

Abstract
Protein folding in the endoplasmic reticulum (ER) is monitored by ER quality control (ERQC) mechanisms. Proteins that pass ERQC criteria traffic to their final destinations through the secretory pathway, whereas non-native and unassembled subunits of multimeric proteins are degraded by the ER-associated degradation (ERAD) pathway. During ERAD, molecular chaperones and associated factors recognize and target substrates for retrotranslocation to the cytoplasm, where they are degraded by the ubiquitin–proteasome machinery. The discovery of diseases that are associated with ERAD substrates highlights the importance of this pathway. Here, we summarize our current understanding of each step during ERAD, with emphasis on the factors that catalyse distinct activities.
The eukaryotic cell environment is chemically complex and crowded, with macromolecules occupying 20−40% of the cell volume1,2. Most proteins attain their three-dimensional conformations in this environment, a process that is required for their proper function. However, protein folding is inherently error prone because the folding energy landscape for a polypeptide might include several off-pathway non-native intermediate states in addition to the state occupied by the native conformation3. Moreover, spontaneous errors during transcription and translation, genetic mutations, toxic compounds and cellular stresses, such as increased temperature and osmotic stress, can compromise folding efficiency and/or rate. Finally, the chemical environment in the cell might not match the conditions that are required for the efficient folding of a given polypeptide. Not surprisingly, protein misfolding can lead to a host of diseases, such as cystic fibrosis, antitrypsin deficiency and protein aggregation diseases (such as Huntington's, Parkinson's, Alzheimer's and prion-associated diseases). To prevent misfolding-induced toxicity, aberrant proteins are often destroyed4.
The folding of secreted proteins poses a specific problem. Approximately one-third of all proteins in eukaryotes are targeted to the secretory pathway, and the first compartment encountered by this diverse substrate ensemble is the endoplasmic reticulum (ER)5,6. The ER contains molecular chaperones that assist in protein folding, and also contains unique enzymes that maintain an oxidizing environment relative to the cytoplasm and catalyse co- and post-translational modifications.
To ensure that the ER assembly line manufactures products that meet the needs of the cell, secreted proteins are subject to ER quality control (ERQC)7. The primary mediators of ERQC are molecular chaperones that not only sample and help polypeptides to fold but also evaluate the conformations of their substrates. If a polypeptide has attained its native conformation, it might be targeted to its final destination. If folding is delayed or an illegitimate conformation arises, the substrate is either subjected to additional folding cycles or is selected for a process termed ER-associated degradation (ERAD)8. If, however, the concentration of these potentially toxic protein species increases, compensatory pathways are induced.
Here, we present a brief historical overview of the ERAD pathway and focus on individual steps during ERAD: namely substrate recognition, targeting, retrotranslocation, ubiquitylation and proteasomal degradation. We discuss how ERAD efficiency is regulated and describe the intersection between the ERAD pathway, autophagy and the unfolded protein response (UPR), which can lead to apoptosis if unmitigated. Finally, we briefly survey the relationship between ER-protein folding, ERAD and protein transport to later compartments of the secretory pathway. As these fields have rapidly expanded, this review highlights select reports in the literature and emphasizes open questions.
ER degradation: an historical perspective
The existence of an ER-associated degradation pathway arose from studies by Klausner and colleagues9 who investigated the turnover of subunits in the heptameric T-cell receptor (TCR) complex. TCR subunits are synthesized in non-stoichiometric ratios, and assembly into a hetero-oligomeric complex is essential for trafficking beyond the ER. However, it was found that isolated α and μ subunits were degraded in a non-lysosomal, pre-Golgi compartment. Two models were proposed to explain this observation: either TCR subunits are targeted to an unknown compartment for degradation or a specific protease resides within the ER10. Initially, the degradation of a misfolded form of the yeast vacuolar protease carboxy-peptidase Y (also known as Cpy*) was also thought to occur by one of these mechanisms11. In fact, a protein resident of the mammalian ER, denoted ERP60, was isolated and proposed to be the quality-control protease12.
The first hint that ER-protein degradation might instead require a cytoplasmic factor emerged from the work of Sommer and Jentsch13. They discovered that a thermosensitive mutation in the gene that encodes Sec61, the ER-embedded translocation channel, was rescued by the disruption of UBC6, which encodes a ubiquitin-conjugating enzyme in the ER membrane in budding yeast. This suggested that the cytoplasmic proteasome degrades misfolded ER-membrane proteins because ubiquitin-conjugating enzymes, or E2s, are required for the ubiquitylation of proteasome-targeted substrates (see below). Support for this model emerged from subsequent studies on the human cystic fibrosis transmembrane regulator (CFTR), a plasma membrane chloride channel that, when mutated, leads to cystic fibrosis14,15. Soon thereafter, it was shown in yeast that misfolded soluble proteins in the ER, such as mutant pro-alpha factor and Cpy*, are destroyed by the proteasome; in this case, the substrate had to be retrotranslocated (or dislocated) from the ER to the cytoplasm8,16,17. Concurrently, the proteasome was shown to be required for the regulated degradation of yeast hydroxymethylglutaryl coenzyme A reductase18, the enzyme that catalyses the rate-limiting step in cholesterol biosynthesis, and for the human cytomegalovirus (HCMV)-induced degradation of the major histocompatibility class I (MHCI) protein19. More recent data indicate that bacterial agents, such as cholera toxin, Shiga toxin and ricin, also use elements of the ERAD pathway to intoxicate host cells20.
A large number of components that are required for ERAD have been identified through both genetic and biochemical approaches21-23. Based on these efforts, it has also become clear that ERAD substrates are recognized, targeted, retrotranslocated, polyubiquitylated and then degraded by the 26S proteasome (FIG. 1). Individual components have been found to catalyse unique steps during ERAD, and most are conserved from yeast to humans (TABLE 1).
Figure 1. A step-by-step illustration of endoplasmic reticulum-associated degradation.

a | Protein recognition. Misfolded proteins containing cytoplasmic, intramembrane or endoplasmic reticulum (ER)-luminal lesions are recognized by cytoplasmic and luminal chaperones and associated factors, such as 70 kDa heat-shock protein (Hsp70)-family members, calnexin and calreticulin, and protein disulphide isomerases. b | Protein targeting. ER-associated degradation (ERAD) substrates are targeted to the retrotranslocation machinery (the retrotranslocon) and/or to E3 ligases. c | Retrotranslocation initiation. Substrate retrotranslocation into the cytoplasm might be initiated in part by the cell-division cycle-48 (Cdc48) complex; other components, such as molecular chaperones or the proteasome, might also be required for this step. The energy derived from ATP hydrolysis by Cdc48, which is a AAA+ATPase, is coupled to retrotranslocation. d | Ubiquitylation and further retrotranslocation. As proteins exit the retrotranslocon they are polyubiquitylated by E3 ubiquitin ligases. This promotes further retrotranslocation and is aided by cytoplasmic ubiquitin-binding protein complexes. e | Proteasomal targeting and degradation. Once a polyubiquitylated substrate is displaced into the cytoplasm, it is recognized by receptors in the 19S cap of the 26S proteasome. De-ubiquitylating enzymes (not shown) remove the polyubiquitin tag, and peptide N-glycanase (not shown) might also be required for efficient degradation. The substrate is then threaded into the 20S catalytic core of the proteasome where it is broken down into peptide fragments. Ubiquitin that is generated by this process can be recycled for subsequent rounds of modification.
Table 1.
Select components that are required for ERAD
| Component | Location | Yeast | Mammals* |
|---|---|---|---|
| Recognition | |||
| Hsp70 family | ER | BiP (also known as Kar2) | BIP (also known as GRP78) |
| Cytoplasm | Ssa1 | HSP70 and HSC70 | |
| Hsp40 family | ER | Scj1 and Jem1 | ERDJ1−5 and p58IPK |
| Cytoplasm | Ydj1 and Hlj1 | HDJ1−2, HSJ1 and Cys string protein | |
| Nucleotide-exchange factor | ER | Sil1 (also known as Sls1) and Lhs1 | SIL1, GRP170 and BAP |
| Cytoplasm | Snl1, Fes1 and Sse1 | BAG1−2, HSPBP1 and HSP110 | |
| Small Hsps | Cytoplasm | Hsp26 and Hsp42 | α-Crystallin |
| Hsp90 family | ER | Unknown | GRP94 |
| Cytoplasm | Hsp82 and Hsc82 | HSP90 | |
| Calnexin | ER membrane | Cne1 | Calnexin |
| Calreticulin | ER | Unknown | Calreticulin |
| Protein disulphide isomerase | ER | Pdi1 and Eps1 | PDI, ERP29, ERP57, ERP72 and ERDJ5 |
| Targeting | |||
| α-Mannosidase-like‡ | Possibly ER or ER membrane | Htm1 (also known as Mnl1) | EDEM1−3 |
| Mannose-6-phosphate receptor-like‡ | ER | Yos9 | OS9 and XTP3-B |
| UBL domain containing | ER membrane | Usa1 | HERP |
| Retrotranslocation | |||
| Sec61 complex | ER membrane | Sec61 complex and Ssh1 complex | Sec61 complex |
| Derlins | ER membrane | Dfm1 and Der1 | Derlin-1−3 |
| Regulators? | ER membrane | Usa1 | HERP |
| Ubx2 | Unknown | ||
| Unknown | VIMP, BAP31 and SVIP | ||
| Ubiquitylation | |||
| E1 ubiquitin-activating enzyme | Cytoplasm | Uba1 | UBE1 |
| E2 ubiquitin-conjugating enzyme | ER membrane | Ubc6 | UBC6e |
| Membrane associated | Ubc7–Cue1 complex | UBCH7 (also known as UBC7) | |
| Cytoplasm | Not established | UBCH5 | |
| E3 ubiquitin ligase | ER membrane | Hrd1–Hrd3 complex | HRD1–SEL1L complex |
| Doa10 | TEB4 (also known as MARCH IV) | ||
| Unknown | GP78 and RMA1 (also known as RNF5) | ||
| Cytoplasm | Rsp5 | NEDD4−2 | |
| Unknown | Parkin, CHIP and SCFFBX2 or SCF2FBS2 | ||
| E4 chain-extension enzyme | Cytoplasm | Ufd2 | UFD2a |
| Proteasomal targeting and degradation | |||
| Cdc48 complex | Membrane associated | Cdc48–Ufd1–Npl4 | p97–UFD1–NPL4 |
| UBL and UBA domain containing | Cytoplasm | Rad23 and Dsk2 | RAD23 |
| Deglycosylating enzyme | Possibly membrane associated or cytoplasm | Png1 | Peptide N-glycanase |
| De-ubiquitylating enzyme | Cytoplasm | Unknown | Ataxin-3 |
| Ubiquitin receptor | 19S proteasomal cap | Rpn10 | RPN10 (also known as S5a) |
| Rpn13 | RPN13 | ||
| Rpt5 | RPT5 (also known as TBP1 or S6) | ||
Note that the roles of some mammalian factors in ERAD have not been conclusively established.
