

Abstract
The epoxygenase metabolite, 11,12-epoxyeicosatrienoic acid (11,12-EET), has renal vascular actions. 11,12-EET analogs have been developed to determine the structure activity relationship for 11,12-EET and as a tool to investigate signaling mechanisms responsible for afferent arteriolar dilation. We hypothesized that 11,12-EET mediated afferent arteriolar dilation involves increased phosphoprotein phosphatase 2A (PP2A) and large-conductance calcium activated K+ (KCa) channels. We evaluated the chemically and/or metabolically stable 11,12-EET analogs: 11,12-EET-N-methylsulfonimide (11,12-EET-SI), 11-nonyloxy-undec-8(Z)-enoic acid (11,12-ether-EET-8-ZE), and 11,12-trans-oxidoeicosa-8(Z)-eonoic acid (11,12-tetra-EET-8-ZE). Afferent arteriolar responses were assessed. Activation of KCa channels by 11,12-EET analogs were established by single cell channel recordings in renal myocytes. Assessment of renal vascular responses revealed that 11,12-EET analogs increased afferent arteriolar diameter. Vasodilator responses to 11,12-EET analogs were abolished by K+ channel or PP2A inhibition. 11,12-EET analogs activated renal myocyte large-conductance KCa channels. 11,12-EET analogs increased cAMP by 2-fold and PP2A activity increased 3-8 fold in renal myocytes. PP2A inhibition did not significantly affect the 11,12-EET analog mediated increase in cAMP and PP2A increased renal myocyte KCa channel activity to a much greater extent than PKA. These data support the concept that 11,12-EET utilizes PP2A dependent pathways to activate large-conductance KCa channels and dilate the afferent arteriole.
Keywords: epoxyeicosatrienoic acids, endothelium-derived hyperpolarizing factor, vascular smooth muscle, CYP450 metabolites
Introduction
Over the past decade considerable interest has focused on the arachidonic acid CYP450 pathway. Substantial evidence has accumulated demonstrating that monooxygenase metabolites are involved in the regulation of epithelial transport and vascular smooth muscle cell function [3,16,34,38]. Afferent arterioles are the primary preglomerular vessels that contribute to glomerular capillary hemodynamics [16,30]. Epoxyeicosatrienoic acids (EETs) dilate preglomerular arterioles by activating large-conductance calcium-activated K+ (KCa) channels and hyperpolarizing smooth muscle cells [5,21,48]. Alterations in afferent arteriolar function and regulation of the CYP450 pathway contribute to renal pathologies [16,20]. The kidney and renal microvascular formation of arachidonic acid CYP450 metabolites is altered in hypertension, hepatorenal syndrome, diabetes and pregnancy, and it is likely that changes in epoxygenase metabolites contribute to the abnormalities in vascular function that are associated with these conditions [3,16,34,38]. Although the importance of the vascular epoxygenase pathway is now well recognized, many aspects concerning EET cell-signaling mechanisms remain unresolved.
Epoxygenase metabolites have been identified as EDHFs in the kidney whereas their metabolic breakdown product, dihydroxyeicosatrienoic acids (DHETEs) are devoid of renal vascular activity [16,20]. EETs act as vasodilators in a number of vascular beds by activating KCa channels resulting in vascular smooth muscle cell hyperpolarization [1,2,12,13,14]. Moreover, 11,12-EET and 14,15-EET are the most active epoxides on the renal microvasculature with 11,12-EET being more potent than 14,15-EET [5,16,20]. It is now becoming clear that 11,12-EET and 14,15-EET are the primary epoxide metabolites that possess vascular biological activity. As for the renal vasodilator actions of 11,12-EET, one possible mechanism by which 11,12-EET may decrease afferent arteriolar resistance is activation of protein kinase A (PKA) [18]. There is also evidence that supports a role for ADP ribosylation of coronary artery large-conductance KCa channels as a mechanism responsible for vasodilation in the heart [25,26]. Besides the EET cell-signaling mechanisms, very little is known about the structure requirement for EET activity. A recognition/binding domain map for 11,12-EET mediated inhibition of tumor necrosis factor-α (TNF-α) induced vascular cell adhesion molecule-1 (VCAM-1) expression in cytokine activated human endothelial cells was recently described by J.R. Falck et al. [11]. Nevertheless, the structural features of 11,12-EET that determine vasodilator properties are not known. Therefore, the aims of the present study were to identify afferent arteriolar cell signaling pathways involved in the vasodilator response to 11,12-EET and to identify structural requirements essential for smooth muscle cell relaxation.
