhibit RAS activation by targeting its posttranscriptional modification have so far yielded little success as these compounds also affect cellular targets/pathways other than RAS. On the other hand, novel cell-based screens that target the Achilles' heel of PC (the dependence of PC cell's survival on oncogenic RAS) may lead to the identification of effective inhibitors for PC. Recently, a chemical genetic screening strategy has been developed that involves searching for synthetic lethal antitumor agents that selectively kill tumor cells with specific genotypes such as, the presence of a specific oncoprotein or the loss of a specific tumor suppressor (6;20).
In this report, we developed a chemical genetic screen using an oncogenic KRAS-based human pancreatic ductal epithelia (HPDE) cancer model. This cell-based assay allowed us to search through existing libraries of small chemical compounds to identify mechanism-based inhibitors that show selective lethality in the presence of oncogenic KRAS alleles. Those compounds with KRASV12-selective lethality may serve as leads for subsequent development of clinically-effective drugs with a favorable therapeutic index for PC.
3. Materials and Methods
3.1. Cell lines
Primary human pancreatic ductal epithelial cell (HPDE-c7) immortalized by E6/E7 genes of human papilloma virus (HPV)-16 virus was a gift from Dr. Ming S. Tsao. Stable HPDE-c7-KRASV12 cell line was generated from HPDE-c7 by the addition of oncogene KRASV12 using retroviral vector (18). Parental HPDE-E6E7c7 and HPDE-c7-KRASV12 cells are maintained in keratinocyte-SFM medium supplemented by bovine pituitary extract and epidermal growth factor (Gibco-BRL) at 37 °C and 5% CO2.
3.2. Compound libraries
Small chemical compounds from the Diversity set (1990 compounds), Mechanistic set (879 compounds), Challenge set (57 compounds), and Natural Product set (235 compounds) were obtained from the Open Chemical Repository of National Cancer Institute Developmental Therapeutics Program (DTP). Compounds were supplied in DMSO in 96-well polypropylene plates and stored at -80°C. Replica daughter plates were generated by diluting original stock plates 10-fold in DMSO and used for screening.
3.3. Screening
Assay plates were prepared by seeding cells in 96-well plates (4000 cells/well in 100 μl) using a repetitive dispenser. Columns 2-11 were treated with compounds from a daughter library plate. The final compound concentration in assay plates was 10 μM. Columns 1 and 12 were treated with vehicle and 1 μM Taxol as negative and positive controls, respectively. The assay plates were incubated for 24-48 hrs at 37°C in a humidified incubator containing 5% CO2 and processed for cell viability assay, as described below.
3.4. Alamar Blue cell viability assay
A 100 μl of the cell suspension was seeded per well in duplicates in 96-well microtiter plates (Corning, NY) and treated with compounds. An aliquot of 10 μl of Alamar Blue was added to each well and the plates were incubated for another 4 hrs. Following the incubation, fluorescence intensity was monitored using a SpectraMax M2 microplate reader (Molecular Devices) with excitation and emission wavelengths set at 530 and 590 nm, respectively.
3.5. Data Analysis
Mean relative fluorescence units for untreated cells (RFUuntreated cell) were calculated by averaging columns 1 (wells treated with vehicle). Percentage inhibition of each well was calculated as ((1 − RFU/RFUuntreated cell) ·100). Compounds causing at least 50% inhibition of Alamar Blue signal in the primary screen will be tested for selectivity toward KRAS transformed HPDE cells using HPDE-c7-KRASV12 and parental HPDE-c7 cells in parallel in 2-fold concentration dilution series to determine the dose response curves. The quality of the Alamar Blue screen assay was determined by the Z' score as described previously (31).
4. Results
4.1. Oncogenic transformation of HPDE by KRASV12
To target oncogenic KRAS in pancreatic cancer cells, we established an oncogenic KRAS-based human PC model using an immortalized primary HPDE cell line, HPDE-c7. This well-characterized cell line is a near diploid HPDE cell line originally derived from a normal pancreas. While immortalized by E6/E7 genes of human papilloma (HPV)-16 virus, HPDE-c7 is non-tumorigenic and incapable of inducing tumor growth in nude mice (26; 27). Stable expression of KRASV12 in HPDE-c7 cells, using a retroviral expression vector, led to transformation of the cell line. The resultant cells, HPDE-c7-KRASV12, expressed increased levels of total RAS protein, showed high RAS-GTP activity (Figure 1A), and were grown anchorage-independently in soft agar (Figure 1B). In addition, expression of KRASV12 in HPDE-c7 cells also led to an increased activation of its downstream effectors, such as MAPK and AKT. The phospho-MAPK and phospho-AKT levels were enhanced in the HPDE-c7-KRASV12 cells compared with the parental cells (Figure 1C). These observations are in complete agreement with results obtained from an independently established KRAS human PC model using the same HPDE-c7 parental cell line (28).