Lectin-like proteins.
BAG, B-cell lymphoma-2-associated athanogene; BIP, immunoglobulin binding protein; Cdc48, cell-division cycle protein-48; CHIP, C terminus of HSC70-interacting protein; EDEM, ER degradation-enhancing α-mannosidase-like lectin; ER, endoplasmic reticulum; ERAD, ER-associated degradation; ERP, ER protein; FBX, F-box only protein; FBS, F-box/SEC7 protein; HERP, homoCys-responsive ER-resident protein; HSC, heat-shock cognate; Hsp, heat-shock protein; SCF, S-phase-kinase-associated protein-1–cullin–F-box complex; UBA, ubiquitin association; UBL, ubiquitin-like; VIMP, valosin-containing protein-interacting membrane protein.
Substrate recognition
Potential ERAD substrates include soluble and integral membrane proteins, polypeptides that have failed to become post-translationally modified or are otherwise damaged or misfolded, and unassembled members of multiprotein complexes. With few exceptions, it remains mysterious how ERAD substrates are selected from proteins that are properly folded or that are on the correct folding pathway; thus, many proteins might be mistakenly targeted for ERAD. In turn, increasing evidence indicates that a large percentage of some ‘wild-type’ proteins are destroyed by the ERAD pathway24. Overall, it is better that questionable proteins in the ER are degraded because the cell cannot afford to risk the acquisition of toxic aggregates.
Hydrophobic patches: is being ‘oily’ sufficient for ERAD?
In the native conformation, hydrophobic patches are usually buried within the interior of soluble proteins in order to maintain the lowest energy state3. However, these patches might become exposed in the unfolded state, which can lead to aggregation. To minimize this event, molecular chaperones, such as members of the 70 kDa heat-shock protein (Hsp70) family, bind to short polypeptide motifs with hydrophobic properties. Binding and release of Hsp70-family substrates are ATP dependent and are regulated by both Hsp40-family co-chaperones and dedicated nucleotide-exchange factors (NEFs) (BOX 1). As might be anticipated, an ER-luminal Hsp70-family member — immunoglobulin binding protein (BIP, also known as GRP78) — and in some cases ER-resident Hsp40-family members, associate with several ERAD substrates and maintain their solubility25-28. It is currently unclear whether the BIP-associated NEFs have a role during ERAD, and whether BIP and/or Hsp40-family co-chaperones are required for the recognition of every ERAD substrate.
By analogy, membrane proteins with large cytoplasmic domains might be expected to bind to cytoplasmic Hsp70s and Hsp40s. Consistent with this hypothesis, these chaperones facilitate the ERAD of CFTR in both yeast29,30 and mammals31,32; CFTR contains two large, cytoplasmically exposed nucleotide-binding domains. By contrast, the cytoplasmic NEFs in mammalian cells, HSP-binding protein-1 (HSBP1) and B-cell lymphoma-2 (BCL2)-associated athanogene-2 (BAG2), negatively regulate CFTR degradation33,34. These data are consistent with the fact that the ATP-bound Hsp70s have a low affinity and a high release rate for peptide substrates (BOX 1).
Based on these and other results (see below), it is likely that Hsp70s have an important, general role during ERAD substrate selection. In one view, prolonged interaction between an ERAD substrate and an Hsp70 might be sufficient to recruit a ubiquitin ligase, or E3; once the substrate becomes polyubiquitylated, ERAD can be ensured. Consistent with this model, BIP resides in an E3-containing multiprotein complex in the ER membrane35, and cytoplasmic Hsp70 family members recruit a cytoplasmic E3 that ubiquitylates CFTR36. The fundamental importance of BIP is emphasized by the observation that BIP-mutant knock-in mice show defects that are consistent with compromised ERQC and display profound defects in brain development37.
The selection of N-linked glycans for ERAD
Most proteins that translocate into the ER are co-translationally modified with an N-linked oligosaccharide (GlcNAc2-Man9-Glc3, of which GlcNAc is N-acetylglucosamine, Man is mannose and Glc is glucose) that is added onto an Asn in a consensus Asn-X-Ser/Thr motif (in which X represents any amino acid) (FIG. 2). The subsequent removal of terminal glucose residues by glucosidases, and facilitated folding by the lectin-like chaperones calnexin and calreticulin results in a glycoprotein that contains a GlcNAc2-Man9 moiety. Proteins with this sugar are competent for ER exit and can transit to their final destinations. If instead the glycoprotein contains hydrophobic patches or is in a molten globule-like state, it is recognized by UDP-glucose:glycoprotein glucosyltransferase (UGGT)38,39, which adds a glucose molecule to the N-linked glycan. The resulting monoglucosylated, non-native species re-enters the calnexin–calreticulin cycle40. Whether UGGT can directly detect terminal misfolding and target substrates for ERAD is currently unknown. Moreover, some ER-retained species are modified by mannosidases, which might act as timers for glycoprotein degradation and thus prevent glycoproteins from becoming permanently trapped in a re-glucosylation and folding cycle41. However, it is important to note that only a select number of ERAD substrates have been shown to require these events and it remains contentious whether ERAD occurs only after a specific number of mannoses have been trimmed.
Figure 2. N-linked glycosylation and the degradation of glycosylated proteins.

Proteins that enter the endoplasmic reticulum (ER) are often modified by the addition of a GlcNAc2-Man9-Glc3 glycan to the side-chain nitrogen of Asn residues in the consensus Asn-X-Ser/Thr motif. First, the translocon-associated oligosaccharyl transferase (OST) complex co-translationally transfers GlcNAc2-Man9-Glc3 glycans from dolichol to substrate proteins. Next, glucosidase-I and glucosidase-II sequentially remove two terminal glucoses, generating monoglucosylated substrates that are recognized by calnexin and calreticulin through their carbohydrate-binding globular domains (calreticulin is a soluble protein and is not shown). The interaction with calnexin and calreticulin facilitates folding. ERP57, a protein disulphide isomerase homologue that is associated with the arm domain of calnexin and calreticulin, catalyses disulphide bond formation. Following release from the calnexin–calreticulin cycle, the final glucose is trimmed by glucosidase-II. If glycoproteins have adopted their native conformations, they can be demannosylated (denoted by the use of parentheses around the mannoses) by ER mannosidases I and II (ER man-I and man-II) and exit the ER through coatomer protein complex-II vesicles. However, the folding of some glycoproteins requires multiple rounds of association with calnexin–calreticulin. Such proteins are reglucosylated by UDP-glucose:glycoprotein glucosyltransferase (UGGT), which recognizes non-native states and transfers a glucose from UDP-glucose to the N-linked GlcNAc2-Man9 glycan. Re-monoglucosylation promotes re-entry into the folding cycle. Terminally misfolded glycoproteins might also be targeted for ER-associated degradation (ERAD) by calnexin and calreticulin or by other ERAD-requiring components. EDEM, ER degradation-enhancing α-mannosidase-like lectins; Glc, glucose; GlcNAc, N-acetylglucosamine; Man, mannose.
Additional features of the pathway for glycoprotein selection during ERAD are noteworthy. First, the number and position of glycans in glycoproteins might be optimized for entry into or exit from the calnexin–calreticulin cycle42-45. Second, calnexin and calreticulin bind to glycosylated and non-glycosylated proteins — mutation of the lectin-like domain of calnexin or calreticulin has no apparent effect on chaperone function in cultured cells46. Third, there is crosstalk between BIP and the calnexin–calreticulin system. For example, some substrates interact sequentially with BIP and calnexin–calreticulin in the cell47, whereas calnexin and BIP synergistically suppress the aggregation of a non-glycosylated substrate in vitro48. Furthermore, BIP can compensate for the absence of the calnexin–calreticulin cycle by binding to glycosylated substrates with which it does not normally interact49. In some cases, the two chaperone systems even have unique effects on the fate of a substrate50-53. Furthermore, budding yeast lack UGGT but exhibit most other aspects of glycoprotein quality control and ERAD.
Lectin-like domain.
Lectins are sugar-binding proteins that either bind to soluble carbohydrate molecules or to carbohydrate moieties in glycoproteins. Lectin-like domains are found in a wide range of proteins that are involved in protein–protein, protein–lipid and protein–nucleic acid interactions.
Disulphide bond formation
The ER environment is more oxidizing than the cytoplasm, and this favours the formation of disulphide bonds. This process is catalysed by protein disulphide isomerases (PDIs), which possess thiol oxidoreductase activity (BOX 1). Besides enabling the de novo formation of disulphide linkages, PDIs also isomerize non-native disulphide bonds, thus facilitating the acquisition of the native state. To date, 19 PDI homologues have been identified in the human ER, which suggests that these enzymes exhibit substrate specificity, that their expression is regulated and/or that they possess unique oxidizing potentials in vivo.
Mammalian PDI participates in the ERAD of several substrates and can work in tandem with BIP50,51. PDI enables the retrotranslocation of cholera toxin and the simian virus-40 (SV40) polyoma virus, both of which use the endocytic pathway to invade host cells54,55. ERP57, a mammalian PDI homologue, associates with calnexin and calreticulin and is involved in glycoprotein quality control56. ERDJ5 also helps to retrotranslocate the SV40 virion55. Interestingly, a yeast PDI homologue, Pdi1, recognizes a Cys-free model ERAD substrate and catalyses its degradation57. One recently characterized human PDI homologue, ERDJ5, contains four canonical thioredoxin-like active-site Cys-X-X-Cys motifs and is a BIP cofactor28. ERDJ5 regulates the degradation of null Hong Kong (NHK), a disease-causing α1-antitrypsin variant, by accelerating the formation of degradation-competent NHK monomers from disulphide-linked dimers58.
Because of the diversity of this enzyme family and the diversity of potential substrates, many questions remain unanswered. For example, does ERAD require the formation of disulphide bonds between PDIs and free Cys residues in substrates, as suggested for ERDJ5, or is polypeptide recognition sufficient for ERAD, as is the case for yeast Pdi1? In addition, does the retrotranslocation of oxidized substrates require the reduction of all disulphide bonds? For some substrates, completely reduced forms cannot be identified prior to degradation50,59. However, enhanced degradation efficiencies have been noted in cells that have been treated with reducing agents60. The observed increase in degradation might be indirect because reducing agents also induce the UPR (BOX 2), which results in the upregulation of some components required for ERAD61.