Experimental Procedures
Animals
All experiments involving animals were carried out according to the guidelines of Medical College of Georgia Institutional Animal Care and Use Committee. Male Sprague-Dawley rats weighing 351 ± 9g (Charles River Laboratories) were used for this study. The rats were housed in separate cages, maintained in a temperature and light-controlled room and had access to standard chow and drinking water. Tissue harvesting was conducted under sterile conditions in pentobarbital (40mg/kg i.p.) anaesthetized animals. Animals were euthanized after tissue harvesting.
Renal microvascular diameter responses
Experiments were peformed using the in vitro perfused juxtamedullary nephron preparation as described previously [17]. The isolated kidney was perfused with a reconstituted red blood cell containing solution and renal artery perfusion pressure was set to 100mmHg. A single blood vessel was chosen from each kidney for each experiment. Following a 20min equilibration period, an afferent arteriole, efferent arteriole or vasa recta vessel was chosen for study and control vascular diameter determined. Norepinephrine (0.5μM) was added to the preparation to elevate vascular tone. Following norepinephrine treatment, the afferent arteriole, efferent arteriole or vasa recta vessel diameter response to the N-methylsulfonimide analog of 11,12-EET (11,12-EET-SI) was determined. Stock solutions of 11,12-EET analogs in ethanol were kept in sealed vials and stored at −80 C until the experiment. Immediately before use the stock solution was added to the superfusion solutions and the final concentration of ethanol vehicle was <0.05% (vol/vol). We have previously demonstrated that this concentration of ethanol does not alter afferent arteriolar diameter or channel activity [6,7,13,29]. 11,12-EET-SI was designed to resist eserification and β-oxidation while retaining full biological activity [18]. Afferent arteriolar diameters were measured at 15s intervals using a digital image-shearing monitor. The image-shearing device is accurate to within 0.2% of the screen width or 0.2μm and measurements reproducibility is within 0.5μm. Steady-state diameter was attained by the end of two minutes and the average diameter at three to five minutes for each 11,12-EET-SI concentration was utilized for statistical analysis.
In additional series of experiments, the afferent arteriolar diameter responses to 11,12-EET analogs were determined. A list of the 11,12-EET analogs tested is provided in Table 1. Once the activity of the 11,12-EET analogs was assessed, the contributions of various signal transduction pathways to the 11,12-EET afferent arteriolar dilator responses were investigated. The contribution of protein phosphatase 2A (PP2A) to the 11,12-EET dilator responses was determined using the PP2A inhibitor, okadaic acid (10nM) [15,29,44]. Tetraethylammonium (1mM) [42,44], apamin (1μM) [37,40,42], iberiotoxin (100nM) [24,40], charybdotoxin (10nM and 100nM) [37,40,42,43] and TRAM-34 (1μM) [27] were used to determine the K+ channel contribution. Afferent arteriolar diameter responses were evaluated using the following protocol. After the addition of norepinephrine, 11,12-EET analogs were added to the preparation and concentration diameter response curves determined. After a recovery period, K+ channel or PP2A inhibitors were added to the preparation for 20 minutes. 11,12-EET analog concentration diameter response curves were repeated in the presence of the inhibitors. No differences in the repeat afferent arteriolar diameter responses to 11,12-EET analogs (n=3-4) were observed in time control experiments.
Table 1.
Structures and names of 11,12-epoxyeicosatrienoic (EET) analogs.