Figure. 1.
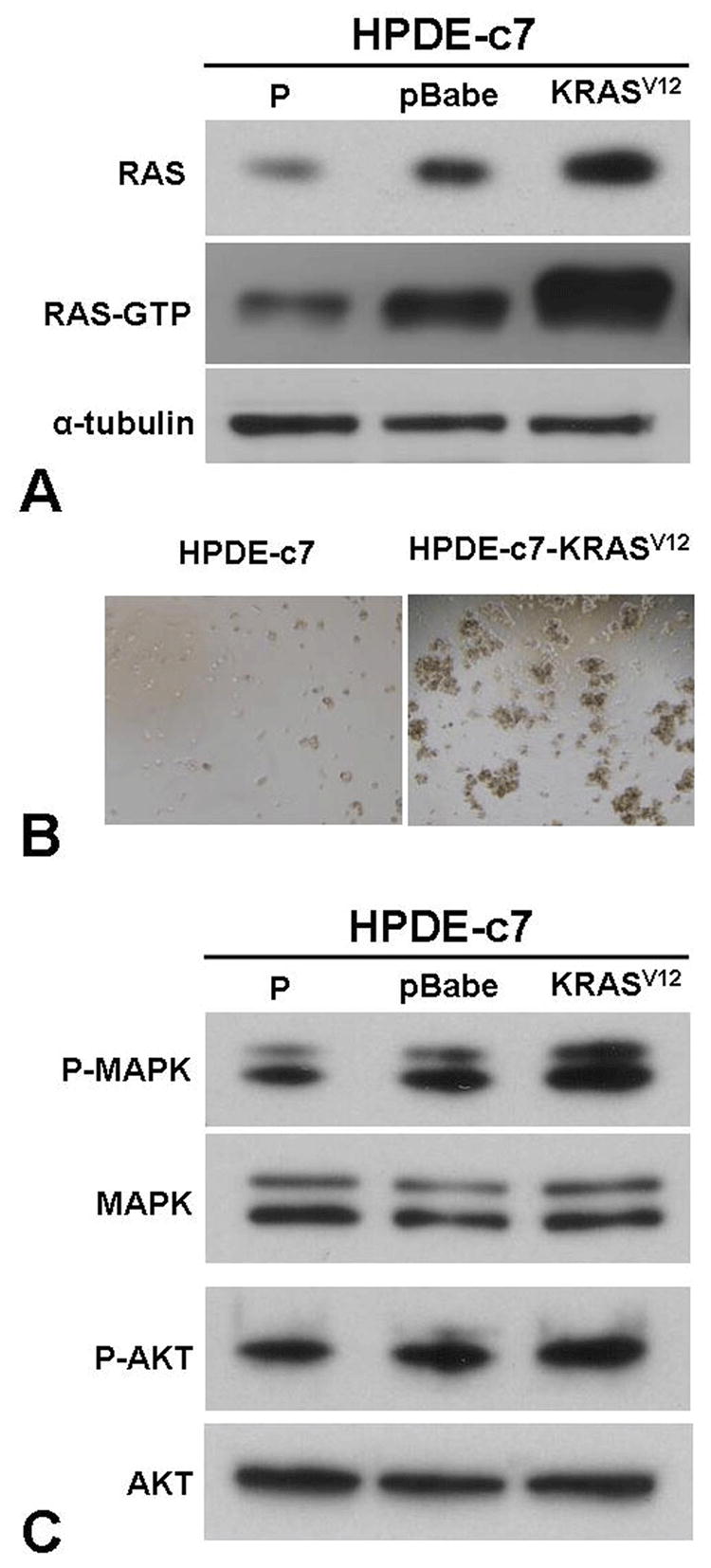
Oncogenic transformation of HPDE-c7 cells induced by oncogenic KRASV12. (A) Total cellular RAS and GTP-bound RAS levels in HPDE-c7 (P), HPDE-c7-pBabe (pBabe) and HPDE-c7-KRASV12 (KRASV12) cells measured by immunoblotting and RAS-GTP pull-down assay, respectively. (B) The in vitro tumorigeneicity of HPDE-c7 and HPDE-c7-KRASV12 cells as measured by soft agar colony formation assay. (C) Levels of phosphorylated MAPK (P-MAPK) and AKT (P-AKT) in HPDE-c7 (P), HPDE-c7-pBabe (pBabe) and HPDE-c7-KRASV12 (KRASV12) cells as monitored by immunoblotting analyses using anti phospho-MAPK and anti phospho-AKT antibodies.