Assembly of oligomeric structures and ERAD escape
The folding of individual subunits of multimeric complexes can occur either prior to the acquisition of the quaternary structure62,63 or after subunit oligomerization64. Whether assembly enhances conformational stability or results in a transport-competent state for most multiprotein complexes remains ill-defined: ERAD might ensue from a folding defect or from prolonged ER retention. Assembly can also mask peptide signals that act as determinants of ER retention or degradation65-67.
Box 1 | Select molecular chaperones involved in ER quality control.

The endoplasmic reticulum (ER) possesses a unique folding environment, which is maintained by the concerted action of distinct families of molecular chaperones.
The 70 kDa heat-shock protein (Hsp70)-family chaperones are highly expressed in most eukaryotic organelles. Hsp70s contain an N-terminal ATPase domain followed by a substrate-binding domain, which contains a hydrophobic cleft for binding to client proteins, and a C-terminal lid. ATP hydrolysis is essential for Hsp70 function and is stimulated by Hsp40s and other J-domain-containing proteins or by peptide substrates (see figure). The resultant ADP-bound state possesses high affinity and low release rates for substrates, which prevents substrate aggregation.
Hsp40s and other J-domain-containing proteins act as co-chaperones for Hsp70s, although chaperone-like roles have also been described for some Hsp40s. The predominant feature of these proteins is the presence of an ∼70 amino-acid J-domain, which interacts with the ATPase domain of Hsp70s and stimulates ATP hydrolysis (see figure). Hsp40s might also contain distinct regions for substrate binding, zinc chelation and/or dimerization.
The Hsp70 hydrolytic cycle might be further augmented by the catalysed release of ADP, as performed by nucleotide-exchange factors (NEFs; see figure). Several classes of NEFs have been identified, including the Hsp110s, which belong to the Hsp70 superfamily, B-cell lymphoma-2-associated athanogenes (BAGs) and GRPE-like proteins (see the main text).
Calnexin and calreticulin are carbohydrate-binding lectins that are required for the folding and quality control of glycosylated proteins. Both bind to the N-linked core glycan of monoglucosylated substrates and might also bind to the hydrophobic polypeptide backbone of substrates.
During protein folding in the ER, protein disulphide isomerases participate in the oxidation and/or isomerization of disulphide bonds. These proteins are characterized by the presence of one or more thioredoxin-like domains with active-site Cys-X-X-Cys motifs (in which X represents any amino acid).
Thioredoxin-like.
Thioredoxins are disulphide-containing proteins that regulate the redox status of the cell and have a role in diverse oxidative cellular processes. The thioredoxin-like domain typically adopts a two- or three-layer β-sandwich structure and contains a conserved Cys-X-X-Cys active-site motif.
The homotrimerization of the influenza virus haemagglutinin protein within the ER provides an example in which monomer folding occurs prior to oligomerization63. Haemagglutinin monomers, which fold cotranslationally and adopt a stable disulphide-bonded conformation, assemble into a homotrimeric complex prior to ER exit. A second example is the oligomerization of subunits of the heptameric TCR complex. In this case, acquisition of the quaternary structure enhances the stability of individual TCR subunits, which are otherwise rapidly turned over by ERAD owing to the exposure of basic residues in the ER membrane68. Another well-studied example in which some of these phenomena are evident is the assembly of secreted immunoglobulin, which requires the formation of disulphide bonds between heavy chains (HCs) and light chains (LCs). In plasma cells that lack LCs, HCs are either maintained in a partially folded, assembly competent state by binding to BIP, or are degraded by ERAD65,69,70. The HCs also contain a C-terminal Cys, which is exposed in the unassembled state and might act as a degradation determinant65. Degradation prevents immunoglobulin subunit aggregation, which can occur through the incorrect formation of disulphides.
Substrate targeting
Soluble ERAD substrates must first be selected for retrotranslocation to the cytoplasm because the enzymes required for ubiquitylation reside in this compartment (BOX 3). For ERAD substrates that reside in the ER membrane, ubiquitylation precedes or can occur concomitant with retrotranslocation.
Distinct yeast ERAD-C, ERAD-L and ERAD-M pathways
Accumulating evidence suggests that the location of a misfolded lesion dictates the factors that are required for ERAD substrate targeting. Thus, proteins with lesions in the cytoplasmic, luminal and membrane-spanning domains follow the ERAD-C, ERAD-L and ERAD-M pathways, respectively35,71-74. ERAD-C substrates use a complex that contains the Doa10 ubiquitin ligase. Substrates that follow the ERAD-L pathway — either soluble or integral membrane proteins that contain a luminal lesion — interact with the Hrd1 ubiquitin ligase complex. Much less is known about the ERAD-M pathway, but its substrates also seem to be ubiquitylated by the Hrd1 complex.
It is important to point out that the ERAD-C, ERAD-L and ERAD-M pathways have only been defined in yeast. As mammals possess a more elaborate repertoire of ERAD-requiring components (TABLE 1), it is possible that distinctions between the pathways became blurred as higher eukaryotes evolved larger numbers of and more complex secretory-pathway residents. Even in yeast, certain membrane substrates require both the Doa10 and Hrd1 ubiquitin ligases, and hence the ERAD-C and ERAD-L pathways can overlap72,75,76. This probably serves to increase degradation efficiency and allows the cell to compensate for substrate overload in one or the other pathway.
The coupling of recognition and targeting
Substrate recognition and targeting can become indistinguishable because ERAD substrates might not be passed between distinct recognition and targeting complexes. Recent observations suggest that factors required for recognition reside within multiprotein complexes that are also essential for targeting. For example, in yeast, BiP is tethered to the ER membrane by virtue of its interaction with an integral membrane partner, Sec63. In turn, Sec63 resides in a multiprotein ensemble in the yeast ER membrane that includes Sec61 (REF. 77) — a candidate for the retrotranslocation channel (see below). Thus, a complex containing BiP and two associated Hsp40s in the ER27 might recognize and then direct selected ERAD substrates to the ER channel.
In mammals, a recent study implicated a transmembrane UBL domain-containing protein, homoCys-responsive ER-resident protein (HERP), as a receptor for non-glycosylated BIP substrates59. As HERP co-precipitates with derlin-1, which is another candidate for the retrotranslocation channel78, and with ubiquitylated proteins and the 26S proteasome, this targeting factor might bridge the ER-recognition machinery to the cytoplasmic ubiquitin–proteasome system. A similar function can be attributed to the Hrd1 complex, which contains ER-luminal chaperones and other targeting components tethered both to the membrane-integrated E3 ligase and to a cytoplasmic factor that drives substrate extraction35,73,74,79 (see below).
Recently, several ER-resident targeting factors that recognize glycoproteins have been characterized in both yeast and mammals. These include the ER degradation-enhancing α-mannosidase-like lectins (EDEMs) (TABLE 1) and lectins that contain mannose-6-phosphate receptor-like domains, such as Yos9 (in yeast) and OS9 and XTP3-B (in mammals). It is thought that these factors deliver ERAD substrates to the retrotranslocation channel80.
Box 2 | The unfolded protein response.
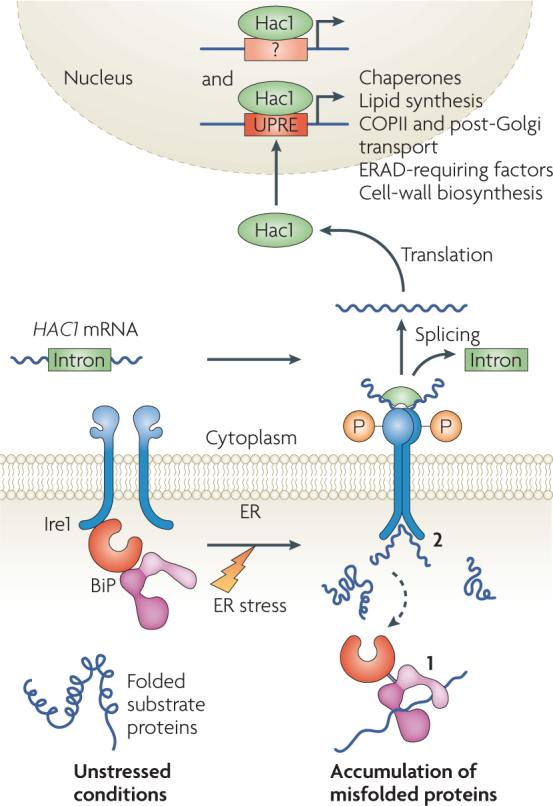
Misfolded protein accumulation in the endoplasmic reticulum (ER) can lead to the induction of the unfolded protein response (UPR), which reduces ER-protein load by several mechanisms (see the main text).
In yeast, the sole UPR transducer is inositol-requiring protein-1 (Ire1), an ER-localized transmembrane Ser/Thr kinase and site-specific endoribonuclease. Under unstressed conditions, immunoglobulin binding protein (BiP) binds to Ire1 in the ER lumen and maintains the enzyme in an inactive state (see figure). When the ER is stressed, BiP can be titrated away to bind to misfolded substrates, in turn activating Ire1 (see figure, step 1). Ire1 also dimerizes and might directly bind to misfolded proteins owing to the formation of a peptide-binding pocket in the ER-luminal domain, thus resulting in activation (see figure, step 2).
Ire1 activation involves the transphosphorylation of its cytoplasmic domain, which triggers endoribonuclease activity and splices an intron in the mRNA that encodes Hac1 (homologous to ATG6/CREB), a dedicated UPR transcriptional activator. The processed mRNA is re-ligated by a tRNA ligase, Trl1, and translated. Hac1 then translocates into the nucleus, binds to UPR elements (UPREs) and possibly other sequences in the promoter region of target genes, and upregulates their expression.
In higher eukaryotes, the UPR consists of three transducers, IRE1, PERK and activating transcription factor-6 (ATF6) (not shown)157. IRE1 functions in a manner that is identical to its yeast homologue. ER-stress-activated PERK is a transmembrane kinase that phosphorylates the α-subunit of the eukaryotic translation initiation factor-2 (eIF2α), and thus inhibits protein translation. ATF6 traffics to the Golgi under conditions of ER stress. Here, it is proteolytically processed by the S1P and S2P intramembrane proteases to release the ATF6-fragment transcription factor. This fragment translocates into the nucleus and upregulates target genes. In each case, BIP is also required for transducer activation. Interestingly, the IRE1 branch of the UPR seems to be anti-apoptotic, whereas persistent PERK signalling might trigger apoptosis158. COPII, coatomer protein complex-II; ERAD, ER-associated degradation.