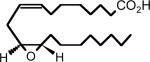 |
|
| Full name: 11,12-EET-methylsulfonimide | Full name: 11,12-trans-oxido-eicosa-8(Z)-enoic acid |
| Abbreviation: 11,12-EET-SI | Abbreviation: 11,12-tetra-EET-8-ZE |
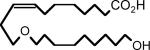 |
|
| Full name: 11-nonyloxy-undec-8(Z)-enoic acid | Full name: 11-(9-hydroxy-nonyloxy)-undec-8(Z)-enoic acid |
| Abbreviation: 11,12-ether-EET-8-ZE | Abbreviation: 11,12-ether-EET-8-ZE-OH |
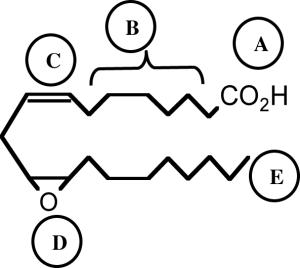 | |
| Recognition/binding domain map for 11,12-EET. The structural requirement for 11,12-EET to cause afferent arteriolar dilation includes five general regions: A.) ionic attraction of the carboxylate at carbon-1; B.) the Δ-5,6 olefin region is lipophilic and makes a minor contribution to recognition; C.) the Δ8,9-olefin double bond; D.) an epoxide positioned between C(11) and C(12); E.) a terminal lipophilic pocket. | |
Isolation of renal myocytes
Male Sprague-Dawley rats were anesthetized with pentobarbital, and the abdominal cavity exposed to permit cannulation of the abdominal aorta via the superior mesenteric artery. Renal microvessels were isolated according to a method described previously [17]. Briefly, the kidneys were infused with a physiological salt solution composed of 0.1mM CaCl2, 125.0mM NaCl, 5.0mM KCl, 1.0mM MgCl2, 10.0mM glucose, 20.0mM HEPES (100μM Ca2+ PSS) and 6% bovine serum albumin and the renal microvessels were separated from the rest of the cortex with the aid of sequential sieving, a digestion period and collection under a stereomicroscope. Single cell myoytes were isolated by the previously described procedure [7,48]. The rema; microvessels were incubated at 37°C in a solution containing 10mg papain, 3mg dithiothereitol, and 0.02% bovine serum albumin for 30 minutes. Then the vessels were gently triturated and the solution was removed after centrifuging at 500 rpm for 15 minutes. The pellet was resuspended in high potassium medium containing (mM): 140 KCl, 10 MgCl2, 0.1 CaCl2, 10 HEPES and 30 glucose (pH 7.4; 22-25°C).
Patch-clamp studies
Single myocytes were isolated and potassium channel activity was recorded from cell-attached patches as previously described [7]. Briefly, for cell-attached patches several drops of cell suspension were placed in a recording chamber containing (mM): 140 KCl, 10 MgCl2, 0.1 CaCl2, 10 HEPES and 30 glucose (pH 7.4; 22-25°C). Activity of single potassium channels was recorded in cell-attached patches by filling the patch pipette (2-5 MΩ) with Ringer solution (mM): 110 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2 and 10 HEPES. In experiments recording potassium channel activity of inside-out patches the bathing solution exposed to the cytoplasmic surface of the membrane consisted of the following (mM): 60 K2SO4, 30 KCl, 2 MgCl2, 0.16 CaCl2, 1 BAPTA (pCa 7), 10 HEPES, 5 ATP, and 10 glucose (pH 7.4; 22 – 25°C). The pipette solution was the same Ringer's solution described above. In another set of inside-out patch experiments single-channel current-voltage relationships were measured under symmetrical potassium (140mM) conditions, and the conductance was calculated from the slope of the current-voltage relationship fitted by linear regression. Average channel activity (NPo) in patches with multiple large-conductance KCa channels was determined as described previously [7].
Renal microvascular cell cultures
Renal microvascular smooth muscle cells were harvested as described above. The smooth muscle cells were cultured at 37 C in a growth medium containing Dulbecco's modified medium, 10% fetal calf and an antibiotic mixture of penicillin (100U/ml) and streptomycin (100mg/ml) serum. Relatively pure (>95%) renal myocyte cultures were confirmed by immunofluorescence staining with α-actin antibodies. All passages were performed with a disposable cell scraper and only renal microvascular smooth muscle cells of less than six passages were used. Renal myocytes were serum starved for 24 hours prior to experiments. Confluent renal vascular smooth muscle cells were treated with 11,12-EET analogs and inhibitors for the indicated time intervals.
PP2A activity assay
PP2A activity was determined in cultured renal microvascular smooth muscle cells with the Ser/Thr phosphatase activity assay by Promega (Madison, WI). Briefly, cell lysates were incubated with 4μg of anti-PP2A antibodies at 4°C (Upstate, Lake Placid, NY). After 3 hours, protein A/G plus agarose was added and allowed to form a complex for one hour at 4°C. Immuno-complexes were washed three times with PP2A buffer (250mM imidazole, 1mM EGTA, 0.1% β-mercaptoethanol and 0.5mg/ml BSA). Phosphatase activity was assayed by suspending the final pellet in 15ml PP2A buffer containing 1mM phophopeptide substrate RRA(pT)VA for 15 minutes at 24°C. The reaction was stopped by the addition of 50μl molybdate dye solution. The supernatants were transferred to a plate and absorbance was read at 600nm.