4.2. Development of a cytotoxic assay suitable for high-throughput screen
We selected Alamar Blue-based cytotoxicity assay as our primary assay because Alamar Blue is a non-toxic metabolic indicator of viable cells that becomes fluorescent upon mitochondrial reduction and the assay does not involve washing steps. It is, therefore, easier to adapt to a high-throughput format for handling large volume screening (26). To establish the optimal assay conditions, we performed the assay under various cell-plating densities and incubation times. As shown in Figure 2, the assay exhibited wide dynamic ranges for both cell plating density and incubation time. The relative fluorescence readout displayed excellent linear dependence on cell density (0.3 to 22.5×103 per well) and time up to 6 hrs. Based on these results, optimal assay conditions were established using 4000 cells/well and 4 hrs incubation time for subsequent screenings. DMSO titration studies suggest that our cell viability assay can tolerate up to 0.5% of DMSO. The robustness and reproducibility of the assay were tested using 0.1% DMSO and Doxorubicin (10 μM) as vehicle and positive controls. An average Z' score of 0.79 was obtained from measurements across the entire plate, which demonstrated robustness of this assay. Similar results were obtained with measurements at various times on different machines, suggesting minimal day-to-day and machine-to-machine variations for our assay. This assay was also adapted to a 384-well format at the San Diego Chemical genomic Center, and an average Z' score of 0.75 was obtained. When our assay was tested using known cytotoxic compounds, such as Doxorubicin or Taxol, reproducible and dose-dependent responses were observed for both HPDE-c7 and HPDE-c7-KRASV12 cells.
Figure. 2.

Time and cell-density dependence of Alamar Blue cell viability assay. Relative Alamar Blue fluorescence signal of HPDE cells at various plating densities as a function of incubation time after the addition of Alamar Blue reagent.
4.3. Identification of synthetic lethal compounds selectively against mutant KRAS expressed cells
Using the Alamar Blue based cytotoxic assay, we then carried out a median-throughput chemical screen to identify compounds that selectively kill KRASV12 transformed cells. The process consists of a three-step screening. Initially, compounds were evaluated in the HPDE-c7-KRASV12 cell line at a single concentration of 10 μM. After screening four compound libraries, the NCI diversity set (1,990 compounds), Natural product set (235 compounds), Mechanistic set (879 compounds), and Challenge set (57 compounds), we identified a total of 580 compounds that showed >50% growth inhibition activity against the HPDE-c7-KRASV12 cells at 10 μM (Figure 3). These compounds were then subjected to the second-step of selectivity screen, in which each compound was tested against both HPDE-c7 and HPDE-c7-KRASV12 cell lines in parallel at four different concentrations: 20, 10, 5 and 2.5 μM (Figure 4A). If a specific compound completely inhibited the growth/survival of both cell lines at 2.5 μM, then further dilutions at lower testing concentrations were used (Figure 4B). 24 compounds that inhibited the HPDE-c7-KRASV12 cells 20% more than the control cells at two or more of the four concentrations were selected. These 24 compounds were further evaluated at various concentrations in triplicate to determine their IC50 against HPDE-c7 and HPDE-c7-KRASV12 cell lines, respectively. IC50 values of each compound were determined and used to calculate the selectivity index (IC50 of HPDE-c7 cells/IC50 of HPDE-c7-KRASV12 cells). One compound, SLI501, with a selectivity index >2 was identified. The activity profile of SLI501 in HPDE-c7 and HPDE-c7-KRASV12 cell lines was shown in Figure 5. SLI501 has an IC50 value of 3.6 μM in HPDE- c7-KRASV12 cells and an IC50 value of 9.0 μM in HPDE-c7 cells.
Figure. 3.