UBL domain.
(Ubiquitin-like domain). A non-enzymatic domain that resembles ubiquitin in structure. UBL domain-containing proteins might have a role in the recruitment of ubiquitylated substrates to the 26S proteasome.
EDEM1 facilitates the degradation of the NHK α1-antitrypsin mutant, and based on its interaction with calnexin, the lectin might receive substrates from the calnexin cycle81-83. Two homologues, EDEM2 (REFS 84,85) and EDEM3 (REF. 86), probably function in a similar way. In keeping with a role for the EDEMs in substrate targeting, EDEM1 associates with derlin-2 and derlin-3 (REF. 86), which are retrotranslocation channel candidates. Further supporting these observations, a yeast EDEM homologue has been implicated in the turnover of ERAD-L substrates71. It is unclear whether the EDEMs exhibit substrate specificity, whether every EDEM homologue acts as a mannosidase and whether binding to a distinct mannose-trimmed protein species is essential for substrate selection.
Yos9 was uncovered in a genetic screen to identify components that participate in glycoprotein turnover87. Soon after, three groups reported that Yos9 functions in the ERAD of soluble and membrane-bound glycoproteins that contain luminal lesions88-90. Yos9 was then shown to bind to misfolded substrates, even those that lack glycans, which is suggestive of a chaperone-like activity for this targeting factor. Yos9 also forms a stable complex with BiP and resides in the ERAD-L Hrd1 complex, possibly regulating the selectivity of Hrd1 for misfolded substrates35,73,91.
Two mammalian Yos9 homologues, OS9 and XTP3-B, were recently characterized. OS9 interacts with an ER-luminal Hsp90 homologue, 94 kDa glucose-regulated protein (GRP94), and detects the NHK α1-antitrypsin variant for delivery to the mammalian HRD1 complex92. Short hairpin RNA (shRNA)-mediated knockdown of either OS9 or GRP94 slowed the degradation of the anti-trypsin mutant; XTP3-B seemed to have a lesser role. By contrast, other studies suggested that XTP3-B might prevent the aggregation of NHK α1-antitrypsin and help link the BIP recognition machinery to the HRD1 complex93. It will be important to determine whether members of the Yos9 family exhibit diverse substrate specificities, and what the relative contributions are between lectin-mediated binding and the observed chaperone-like activity.
The ER-resident factors described above do not contribute to the ERAD targeting of membrane substrates that present misfolded domains to the cytoplasm and that contain short or folded luminal segments. Instead, cytoplasmic Hsp70s and Hsp40s mediate the interaction between model membrane substrates and the Doa10 ubiquitin ligase in yeast76. Furthermore, a growing number of soluble proteins in the cytoplasm and in the nucleus require select components of the ERAD machinery for their turnover94,95; in one case yeast Hsp70 and Hsp40 chaperones were found to mediate this process96. Thus, did the ERAD machinery for membrane substrates usurp pre-existing components of a cytoplasmic quality control machinery at the ER, or have some cytoplasmic and nuclear substrates evolved to undergo ERAD?
The retrotranslocon
Initial evidence suggested that the Sec61 translocation channel has a secondary role as the retrotranslocation channel (the retrotranslocon). First, the human Sec61 complex interacted with MHCI molecules during their delivery to the proteasome in a process that is catalysed by a HCMV gene product19. Second, subunits of the Sec61 complex were found to bind to various ERAD substrates en route to degradation. Third, the pre-binding of ribosome-nascent chain complexes to the Sec61 channel in ER-derived vesicles abrogated the retrotranslocation of cholera toxin97. Fourth, mutations in the gene that encodes yeast Sec61 slowed the ERAD of soluble and membrane substrates98,99 and interactions were noted between proteasome subunits and Sec61 (REF. 100). Furthermore, the formation of a disulphide bond between yeast Sec61 and an ERAD substrate that was trapped prior to degradation suggested that this component of the translocation channel intimately associates with an ERAD substrate during degradation101. If the Sec61 channel is indeed the retrotranslocon, or forms a channel component, it will be interesting to define whether unique subpopulations are dedicated to retrotranslocation and translocation. It might also be that the differential binding of Sec61 partners re-engineers the channel for the opposing functions of translocation and retrotranslocation.
Box 3 | The ubiquitin–proteasome system.

Ubiquitin (Ub), a 76 amino-acid peptide, is covalently attached to ε-amino groups of Lys residues in substrates through an isopeptide bond. Ub itself contains several Lys residues, but the covalent linkage through Lys48 seems to be a hallmark for proteasome-mediated degradation. Degradation also requires at least a tetra-Ub chain.
Ub conjugation first requires an E1 Ub-activating enzyme. The C-terminal Gly of Ub is adenylated by the E1 and then displaced following the nucleophilic attack of a conserved Cys residue in the E1, resulting in a thioester linkage between the E1 and Ub (see figure). The next step involves the transfer of Ub to an E2 Ub-conjugating enzyme through the formation of another thioester linkage. The covalent attachment of Ub to substrates is catalysed by E3 Ub ligases, such as RING, U-box and HECT domain-containing proteins. The RING and U-box domain E3s facilitate the transfer of Ub from the E2 to selected substrates (see figure, step 1). The HECT domain E3s are covalently coupled to Ub by a thioester bond in the HECT domain. The subsequent interaction with substrates is required for Ub modification (see figure, step 2).
Once polyubiquitylated, a substrate can be targeted to the 26S proteasome and degraded. The 26S proteasome is built from two 19S caps and a 20S catalytic core. The 19S cap contains 19 subunits (regulatory particle ATPase-1 (RPT1)– RPT6 and regulatory particle non-ATPase-1 (RPN1)–RPN13) and peripheral factors. The 19S cap contains Ub receptors and removes Ub from substrates via the action of de-ubiquitylating enzymes (Dubs), either by sequential trimming or by en bloc removal of the poly-Ub chain. The substrate is then delivered into a central pore in the 20S core, which is composed of 28 subunits. The central cavity is lined by duplicate enzymes that possess trypsin-like (β2 subunit), chymotrypsin-like (β5 subunit) and post-glutamylpeptide hydrolyzing (β1 subunit) activities.
The derlin family of proteins are alternative candidates for the retrotranslocation channel. Yeast Der1 is a component of the Hrd1 ERAD-L complex through its interaction with a UBL-domain-containing protein, U1 SNP-associating protein-1 (Usa1). Interestingly, Usa1 is the yeast HERP homologue, which further suggests that the Hrd1 complex links early and late events during ERAD (that is, recognition and retrotranslocation events, respectively)73. In line with this view, derlin-1, one of three derlin homologues in human cells, interacts with components of the ubiquitylation and targeting machinery, and partakes in the HCMV-catalysed turnover of MHCI102,103, the dislocation of the SV40 virion55 and the retrotranslocation of a model ERAD substrate in vitro in semi-reconstituted ER-derived vesicles104.
The E3 ubiquitin ligases that are required for ERAD in yeast, Doa10 and Hrd1, are multi-spanning membrane proteins and are members of large protein complexes that include substrate-recognition, targeting and retrotranslocation components. Hence, it has been proposed that the E3s either form or are integral components of retrotranslocation channels105,106. Housing the ubiquitylation and retrotranslocation activities in a single enzyme might be the most efficient way to target ERAD substrates to the proteasome. Direct tests of this model are actively being pursued.
Other models have been put forth to describe how some ERAD substrates are retrotranslocated, particularly those that are large and might not completely unfold. For example, the retrotranslocon might form transiently from one or many components. It has also been postulated that proteins exit the ER through the formation of lipid droplets107. However, the search for a single retrotranslocon has been confounded by the observation that each of the factors mentioned above exhibit substrate specificity. Or, the proteasome might clip or shave cytoplasmic domains from membrane proteins, and the resulting intramembrane and luminal domains might simply be transported to different cellular compartments for degradation or become substrates for other, ill-defined proteases. Therefore, a channel might not always be necessary during ERAD.
Ubiquitylation and proteasome degradation
Most ERAD substrates are ubiquitylated prior to proteasome targeting. Protein ubiquitylation, and thus proteasome-dependent degradation, requires the action of an E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes and E3 ubiquitin ligases (BOX 3). In select cases, E4 ubiquitin-chain-extension enzymes have also been shown to facilitate ERAD76,108,109. The importance of chain extension during ERAD might derive from the fact that the polyubiquitin appendage must reach a crucial length before a substrate can be retrotranslocated110.
Tagging proteins for degradation
In yeast, the Doa10 and Hrd1 ligases have been implicated in the degradation of every studied ubiquitylated ERAD substrate. Both enzymes contain catalytic RING domains and exhibit somewhat different preferences for unique E2 ubiquitin-conjugating enzymes: Doa10-dependent ubiquitylation requires Ubc6 and Ubc7 (REF. 105), whereas Hrd1 uses Ubc7 (REF. 111). These E2s are also membrane-associated, but through different means. Ubc6 is an integral ER-membrane protein, whereas Ubc7 is held at the ER through interaction with an ER-membrane adaptor, coupling of ubiquitin conjugation to ER degradation protein-1 (Cue1), which is also required for ERAD112. In one specific case, an overexpressed ERAD substrate required an alternative HECT domain-containing E3 known as reverses SPT-phenotype protein-5 (Rsp5)113.
Mammalian orthologues of Hrd1 (REF. 114) and Doa10 (REF. 115) have also been identified, and perhaps not surprisingly the repertoire of E3s implicated in ERAD is greatly expanded in higher organisms (TABLE 1). Some of these enzymes have a role in the quality control of disease-related proteins. For example, F-box only protein-2, the substrate recognition component of an SCF E3 complex, binds to glycosylated substrates and affects the stability of the α-subunit of TCRα116. CFTR degradation, however, requires the sequential activity of two E3s: one is membrane-associated and acts co-translationally (RING-finger protein with membrane anchor-1 (RMA1)) and the other is a cytoplasmic E3 that acts post-translationally (C terminus of HSC70-interacting protein (CHIP))117. GP78, another integral membrane E3, also cooperates with RMA1 to target mutant CFTR for ERAD118, and interacts with p97 (REF. 119), which helps to provide the driving force for membrane extraction (see below). Finally, an E3 known as parkin works with UBC6 and UBC7 homologues and ubiquitylates an aggregation-prone G-protein-coupled receptor that otherwise triggers cell death120. Subsequent studies showed that CHIP and parkin function together in a Hsp70-containing complex121. These data emphasize the fact that ERAD substrates exhibit promiscuity when choosing E3 partners, and that E3 complexes exist and can complement one another's activities.