Cyclic AMP (cAMP) and Cyclic GMP (cGMP) assays
An enzyme immunometric assay kit (Biomol Pep Tag Assay, Plymoth Meeting, PA) that included all reagents and antibodies was used to measure cAMP and cGMP levels. Briefly, cultured cells were exposed to 1μM 11,12-EET analogs, forskolin 10μM, or sodium nitroprusside 10μM for 30 minutes at 37 C in the presence of 0.1mM isobutylmethylxantine (IBMX). The cells were rapidly washed and lysed by adding 0.1N HCl and kept on ice for 15 minutes. The concentration of cAMP or cGMP in the cell lysate was determined by colorimetric analysis.
Drugs
11,12-EET analogs were synthesized in the laboratory of Dr. J.R. Falck as previously described [11]. Norepinephrine was acquired from Sanofi/Winthrop Pharmaceuticals (San Diego, CA). Papain was obtained from Worthington Biochemical Corp. (Lakewood, NJ). All other chemicals and reagents were obtained from Sigma Chemical (St. Louis, MO).
Statistical Analysis
All data were expressed as means ± SE. Statistical significance between two groups was evaluated with Student's t-test, and a one-way ANOVA was performed to evaluate significance between multiple groups. A p value < 0.05 was considered to reflect a significant difference.
Results
Renal arteriolar responses to 11,12-EET analogs
Previous studies have determined that the functional actions and signaling mechanisms of the sulfonimide analogs of 11,12-EET and 14,15-EET were identical to the actions of the endogenous epoxides [4,10,11,13,18]. As previously reported [10,20], 11,12-EET-SI increased the preglomerular afferent arteriolar diameter by 17±2% (Figure 1). The postglomerular efferent arterioles also dilated in response to 11,12-EET-SI; however, this vascular response was weak compared to the afferent arteriolar dilation. Efferent arteriolar diameter increased by 9 ± 1% in response to 0.1μM 11,12-EET-SI. In contrast to the arterioles, the descending vasa recta were unresponsive to 11,12-EET-SI. Subsequent studies evaluating 11,12-EET analogs to determine structural requirements for activity were conducted on afferent arterioles because this preglomerular arteriole was the most responsive renal microvascular segment.
Figure 1.

Renal vascular responses to the N-methylsulfonimide analog of 11,12-EET (11,12-EET-SI). In the presence of 0.5μM norepinephrine, control afferent arteriolar diameter averaged 21.1 ± 1.2μm (n=6), efferent arteriolar diameter averaged 19.3 ± 2.2μm (n=5), and descending vasa recta diameter averaged 15.3 ± 2.1μm (n=5) prior to 11,12-EET-SI administration. * Significant difference from control diameter.
Initial studies were performed to determine the afferent arteriolar response to various 11,12-EET analogs. Afferent arteriolar diameter averaged 22 ± 1μm at a renal perfusion pressure of 100mmHg and decreased to 18 ± 1μm in response to 0.5μM norepinephrine. The increase in diameter from the norepinephrine-constricted control state was determined. 11,12-EET analogs, 11,12-tetra-EET-8-ZE and 11,12-ether-EET-8-ZE also dilated the afferent arteriole to the same extent as 11,12-EET-SI. Afferent arteriolar diameter increased by 18 ± 1% (n=22), 21 ± 2% (n=33) and 17 ± 1 % (n=18) in response to 1μM 11,12-EET-SI, 11,12-ether-EET-8-ZE and 11,12-tetra-EET-8-ZE, respectively. Although the afferent arteriolar diameter increased in response to 11,12-ether-EET-8-ZE, hydroxylation of C(20) significantly decreased activity of this 11,12-EET analog. Afferent arteriolar diameter increased by 3 ± 2% (n=4) in response to 1μM 11,12-ether-EET-8-ZE-OH. Thus, 11,12-EET-SI, 11,12-ether-EET-8-ZE and 11,12-tetra-EET-8-ZE were utilized to investigate afferent arteriolar cell signaling mechanisms that could contribute to vasodilation.