Cytotoxic effects of all compounds on HPDE-c7KRASV12 cells. HPDE-c7-KRASV12 cells were treated with individual compounds at 10 μM for 48 hrs. Cell viability was measured using Alamar Blue assay and normalized to the vehicle control treatment.
Figure. 4.
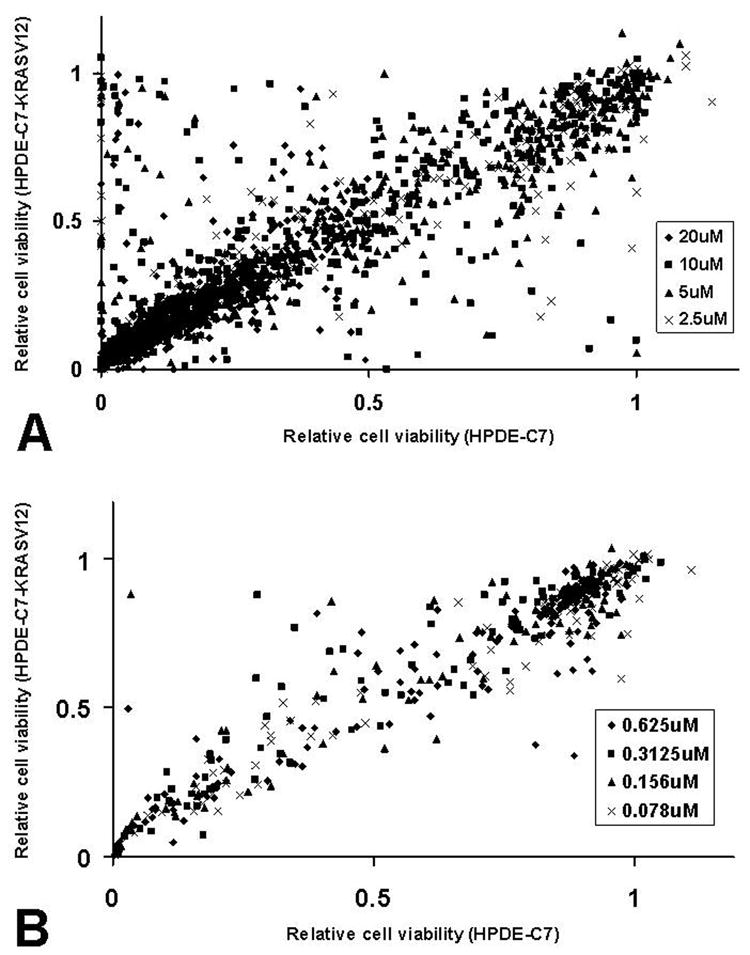
Synthetic lethal screening using HPDE-c7 and HPDE-c7-KRASV12 cells. HPDE-c7 and HPDE-c7-KRASV12 cells were treated with individual compounds at various high (A) or low (B) concentrations in parallel. Cell viability was determined by Alamar Blue assay and normalized to the vehicle control treatment. Data were plotted as relative cell viability of HPDE-c7-KRASV12 vs relative cell viability of HPDE-c7.
Figure. 5.

Cell viability of HPDE-c7 and HPDE-c7-KRASV12 cells as a function of SLI501 concentrations. HPDE-c7 (closed circle) and HPDE-c7-KRASV12 (open circle) cells were treated with various concentrations of compound SLI501 for 48 hrs. Cell viability was determined by Alamar Blue assay and normalized to the vehicle control treatment. Insets: Cell images of HPDE-c7 and HPDE-c7-KRASV12 cells after treatment with 5.0 μM of SLI501 for 48 hrs.
5. Discussion
Although PC is now a well-characterized neoplasm at the genetic level, the prognosis of PC patients is still very poor. This is largely due to the aggressive nature of this disease and the lack of effective therapeutic agents. The refractory nature of PC clearly warrants the need for novel drug discovery research that specifically targets PDA. Molecularly-targeted drug discovery has recently matured as an effective approach for developing cancer therapeutics. This approach directly targets the genetic makeup that is essential for the oncogenic transformation in cancer, but not for normal cells. Targeted therapeutics should be more effective and tolerated with minimal on-target toxicity when compared to traditional cytotoxic compounds. Oncogenes or tumor suppressor genes identified in cancers can be chosen as targets if they prove to be critical for cancer development and survival (3;28). Both structure-based rational design and mechanism-based screening were employed to develop drug candidates (2;3;15;29). One of the most successful examples of molecularly-targeted anticancer therapeutics is Gleevec, an effective, FDA-approved treatment of chronic myelogenous leukemia (CML) (5). Gleevec inhibits the breakpoint cluster region-abelsen kinase (BCR-ABL) oncogene which is frequently found in CML and is a major driver for CML initiation and progression (7).