Cytoplasmic extraction and proteasomal targeting
Once a protein has become polyubiquitylated, it must be extracted from the membrane either prior to or during proteasome targeting. Although the proteasome is sufficient in a few cases to retrotranslocate substrates122,123, another cytoplasmic protein complex, the cell-division cycle-48 (Cdc48) complex, has a more pivotal role in substrate retrotranslocation in both yeast and mammals124. In yeast, the complex consists of Cdc48, which is a hexameric AAA+ ATPase, and two associated factors, Ufd1 and Npl4. In mammals, the Cdc48 homologue p97 similarly associates with conserved UFD1 and NPL4 homologues. The complex might be recruited to the ER membrane in part through its interaction with the UBX-domain-containing membrane protein Ubx2 (in yeast)125,126 or with valosin-containing protein-interacting membrane protein (VIMP; in mammals)102,125,126. The Cdc48 complex also binds to several other ER-resident components, including GP78, Der1 and the Hrd1 complex35,73,74,126.
RING domain
A Cys-rich tandem zinc-finger domain of 40−60 amino acids that is found in the RING E3 enzymes, a main class of E3 ubiquitin ligases.
HECT domain
(Homologous to E6AP C terminus domain). A domain in the second largest class of E3 ubiquitin ligases. In contrast to RING ligases, HECT-domain ligases form an essential thioester intermediate with ubiquitin as it is transferred from the E2 enzyme to the substrate.
SCF E3 complex
(S-phase-kinase-associated protein-1 (SKP1)–cullin–F-box E3 complex). The third largest class of E3 ubiquitin ligases. The cullin component of the SCF complex forms a scaffold and organizes substrate receptor and E2 recruitment modules at its N and C termini, respectively. Substrates are recruited to cullin by SKP1 and various F-box proteins, which regulate substrate specificity.
AAA+ ATPase
(ATPases associated with diverse cellular activities). An ATP-hydrolyzing enzyme that contains one or two conserved ATP-binding domains, which are in turn comprised of conserved A and B motifs. AAA+ ATPases assemble into oligomeric assemblies (often hexamers) that form a ring-shaped structure with a central pore.
UBX domian
(Ubiquitin-regulatory ‘X’ domain). An ∼80 amino-acid domain that is found at the C terminus of ubiquitin-regulatory proteins. The UBX domain is a general CDC48-interaction module.
It is unknown how the Cdc48 complex first ‘sees’ ERAD substrates, particularly those inside the ER lumen that need to access the cytoplasmic face of the ER. One possibility is that the Cdc48 complex might transiently embed into the putative retrotranslocon and then recognize and pull substrates into the cytoplasm. Another possibility is that the polyubiquitin moiety provides a handle for the complex to initiate ATP-dependent extraction127. It is also not completely clear whether this complex is essential for the ERAD of each and every ubiquitylated ERAD substrate; in fact, recent evidence suggests that the extent to which a membrane protein is embedded in the lipid bilayer might dictate the degree to which Cdc48 complex function is needed128.
As Cdc48 associates with the proteasome cap129, soluble and integral membrane substrates might be transported directly from the membrane-associated Cdc48-containing engine to the proteasome for degradation. However, increasing evidence indicates that distinct Cdc48- and proteasome-interacting factors have important roles prior to substrate degradation130. Members of this family include UBA domain- and UBL domain-containing proteins, which interact both with the proteasome and with ubiquitylated substrates in the Cdc48 complex. Two factors in this class, Rad23 and Dsk2, increase ERAD efficiency108,131. Of interest, one Cdc48- and Rad23-associated protein is a deglycosylating enzyme132,133 that might be needed to ensure that a polypeptide is not sterically hindered from entering the catalytic chamber of the proteasome. It is unknown whether any of these factors are static members of the Cdc48 complex, or whether they bind to and deliver ERAD substrates to the proteasome by acting as mobile escorts.
Ubiquitin receptors that are residents of the 19S proteasome subunit have also been identified. These include regulatory particle non-ATPase-13 (Rpn13), Rpn10 and regulatory particle ATPase-5. Recent data strongly suggest that Rpn13 mediates the highest affinity binding, and its position in the proteasome is consistent with it having a key role during substrate degradation134. Rpn10 is thought to bind to and then drive substrates into the 20S proteasome core for degradation. It is also worth noting that a substantial percentage of proteasomes reside at the surface of the ER membrane135 and are thus ideally positioned to receive and degrade ERAD substrates. This begs the question of whether an ERAD-dedicated population of proteasomes exists.
De-ubiquitylation prior to or during degradation
In addition to ubiquitin binding, another event mediated both by proteasome-associated enzymes and integral proteasome subunits is substrate de-ubiquitylation. Members of this growing family of enzymes can catalyse the en bloc removal of polyubiquitin moieties or catalyse the trimming of the polyubiquitin chain136. Similar to glycan removal, removal of polyubiquitin moieties might be essential for a substrate to enter the proteasome. Polyubiquitin trimming might give proteasome-targeted substrates a second chance to escape degradation. In fact, mutations in a human de-ubiquitylating enzyme known as ataxin-3 result in spinocerebellar ataxia. Consistent with the contribution of ataxin-3 to ERAD, the enzyme binds to the derlin–VIMP complex and the human RAD23 homologue; it also binds to p97 and seems to act downstream of the Cdc48 complex during degradation. In addition, a dominant-negative ataxin-3 mutant induces the UPR and slows the ERAD of TCRα137. Additional de-ubiquitylating enzymes that facilitate ERAD are unidentified, and this remains an important area of investigation.
Interplay between ERAD and other pathways
The inefficient disposal or overproduction of aberrant proteins in the ER can compromise ER and cellular homeostasis. Therefore, ERAD must be regulated. Several studies showed that the transcription of a subset of factors required for ERAD is induced by the UPR61,138-140, which is activated through the directed interaction of misfolded proteins with a transmembrane sensor in the ER and/or through the titration of BiP away from this sensor141 (BOX 2). Other factors induced by the UPR reduce ER stress through more elaborate mechanisms: the volume of the ER expands by upregulated lipid synthesis, the concentration of molecular chaperones and enzymes required for post-translational modifications rises, protein translation and ER translocation decreases, and protein transport through the secretory pathway probably increases, thereby emptying this compartment of potentially toxic polypeptides. UPR induction might also result in the cleavage of ER-associated mRNAs that encode secreted proteins142. Although the individual deletion of genes that encode components of the ERAD or UPR pathways does not compromise the viability of yeast, simultaneous deletions lead to synthetic effects on growth. These data concur with the fact that the ERAD and UPR have complementary roles during ERQC.
Autophagy is another pathway that reduces ER stress, particularly from the threat of protein aggregation. If ERAD efficiency is compromised, substrates that accumulate over time might also aggregate, but in some cases this threat is reduced by autophagy-mediated destruction. During ER-stress-induced autophagy, portions of the ER, along with proteins and protein aggregates, are engulfed in double-membrane structures called autophagosomes and delivered to the lysosome or vacuole for degradation143. Therefore, autophagy serves as a backup for ERAD, at least for aggregation-prone substrates. One substrate for which this is well shown is a mutant, aggregation-prone and disease-causing form of α1-antitrypsin, known as the Z variant (see above for a discussion of the soluble NHK mutant). Although this protein is usually degraded by ERAD, it might accumulate and polymerize in the ER before being delivered for autophagic degradation144. This phenomenon has been recapitulated in cells from genetically engineered mice145 and yeast146. Although not proven, it is likely that the autophagosomes engulf ER fragments that contain the polymerized mutant protein.
UBA domain
(Ubiquitin-association domain). A domain of ∼45 amino acids that adopts a structure comprised of a three α-helical bundle. The domain binds to ubiquitin through a conserved hydrophobic surface patch.
Autophagosome
A double-membrane vesicle that is formed from elements of the cytoplasm and other organelles; it fuses with the vacuole or lysosome, in which the autophagosomal contents are subject to degradation.
The ERAD pathway is affected by and affects other cellular pathways, although the mechanistic basis for cross-pathway communication remains poorly defined. In mammalian cells, for example, cytoplasmic stresses (such as oxidative stress and heat shock) decrease ERAD efficiency147. Furthermore, induction of the heat-shock response in yeast can compensate for ER stress that is caused by the overexpression of Cpy* in cells that lack inositol-requiring protein-1 (Ire1) (REF. 148). Combined with other observations, these data suggest that factors induced by cytoplasmic stresses also reduce the physiological consequences of ER stress and the accumulation of misfolded proteins in the ER.
In the event that ER stress cannot be resolved, the apoptotic pathway can be induced. Several models have been proposed to explain how ER stress is linked to apoptosis. One model involves the cleavage and activation of an ER-membrane-localized caspase, caspase-12 (REF. 149). Caspase-12 might be activated by association with TNF-receptor-associated factor-2 (TRAF2)150, a protein that is recruited to the ER membrane by the cytoplasmic tail of IRE1, one of the UPR transducers in mammalian cells151. The IRE1–TRAF2 complex might in turn recruit and activate the apotosis signal-regulating kinase-1, which signals to downstream effectors, p38 and c-Jun N-terminal kinase152. An alternative model suggests that the ER kinase PERK, another UPR transducer in mammalian cells, upregulates C/EBP-homologous protein (CHOP), a transcriptional repressor that inhibits the expression of pro-survival BCL2 proteins and possibly induces a mitochondrial death-signalling cascade. CHOP overexpression results in cell-cycle arrest and induces apoptosis153. Regardless, the ER-stress signal does seem to be transmitted by the mitochondria, resulting in the induction of the mitochondrial intrinsic apoptotic pathway154.
Coupling folding, ERAD and ER exit
The core principle of ERQC is that only properly folded proteins can be allowed to transit to their final destinations through the secretory pathway. Thus, the ERAD recognition and targeting machineries must read the folded state of the protein. What follows is less clear: how do proteins that pass an ERQC checkpoint find their way to specialized ER-exit sites for transport from the ER? What is the mechanism that underlies the handover of folded proteins from the recognition or targeting machinery to components that select cargo for coatomer protein complex-II (COPII) export vesicles? A recent publication indicated that there is competition between the protein transport and ERAD selection machineries155, suggesting that the decision of whether to transport or degrade a secreted polypeptide might depend on other variables, such as cell type, stress, secretory-protein load and/or signalling processes.
Based on the rate of folding and the stability of a protein, there should be a way to predict whether a polypeptide will fold and transit properly, aggregate or be targeted for ERAD. By analysing the folding efficiency and stability of a large collection of mutant forms of transthyretin — which when mutated can lead to neurodegenerative disorders and cardiomyopathies — a model was formulated to predict the behaviour of a secreted protein by correlating these parameters with ERAD and ER export efficiencies156. In essence, the fate of a protein is determined more by a network of interacting factors and parameters, rather than the presence of a single event or interaction partner. Overall, through continued cellular and biophysical analyses of the behaviour of wild-type and mutant secreted proteins, there is hope that this model can be further refined. In turn, this will help define how chemical chaperones and modulators of protein biogenesis can be used as pharmaceutical agents to treat protein conformational diseases.
Conclusions and remaining questions
Because the term ERAD was coined only ∼13 years ago, one might think that the field is still in its infancy. However, based on the studies evaluated in this review, it should be obvious that the field has matured quickly. Such growth is due to facile genetic attacks and the development of complementary biochemical assays that have resulted in the isolation and characterization of ERAD-affecting factors. The field has also benefited from the recruitment of experts who study chaperone action, protein transport phenomena, the ubiquitin-proteasome system, immunology, toxicology, human genetics, and genomics and proteomics.
Nevertheless, many original questions remain unanswered. What is the nature of the retrotranslocon? Do membrane proteins with unique topologies use diverse ERAD pathways? What degree of functional redundancy or competition is exhibited by factors that act during individual steps in the ERAD pathway? How is the ERAD machinery regulated both hormonally and developmentally, and are there cell-specific proteins required for ERAD? Can the ERAD pathway be modulated pharmacologically to prevent human disease? Overall, based on the pace at which new discoveries have been made, we suspect that even these questions can be answered in the coming years.
Acknowledgements
Work in the laboratory of J.L.B. on ERAD is supported by grants from the National Institutes of Health and the Cystic Fibrosis Foundation. We thank K. Nakatsukasa for insightful discussions and comments on the manuscript, and we apologize to those researchers whose publications we could not include owing to space limitations.
Footnotes
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
UBC6
OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
Alzheimer's | cystic fibrosis | Huntington's | Parkinson's
UniProtKB: http://www.uniprot.org
BAG2 | CFTR | Cpy* | Der1 | Doa10 | ERDJ5 | ERP60 | GRP78 | HERP | Hrd1 | OS9 | Pdi1 | Sec61 | UGGT | Yos9
FURTHER INFORMATION
Jeffrey L. Brodsky's homepage: http://www.pitt.edu/~biohome/Dept/Frame/Faculty/brodsky.htm
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
ONLINE ONLY
- Endoplasmic reticulum (ER)-associated degradation (ERAD) is a secretory protein quality control process that results in the removal of aberrant proteins from the ER.
- ERAD substrates are selected by molecular chaperones that identify proteins that might be unable to fold, that fold slowly or contain a misfolded domain, or those that lack specific protein partners.
- Nearly all ERAD substrates are modified with ubiquitin, a 76 amino-acid peptide that helps target proteins to the proteasome. Specific E3 ubiquitin ligases are required for ERAD and reside in or near the ER membrane.
- ERAD substrates are degraded by the proteasome, a large multicatalytic protease that resides in the cytoplasm. Although integral membrane proteins in the ER can readily access the proteasome, soluble ERAD substrates (that reside within the lumen) must be retrotranslocated or dislocated from the ER to the cytoplasm before they are degraded.
- The ERAD pathway is conserved from yeast to humans, and indeed many of the factors that contribute to this pathway were first identified in the yeast Saccharomyces cerevisiae.
- A growing number of links between the ERAD pathway and human diseases have been identified.
Jeffrey Brodsky holds the Avinoff Chair in the Department of Biological Sciences at the University of Pittsburgh, Pennsylvania, USA. He joined the faculty at Pittsburgh in 1994, after completing his postdoctoral training in the laboratory of Randy Schekman at the University of California, Berkeley, USA.
Shruthi Vembar is a graduate student in the Brodsky laboratory and received her M.Sc. (Hons) degree at Birla Institute of Technology and Science in Pilani, India.
One step at a time: endoplasmic reticulum-associated degradation
Shruthi S. Vembar and Jeffrey L. Brodsky
The quality control process ERAD, endoplasmic reticulum (ER)-associated degradation, results in the removal of aberrant secreted proteins from the ER. Molecular chaperones and associated factors recognize and target substrates for retrotranslocation to the cytoplasm, where they are degraded by the ubiquitin-proteasome machinery.
References
- 1.Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 2001;26:597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 2.Despa F, Orgill DP, Lee RC. Molecular crowding effects on protein stability. Ann. NY Acad. Sci. 2005;1066:54–66. doi: 10.1196/annals.1363.005. [DOI] [PubMed] [Google Scholar]
- 3.Jahn TR, Radford SE. The yin and yang of protein folding. FEBS J. 2005;272:5962–5970. doi: 10.1111/j.1742-4658.2005.05021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 6.Kanapin A, et al. Mouse proteome analysis. Genome Res. 2003;13:1335–1344. doi: 10.1101/gr.978703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nature Rev. Mol. Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 8.McCracken AA, Brodsky JL. Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 1996;132:291–298. doi: 10.1083/jcb.132.3.291. [The development of an in vitro system led to the first demonstration that a mutated soluble protein in the ER could be exported to the cytoplasm en route to its degradation, and the term ERAD was coined to describe this process.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lippincott-Schwartz J, Bonifacino JS, Yuan LC, Klausner RD. Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell. 1988;54:209–220. doi: 10.1016/0092-8674(88)90553-3. [DOI] [PubMed] [Google Scholar]
- 10.Klausner RD, Sitia R. Protein degradation in the endoplasmic reticulum. Cell. 1990;62:611–614. doi: 10.1016/0092-8674(90)90104-m. [DOI] [PubMed] [Google Scholar]
- 11.Finger A, Knop M, Wolf DH. Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur. J. Biochem. 1993;218:565–574. doi: 10.1111/j.1432-1033.1993.tb18410.x. [DOI] [PubMed] [Google Scholar]
- 12.Otsu M, Urade R, Kito M, Omura F, Kikuchi M. A possible role of ER-60 protease in the degradation of misfolded proteins in the endoplasmic reticulum. J. Biol. Chem. 1995;270:14958–14961. doi: 10.1074/jbc.270.25.14958. [DOI] [PubMed] [Google Scholar]
- 13.Sommer T, Jentsch S. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature. 1993;365:176–179. doi: 10.1038/365176a0. [Provided the first link between the ubiquitin–proteasome pathway and the quality control of a mutated membrane protein in the yeast ER.] [DOI] [PubMed] [Google Scholar]
- 14.Jensen TJ, et al. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 15.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin–proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [Together with reference 14, the degradation of a disease-causing, mutated form of the cystic fibrosis transmembrane conductance regulator was found to require the ubiquitin–proteasome system.] [DOI] [PubMed] [Google Scholar]
- 16.Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl Acad. Sci. USA. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiller M, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin–proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [A misfolded form of yeast carboxypeptidase Y (Cpy*) was shown to transit from the ER into the cytoplasm where it was degraded by the proteasome.] [DOI] [PubMed] [Google Scholar]
- 18.Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [Using a yeast genetic screen, these investigators discovered that the degradation of a wild-type, membrane-integrated enzyme in the ER was metabolically regulated by virtue of its being targeted to the 26S proteasome. This indicated that the ERAD pathway could be used to modulate cellular homeostasis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiertz EJ, et al. The human cytomegalovirus us11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [The major histocompatibility class I protein was found to be dislocated from the ER and degraded by the proteasome in cells that express a unique human cytomegalovirus-encoded gene product. This study provided the first indication that the ERAD pathway could be co-opted by a pathogen.] [DOI] [PubMed] [Google Scholar]
- 20.Lord JM, Roberts LM, Lencer WI. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr. Top. Microbiol. Immunol. 2005;300:149–168. doi: 10.1007/3-540-28007-3_7. [DOI] [PubMed] [Google Scholar]
- 21.Sayeed A, Ng DTW. Search and destroy: ER quality control and ER-associated protein degradation. Crit. Rev. Biochem. Mol. Biol. 2005;40:75–91. doi: 10.1080/10409230590918685. [DOI] [PubMed] [Google Scholar]
- 22.Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 23.Nakatsukasa K, Brodsky JL. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic. 2008;9:861–870. doi: 10.1111/j.1600-0854.2008.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varga K, et al. Efficient intracellular processing of the endogenous cystic fibrosis transmembrane conductance regulator in epithelial cell lines. J. Biol. Chem. 2004;279:22578–22584. doi: 10.1074/jbc.M401522200. [DOI] [PubMed] [Google Scholar]
- 25.Knittler MR, Dirks S, Haas IG. Molecular chaperones involved in protein degradation in the endoplasmic reticulum: quantitative interaction of the heat shock cognate protein BiP with partially folded immunoglobulin light chains that are degraded in the endoplasmic reticulum. Proc. Natl Acad. Sci. USA. 1995;92:1764–1768. doi: 10.1073/pnas.92.5.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmitz A, Maintz M, Kehle T, Herzog V. In vivo iodination of a misfolded proinsulin reveals co-localized signals for Bip binding and for degradation in the ER. EMBO J. 1995;14:1091–1098. doi: 10.1002/j.1460-2075.1995.tb07092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong M, Bridges JP, Apsley K, Xu Y, Weaver TE. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol. Biol. Cell. 2008;19:2620–2630. doi: 10.1091/mbc.E07-07-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, et al. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell. 2001;12:1303–1314. doi: 10.1091/mbc.12.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Youker RT, Walsh P, Beilharz T, Lithgow T, Brodsky JL. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol. Biol. Cell. 2004;15:4787–4797. doi: 10.1091/mbc.E04-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meacham GC, et al. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999;18:1492–1505. doi: 10.1093/emboj/18.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of ΔF508-CFTR. Am. J. Physiol. Cell Physiol. 2000;278:C259–C267. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- 33.Alberti S, Bohse K, Arndt V, Schmitz A, Hohfeld J. The cochaperone HspBP1 inhibits the CHIP ubiquitin ligase and stimulates the maturation of the cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell. 2004;15:4003–4010. doi: 10.1091/mbc.E04-04-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arndt V, Daniel C, Nastainczyk W, Alberti S, Hohfeld J. BAG-2 acts as an inhibitor of the chaperone-associated ubiquitin ligase CHIP. Mol. Biol. Cell. 2005;16:5891–5900. doi: 10.1091/mbc.E05-07-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [The use of proteomic technologies and yeast genetics led to a molecular definition of components that constitute the ERAD-L machinery. The resulting data hinted at the existence of a chaperone-initiated pathway that leads to the recognition and ubiquitylation of ERAD-L substrates.] [DOI] [PubMed] [Google Scholar]