One important factor that contributes to the success of Gleevec is our deep understanding of the genetic alternations responsible for the formation of CML. In theory, successful molecularly-targeted drug discovery should be possible for cancers with well-defined genetic alterations such as oncogenic KRAS in PC. One major challenge is to design mechanism-based HTS assays that are tailored for targeting the genetic alteration in a specific cancer. Two recent scientific advances in genetically-defined human cancer cell models and synthetic lethal screening (SLS) using small chemical libraries have significantly facilitated this process. Oncogenic transformation of human primary cells using defined genetic elements allows for the creation of human cancer cell models that recapitulate the genetic and molecular characteristic of human cancers in a tissue-specific manner (10). SLS is a method originally used to search mutations in a second gene that exert lethality to a specific mutation in a model organism such as yeast (8). Adaptation of SLS using small chemicals in place of mutation has recently emerged as a powerful drug discovery tool in the post-genomic era (6;9;11;19;28). A chemical genetic screening coupling genetically-defined cancer models and SLS provides a general approach for screening mechanism-based cancer inhibitors that are selective for specific targets (6;28).
Our genetic defined KRAS-based PC cell model was created from immortalized HPDE cells (18). Using this KRAS-based PC model, we developed a cell-based HTS assay and uncovered a small chemical compound that selectively inhibits the growth/survival of oncogenic KRAS expressing HPDE cells. Because of the nature of SLS, synthetic lethal compounds may not directly affect the activity of the particular oncogene involved; rather they render cells expressing the oncogene at disadvantage for growth/survival. For example, one SLS has led to the identification of a quinazoline analogue, Erastin that preferentially kills transformed human foreskin fibroblast cells expressing SV40 small T and the RASV12 oncoproteins (6). A recent study suggests that Erastin acts through mitochondrial voltage-dependent anion channels (VDACs) and alters the permeability of the outer mitochondrial membrane, causing the generation of oxidative species and subsequent death through an oxidative, non-apoptotic mechanism in cells harbouring oncogenic RAS (30).
The mechanism of action of the compound identified from our SLS is not known. We are in active pursuit of the identity of the molecular target(s) of this compound. In some cases, the protein target of a compound can be identified through affinity chromatography using the compound coupled to a solid phase matrix and mass spectrometry (22;30). Analysis of gene expression profiles using cDNA microarray between the isogenic cell lines after the treatment with the compounds could also provide some clues about the perturbation of signaling pathways that might be responsible for the selectivity (23).
In summary, Our oncogenic KRAS-based PC model allows us to search through existing libraries of small chemical compounds to identify mechanism-based inhibitors with selective lethality in the presence of oncogenic KRAS, the most frequently mutated allele in PC. Those compounds with KRASV12-selective lethality may serve as leads for subsequent development of clinically effective drugs with a favorable therapeutic index for PC.
Acknowledgments
The authors would like to thank Ms. Betty Redd for assistant in manuscript preparation. X. Cheng was supported by National Institute of Health Grant GM066170 and MH076387. Z.J was supported by a training fellowship from the Pharmacoinformatics Training Program of the Keck Center of the Gulf Coast Consortia (NIH Grant No. 1 T90 DK070109-01 and 1 R90 DK071505-01). P.L. Lory and S.R. Gilbertson were supported by the Robert A. Welch Foundation. Y. Chen was supported by the “111 Project” from the Ministry of Education of China and the State Administration of Foreign Expert Affairs of China (No. 111-2-07).
Abbreviations
- HPDE
human pancreatic ductal epithelia
- HTS
High-through-put screening
- PanINs
pancreatic intraepithelial neoplasias
- PC
pancreatic cancer
- PDA
pancreatic ductal adenocarcinoma
- SLS
synthetic lethal screening
Footnotes
Publisher's Disclaimer: This is an, un-copyedited, author manuscript that has been accepted for publication in the Frontiers in Bioscience”. Cite this article as appearing in the Journal of Frontiers in Bioscience. Full citation can be found by searching the Frontiers in Bioscience (http://bioscience.org/search/authors/htm/search.htm) following publication and at PubMed (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed) following indexing. This article may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright holder, the Frontiers in Bioscience. From the time of acceptance following peer review, the full final copy edited article of this manuscript will be made available at http://www.bioscience.org/. The Frontiers in Bioscience disclaims any responsibility or liability for errors or omissions in this version of the un-copyedited manuscript or in any version derived from it by the National Institutes of Health or other parties.