- 36.Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nature Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 37.Mimura N, et al. Altered quality control in the endoplasmic reticulum causes cortical dysplasia in knock-in mice expressing a mutant BiP. Mol. Cell. Biol. 2008;28:293–301. doi: 10.1128/MCB.00473-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caramelo JJ, Castro OA, Alonso LG, De Prat-Gay G, Parodi AJ. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc. Natl Acad. Sci. USA. 2003;100:86–91. doi: 10.1073/pnas.262661199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor SC, Thibault P, Tessier DC, Bergeron JJ, Thomas DY. Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO Rep. 2003;4:405–411. doi: 10.1038/sj.embor.embor797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. J. Biol. Chem. 2008;283:10221–10225. doi: 10.1074/jbc.R700048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fagioli C, Sitia R. Glycoprotein quality control in the endoplasmic reticulum. Mannose trimming by endoplasmic reticulum mannosidase I times the proteasomal degradation of unassembled immunoglobulin subunits. J. Biol. Chem. 2001;276:12885–12892. doi: 10.1074/jbc.M009603200. [DOI] [PubMed] [Google Scholar]
- 42.Hebert DN, Zhang JX, Chen W, Foellmer B, Helenius A. The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J. Cell Biol. 1997;139:613–623. doi: 10.1083/jcb.139.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kostova Z, Wolf DH. Importance of carbohydrate positioning in the recognition of mutated CPY for ER-associated degradation. J. Cell Sci. 2005;118:1485–1492. doi: 10.1242/jcs.01740. [DOI] [PubMed] [Google Scholar]
- 44.Spear ED, Ng DT. Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J. Cell Biol. 2005;169:73–82. doi: 10.1083/jcb.200411136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vanoni O, Paganetti P, Molinari M. Consequences of individual N-glycan deletions and of proteasomal inhibition on secretion of active BACE. Mol. Biol. Cell. 2008;19:4086–4098. doi: 10.1091/mbc.E08-05-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ireland BS, Brockmeier U, Howe CM, Elliott T, Williams DB. Lectin-deficient calreticulin retains full functionality as a chaperone for class I histocompatibility molecules. Mol. Biol. Cell. 2008;19:2413–2423. doi: 10.1091/mbc.E07-10-1055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Hammond C, Helenius A. Folding of VSV G protein: sequential interaction with BiP and calnexin. Science. 1994;266:456–458. doi: 10.1126/science.7939687. [DOI] [PubMed] [Google Scholar]
- 48.Stronge VS, Saito Y, Ihara Y, Williams DB. Relationship between calnexin and BiP in suppressing aggregation and promoting refolding of protein and glycoprotein substrates. J. Biol. Chem. 2001;276:39779–39787. doi: 10.1074/jbc.M107091200. [DOI] [PubMed] [Google Scholar]
- 49.Zhang JX, Braakman I, Matlack KE, Helenius A. Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins with C-terminal truncations. Mol. Biol. Cell. 1997;8:1943–1954. doi: 10.1091/mbc.8.10.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J. Cell Biol. 2002;158:247–257. doi: 10.1083/jcb.200204122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Svedine S, Wang T, Halaban R, Hebert DN. Carbohydrates act as sorting determinants in ER-associated degradation of tyrosinase. J. Cell Sci. 2004;117:2937–2949. doi: 10.1242/jcs.01154. [DOI] [PubMed] [Google Scholar]
- 52.Molinari M, Galli C, Vanoni O, Arnold SM, Kaufman RJ. Persistent glycoprotein misfolding activates the glucosidase II/UGT1-driven calnexin cycle to delay aggregation and loss of folding competence. Mol. Cell. 2005;20:503–512. doi: 10.1016/j.molcel.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 53.Le Fourn V, Siffroi-Fernandez S, Ferrand M, Franc JL. Competition between calnexin and BiP in the endoplasmic reticulum can lead to the folding or degradation of human thyroperoxidase. Biochemistry. 2006;45:7380–7388. doi: 10.1021/bi060415i. [DOI] [PubMed] [Google Scholar]
- 54.Tsai B, Rodighiero C, Lencer WI, Rapoport TA. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 2001;104:937–948. doi: 10.1016/s0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- 55.Schelhaas M, et al. Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell. 2007;131:516–529. doi: 10.1016/j.cell.2007.09.038. [DOI] [PubMed] [Google Scholar]
- 56.Oliver JD, Roderick HL, Llewellyn DH, High S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol. Biol. Cell. 1999;10:2573–2582. doi: 10.1091/mbc.10.8.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gillece P, Luz JM, Lennarz WJ, de La Cruz FJ, Romisch K. Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J. Cell Biol. 1999;147:1443–1456. doi: 10.1083/jcb.147.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ushioda R, et al. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- 59.Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol. Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valetti C, Sitia R. The differential effects of dithiothreitol and 2-mercaptoethanol on the secretion of partially and completely assembled immunoglobulins suggest that thiol-mediated retention does not take place in or beyond the Golgi. Mol. Biol. Cell. 1994;5:1311–1324. doi: 10.1091/mbc.5.12.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Travers KJ, et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [A microarray analysis was conducted to uncover the range of factors and processes that are induced by the unfolded protein response (UPR) in yeast, and genetic techniques showed the complementary nature of the ERAD and UPR pathways.] [DOI] [PubMed] [Google Scholar]
- 62.Doms RW, Keller DS, Helenius A, Balch WE. Role for adenosine triphosphate in regulating the assembly and transport of vesicular stomatitis virus G protein trimers. J. Cell Biol. 1987;105:1957–1969. doi: 10.1083/jcb.105.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Copeland CS, et al. Folding, trimerization, and transport are sequential events in the biogenesis of influenza virus hemagglutinin. Cell. 1988;53:197–209. doi: 10.1016/0092-8674(88)90381-9. [DOI] [PubMed] [Google Scholar]
- 64.Prabakaran D, Kim PS, Dixit VM, Arvan P. Oligomeric assembly of thrombospondin in the endoplasmic reticulum of thyroid epithelial cells. Eur. J. Cell Biol. 1996;70:134–141. [PubMed] [Google Scholar]
- 65.Fra AM, Fagioli C, Finazzi D, Sitia R, Alberini CM. Quality control of ER synthesized proteins: an exposed thiol group as a three-way switch mediating assembly, retention and degradation. EMBO J. 1993;12:4755–4761. doi: 10.1002/j.1460-2075.1993.tb06164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shenkman M, Ehrlich M, Lederkremer GZ. Masking of an endoplasmic reticulum retention signal by its presence in the two subunits of the asialoglycoprotein receptor. J. Biol. Chem. 2000;275:2845–2851. doi: 10.1074/jbc.275.4.2845. [DOI] [PubMed] [Google Scholar]
- 67.Shapira I, Charuvi D, Elkabetz Y, Hirschberg K, Bar-Nun S. Distinguishing between retention signals and degrons acting in ERAD. J. Cell Sci. 2007;120:4377–4387. doi: 10.1242/jcs.011247. [DOI] [PubMed] [Google Scholar]
- 68.Bonifacino JS, Cosson P, Klausner RD. Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell. 1990;63:503–513. doi: 10.1016/0092-8674(90)90447-m. [DOI] [PubMed] [Google Scholar]
- 69.Bole DG, Hendershot LM, Kearney JF. Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J. Cell Biol. 1986;102:1558–1566. doi: 10.1083/jcb.102.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Elkabetz Y, et al. Immunoglobulin light chains dictate vesicular transport-dependent and -independent routes for IgM degradation by the ubiquitin–proteasome pathway. J. Biol. Chem. 2003;278:18922–18929. doi: 10.1074/jbc.M208730200. [DOI] [PubMed] [Google Scholar]
- 71.Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [The development and use of model, modular ERAD substrates led to the first hint that the ERAD pathway could be subdivided into distinct substrate-specific pathways, which were named ERAD-L and ERAD-C.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huyer G, et al. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- 73.Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin–ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [A comprehensive mass spectrometry analysis was conducted to define the components that comprise the machineries required for ERAD-L and ERAD-C, and the existence of an ERAD-M pathway was confirmed.] [DOI] [PubMed] [Google Scholar]
- 74.Gauss R, Sommer T, Jarosch E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006;25:1827–1835. doi: 10.1038/sj.emboj.7601088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kota J, Gilstring CF, Ljungdahl PO. Membrane chaperone Shr3 assists in folding amino acid permeases preventing precocious ERAD. J. Cell Biol. 2007;176:617–628. doi: 10.1083/jcb.200612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 2008;132:101–112. doi: 10.1016/j.cell.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deshaies RJ, Sanders SL, Feldheim DA, Schekman R. Assembly of yeast Sec proteins involved in translocation into the endoplasmic reticulum into a membrane-bound multisubunit complex. Nature. 1991;349:806–808. doi: 10.1038/349806a0. [DOI] [PubMed] [Google Scholar]
- 78.Schulze A, et al. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J. Mol. Biol. 2005;354:1021–1027. doi: 10.1016/j.jmb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 79.Gardner RG, et al. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J. Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kanehara K, Kawaguchi S, Ng DT. The EDEM and Yos9p families of lectin-like ERAD factors. Semin. Cell Dev. Biol. 2007;18:743–750. doi: 10.1016/j.semcdb.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 81.Hosokawa N, et al. A novel ER α-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2:415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299:1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- 83.Molinari M, Calanca V, Galli C, Lucca P, Paganetti P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science. 2003;299:1397–1400. doi: 10.1126/science.1079474. [Together with reference 82, the role of the ER degradation enhancing mannosidase-like lectin, EDEM, was defined in mammalian cells. EDEM was shown to have a key role during the recognition and targeting of ERAD substrates in the ER.] [DOI] [PubMed] [Google Scholar]
- 84.Mast SW, et al. Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology. 2005;15:421–436. doi: 10.1093/glycob/cwi014. [DOI] [PubMed] [Google Scholar]
- 85.Olivari S, Galli C, Alanen H, Ruddock L, Molinari M. A novel stress-induced EDEM variant regulating endoplasmic reticulum-associated glycoprotein degradation. J. Biol. Chem. 2005;280:2424–2428. doi: 10.1074/jbc.C400534200. [DOI] [PubMed] [Google Scholar]
- 86.Hirao K, et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006;281:9650–9658. doi: 10.1074/jbc.M512191200. [DOI] [PubMed] [Google Scholar]
- 87.Buschhorn BA, Kostova Z, Medicherla B, Wolf DH. A genome-wide screen identifies Yos9p as essential for ER-associated degradation of glycoproteins. FEBS Lett. 2004;577:422–426. doi: 10.1016/j.febslet.2004.10.039. [DOI] [PubMed] [Google Scholar]