References
- 1.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bevan P, Ryder H, Shaw I. Identifying small-molecule lead compounds: the screening approach to drug discovery. Trends Biotechnol. 1995;13:115–121. doi: 10.1016/S0167-7799(00)88916-7. [DOI] [PubMed] [Google Scholar]
- 3.Blundell TL. Structure-based drug design. Nature. 1996;384:23–26. doi: 10.1038/384023a0. [DOI] [PubMed] [Google Scholar]
- 4.Caldas C, Hahn SA, Hruban RH, Redston MS, Yeo CJ, Kern SE. Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res. 1994;54:3568–3573. [PubMed] [Google Scholar]
- 5.Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- 6.Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 7.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 8.Forsburg SL. The art and design of genetic screens: yeast. Nat Rev Genet. 2001;2:659–668. doi: 10.1038/35088500. [DOI] [PubMed] [Google Scholar]
- 9.Friend SH, Oliff A. Emerging uses for genomic information in drug discovery. N Engl J Med. 1998;338:125–126. doi: 10.1056/NEJM199801083380211. [DOI] [PubMed] [Google Scholar]
- 10.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 11.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–1068. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 12.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 13.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 14.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- 15.Hurley LH. DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- 16.Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell. 2002;2:25–28. doi: 10.1016/s1535-6108(02)00093-4. [DOI] [PubMed] [Google Scholar]
- 17.Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 18.Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem. 2007;282:14048–14055. doi: 10.1074/jbc.M611089200. [DOI] [PubMed] [Google Scholar]
- 19.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 20.Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell. 2003;4:463–476. doi: 10.1016/s1535-6108(03)00303-9. [DOI] [PubMed] [Google Scholar]
- 21.Klimstra DS, Longnecker DS. K-ras mutations in pancreatic ductal proliferative lesions. Am J Pathol. 1994;145:1547–1550. [PMC free article] [PubMed] [Google Scholar]
- 22.Knockaert M, Gray N, Damiens E, Chang YT, Grellier P, Grant K, Fergusson D, Mottram J, Soete M, Dubremetz JF, Le Roch K, Doerig C, Schultz P, Meijer L. Intracellular targets of cyclin-dependent kinase inhibitors: identification by affinity chromatography using immobilised inhibitors. Chem Biol. 2000;7:411–422. doi: 10.1016/s1074-5521(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 23.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 24.Luttges J, Schlehe B, Menke MA, Vogel I, Henne-Bruns D, Kloppel G. The K-ras mutation pattern in pancreatic ductal adenocarcinoma usually is identical to that in associated normal, hyperplastic, and metaplastic ductal epithelium. Cancer. 1999;85:1703–1710. [PubMed] [Google Scholar]
- 25.Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57:2140–2143. [PubMed] [Google Scholar]
- 26.Nociari MM, Shalev A, Benias P, Russo C. A novel one-step, highly sensitive fluorometric assay to evaluate cell-mediated cytotoxicity. J Immunol Methods. 1998;213:157–167. doi: 10.1016/s0022-1759(98)00028-3. [DOI] [PubMed] [Google Scholar]
- 27.Tada M, Ohashi M, Shiratori Y, Okudaira T, Komatsu Y, Kawabe T, Yoshida H, Machinami R, Kishi K, Omata M. Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology. 1996;110:227–231. doi: 10.1053/gast.1996.v110.pm8536861. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Han H, Von Hoff DD. Identification of an Agent Selectively Targeting DPC4 (Deleted in Pancreatic Cancer Locus 4)-Deficient Pancreatic Cancer Cells. Cancer Res. 2006;66:9722–9730. doi: 10.1158/0008-5472.CAN-05-4602. [DOI] [PubMed] [Google Scholar]
- 29.Workman P. Towards intelligent anticancer drug screening in the post-genome era? Anticancer Drug Des. 1997;12:525–531. [PubMed] [Google Scholar]
- 30.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]