- 88.Bhamidipati A, Denic V, Quan EM, Weissman JS. Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol. Cell. 2005;19:741–751. doi: 10.1016/j.molcel.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 89.Kim W, Spear ED, Ng DT. Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol. Cell. 2005;19:753–764. doi: 10.1016/j.molcel.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 90.Szathmary R, Bielmann R, Nita-Lazar M, Burda P, Jakob CA. Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol. Cell. 2005;19:765–775. doi: 10.1016/j.molcel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 91.Gauss R, Jarosch E, Sommer T, Hirsch C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nature Cell Biol. 2006;8:849–854. doi: 10.1038/ncb1445. [DOI] [PubMed] [Google Scholar]
- 92.Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1–SEL1L ubiquitin ligase complex for ERAD. Nature Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hosokawa N, et al. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1–SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 2008;283:20914–20924. doi: 10.1074/jbc.M709336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Arteaga MF, Wang L, Ravid T, Hochstrasser M, Canessa CM. An amphipathic helix targets serum and glucocorticoid-induced kinase 1 to the endoplasmic reticulum-associated ubiquitinconjugation machinery. Proc. Natl Acad. Sci. USA. 2006;103:11178–11183. doi: 10.1073/pnas.0604816103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Deng M, Hochstrasser M. Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature. 2006;443:827–831. doi: 10.1038/nature05170. [DOI] [PubMed] [Google Scholar]
- 96.Park SH, et al. The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin–proteasome system. Mol. Biol. Cell. 2007;18:153–165. doi: 10.1091/mbc.E06-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmitz A, Herrgen H, Winkeler A, Herzog V. Cholera toxin is exported from microsomes by the Sec61p complex. J. Cell Biol. 2000;148:1203–1212. doi: 10.1083/jcb.148.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- 99.Pilon M, Schekman R, Romisch K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 1997;16:4540–4548. doi: 10.1093/emboj/16.15.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kalies KU, Allan S, Sergeyenko T, Kroger H, Romisch K. The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 2005;24:2284–2293. doi: 10.1038/sj.emboj.7600731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Scott DC, Schekman R. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J. Cell Biol. 2008;181:1095–1105. doi: 10.1083/jcb.200804053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retrotranslocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 103.Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 104.Wahlman J, et al. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell. 2007;129:943–955. doi: 10.1016/j.cell.2007.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kostova Z, Tsai YC, Weissman AM. Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Semin. Cell Dev. Biol. 2007;18:770–779. doi: 10.1016/j.semcdb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ploegh HL. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature. 2007;448:435–438. doi: 10.1038/nature06004. [DOI] [PubMed] [Google Scholar]
- 108.Richly H, et al. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 109.Kohlmann S, Schafer A, Wolf DH. Ubiquitin ligase Hul5 is required for fragment-specific substrate degradation in endoplasmic reticulum-associated degradation. J. Biol. Chem. 2008;283:16374–16383. doi: 10.1074/jbc.M801702200. [DOI] [PubMed] [Google Scholar]
- 110.Jarosch E, et al. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nature Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- 111.Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nature Cell Biol. 2001;3:24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- 112.Biederer T, Volkwein C, Sommer T. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 1997;278:1806–1809. doi: 10.1126/science.278.5344.1806. [DOI] [PubMed] [Google Scholar]
- 113.Haynes CM, Caldwell S, Cooper AA. An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER–Golgi transport. J. Cell Biol. 2002;158:91–101. doi: 10.1083/jcb.200201053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kikkert M, et al. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- 115.Hassink G, et al. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 2005;388:647–655. doi: 10.1042/BJ20041241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yoshida Y, et al. E3 ubiquitin ligase that recognizes sugar chains. Nature. 2002;418:438–442. doi: 10.1038/nature00890. [DOI] [PubMed] [Google Scholar]
- 117.Younger JM, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 118.Morito D, et al. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRΔF508. Mol. Biol. Cell. 2008;19:1328–1336. doi: 10.1091/mbc.E07-06-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhong X, et al. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J. Biol. Chem. 2004;279:45676–45684. doi: 10.1074/jbc.M409034200. [DOI] [PubMed] [Google Scholar]
- 120.Imai Y, et al. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 121.Imai Y, et al. CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol. Cell. 2002;10:55–67. doi: 10.1016/s1097-2765(02)00583-x. [DOI] [PubMed] [Google Scholar]
- 122.Mayer TU, Braun T, Jentsch S. Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. EMBO J. 1998;17:3251–3257. doi: 10.1093/emboj/17.12.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee RJ, et al. Uncoupling retro-translocation and degradation in the ER-associated degradation of a soluble protein. EMBO J. 2004;23:2206–2215. doi: 10.1038/sj.emboj.7600232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jentsch S, Rumpf S. Cdc48 (p97): a ‘molecular gearbox’ in the ubiquitin pathway? Trends Biochem. Sci. 2007;32:6–11. doi: 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 125.Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nature Cell Biol. 2005;7:993–998. doi: 10.1038/ncb1298. [DOI] [PubMed] [Google Scholar]
- 126.Schuberth C, Buchberger A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nature Cell Biol. 2005;7:999–1006. doi: 10.1038/ncb1299. [DOI] [PubMed] [Google Scholar]
- 127.Flierman D, Ye Y, Dai M, Chau V, Rapoport TA. Polyubiquitin serves as a recognition signal, rather than a ratcheting molecule, during retrotranslocation of proteins across the endoplasmic reticulum membrane. J. Biol. Chem. 2003;278:34774–34782. doi: 10.1074/jbc.M303360200. [DOI] [PubMed] [Google Scholar]
- 128.Carlson EJ, Pitonzo D, Skach WR. p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J. 2006;25:4557–4566. doi: 10.1038/sj.emboj.7601307. [Using a novel in vitro system, this study showed that the p97 (Cdc48) complex reliance on ERAD correlates with the membrane stability of the substrate.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Verma R, et al. Proteasomal proteomics: identification of nucleotide-sensitive proteasome-interacting proteins by mass spectrometric analysis of affinity-purified proteasomes. Mol. Biol. Cell. 2000;11:3425–3439. doi: 10.1091/mbc.11.10.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Raasi S, Wolf DH. Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin. Cell Dev. Biol. 2007;18:780–791. doi: 10.1016/j.semcdb.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 131.Medicherla B, Kostova Z, Schaefer A, Wolf DH. A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 2004;5:692–697. doi: 10.1038/sj.embor.7400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li G, Zhao G, Zhou X, Schindelin H, Lennarz WJ. The AAA ATPase p97 links peptide N-glycanase to the endoplasmic reticulum-associated E3 ligase autocrine motility factor receptor. Proc. Natl Acad. Sci. USA. 2006;103:8348–8353. doi: 10.1073/pnas.0602747103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim I, et al. The Png1–Rad23 complex regulates glycoprotein turnover. J. Cell Biol. 2006;172:211–219. doi: 10.1083/jcb.200507149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Husnjak K, et al. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature. 2008;453:481–488. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rivett AJ. Proteasomes: multicatalytic proteinase complexes. Biochem. J. 1993;291:1–10. doi: 10.1042/bj2910001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 137.Wang Q, Li L, Ye Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J. Cell Biol. 2006;174:963–971. doi: 10.1083/jcb.200605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Casagrande R, et al. Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Mol. Cell. 2000;5:729–735. doi: 10.1016/s1097-2765(00)80251-8. [DOI] [PubMed] [Google Scholar]
- 139.Ng DT, Spear ED, Walter P. The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J. Cell Biol. 2000;150:77–88. doi: 10.1083/jcb.150.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nature Cell Biol. 2000;2:379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- 141.Kimata Y, et al. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol. 2007;179:75–86. doi: 10.1083/jcb.200704166. [The induction of the unfolded protein response in yeast was found to require the clustering of Ire1, the UPR sensor, regulation by BiP and probably the direct binding of Ire1 to unfolded polypeptides.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 143.Nair U, Klionsky DJ. Molecular mechanisms and regulation of specific and nonspecific autophagy pathways in yeast. J. Biol. Chem. 2005;280:41785–41788. doi: 10.1074/jbc.R500016200. [DOI] [PubMed] [Google Scholar]
- 144.Perlmutter DH. The role of autophagy in α-1-antitrypsin deficiency: a specific cellular response in genetic diseases associated with aggregation-prone proteins. Autophagy. 2006;2:258–263. doi: 10.4161/auto.2882. [DOI] [PubMed] [Google Scholar]
- 145.Kamimoto T, et al. Intracellular inclusions containing mutant α1-antitrypsin Z are propagated in the absence of autophagic activity. J. Biol. Chem. 2006;281:4467–4476. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- 146.Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human α-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol. Biol. Cell. 2006;17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kelly SM, Vanslyke JK, Musil LS. Regulation of ubiquitin–proteasome system mediated degradation by cytosolic stress. Mol. Biol. Cell. 2007;18:4279–4291. doi: 10.1091/mbc.E07-05-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liu Y, Chang A. Heat shock response relieves ER stress. EMBO J. 2008;27:1049–1059. doi: 10.1038/emboj.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nakagawa T, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 150.Yoneda T, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 151.Urano F, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 152.Nishitoh H, et al. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 153.Barone MV, Crozat A, Tabaee A, Philipson L, Ron D. CHOP (GADD153) and its oncogenic variant, TLS-CHOP, have opposing effects on the induction of G1/S arrest. Genes Dev. 1994;8:453–464. doi: 10.1101/gad.8.4.453. [DOI] [PubMed] [Google Scholar]
- 154.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 155.Kincaid MM, Cooper AA. Misfolded proteins traffic from the endoplasmic reticulum (ER) due to ER export signals. Mol. Biol. Cell. 2007;18:455–463. doi: 10.1091/mbc.E06-08-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, Balch WE. An adaptable standard for protein export from the endoplasmic reticulum. Cell. 2007;131:809–821. doi: 10.1016/j.cell.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 157.van Anken E, Braakman I. Endoplasmic reticulum stress and the making of a professional secretory cell. Crit. Rev. Biochem. Mol. Biol. 2005;40:269–283. doi: 10.1080/10409230500315352. [DOI] [PubMed] [Google Scholar]
- 158.Lin JH, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]