Potassium channel contributions to the afferent arteriole dilator responses to 11,12-EET analogs were determined. Figure 2 depicts the effect of the K+ channel inhibitor, TEA (1mM), on the afferent arteriolar diameter response to 11,12-EET analogs. Afferent arteriolar dilator responses averaged 17% when superfused with 11,12-EET analogs at a dose of 1μM and increases in vessel caliber were attenuated by greater than 90% in the presence of TEA. Inhibition of the large-conductance KCa channel by iberiotoxin also eliminated the afferent arteriolar dilation to 11,12-EET analogs (Figure 3). On the contrary, the small-conductance KCa channel inhibitor apamin did not alter the afferent arteriolar dilation to the three 11,12-EET analogs (Figure 4, response to 11,12-ether-EET-8-ZE shown). Afferent arteriolar diameters increased by 6.9 ± 1.0 %, 12.7 ± 0.9%, and 17.3 ± 1.0% (n=13) in response to 0.01, 0.1 and 1μM of the 11,12-EET analogs before treatment and 6.8 ± 0.7%, 12.9 ± 1.5%, and 17.0 ± 1.6% in response to the 11,12-EET analogs after apamin administration. Charybdotoxin (10nM), an inhibitor of intermediate-conductance KCa and voltage gated K+ (Kv) channels [40,42,43], also did not affect the ability of the afferent arteriolar to dilate in response to 11,12-EET analogs (Figure 4, response to 11,12-ether-EET-8-ZE shown). Afferent arteriolar diameters increased by 7.2 ± 1.3 %, 12.4 ± 1.4%, and 17.4 ± 1.3% (n=14) in response to 0.01, 0.1 and 1μM of the 11,12-EET analogs before treatment and 6.0 ± 0.7%, 12.5 ± 0.8%, and 15.0 ± 1.0% in response to the 11,12-EET analogs after charybdotoxin treatment. A higher dose of charybdotoxin (100nM), that is known to inhibit the large-conductance KCa channel [40,42,43], greatly attenuated the afferent arteriolar dilator response to 11,12-ether-EET-8-ZE. Afferent arteriolar diameters increased by 8.3 ± 1.3 %, 18.7 ± 1.6%, and 27.4 ± 3.6% (n=4) in response to 0.01, 0.1 and 1μM of 11,12-ether-EET-8-ZE before treatment and 3.7 ± 0.8%, 3.2 ± 1.8%, and 2.5 ± 1.3% in response to 11,12-ether-EET-8-ZE after charybdotoxin (100nM) administration. TRAM-34, a more selective inhibitor of intermediate-conductance KCa [27], also did not alter the afferent arteriolar dilator response to 11,12-ether-EET-8-ZE (Figure 4). Lastly, we assessed the effect of PP2A inhibition on the afferent arteriolar dilation to 11,12-EET analogs. Administration of okadaic acid (10nM) attenuated the increase in afferent arteriolar diameter to 11,12-EET analogs by greater than 95% (Figure 5). These results indicate that PP2A and large-conductance KCa channels are involved in the 11,12-EET induced afferent arteriolar dilation.
Figure 2.

Effect of tetraethylammonium (TEA, 1mM) on the afferent arteriolar response to the Nmethylsulfonimide analog of 11,12-EET (11,12-EET-SI, n=6), 11-nonyloxy-undec-8(Z)-enoic acid (11,12-ether-EET-8-ZE, n=6) and 11,12-trans-oxido-eicosa-8(Z)-enoic acid (11,12-tetra-EET-8ZE, n=6). In the presence of 0.5μM norepinephrine, control diameter averaged 18.4 ± 1.3μm and after administration of TEA averaged 18.7 ± 0.8μm. * Significant difference from control diameter. † Significant difference from the control diameter response to the 11,12-EET analog.
Figure 3.

Effect of iberiotoxin (100nM) on the afferent arteriolar response to the Nmethylsulfonimide analog of 11,12-EET (11,12-EET-SI, n=5), 11-nonyloxy-undec-8(Z)-enoic acid (11,12-ether-EET-8-ZE, n=5) and 11,12-trans-oxido-eicosa-8(Z)-enoic acid (11,12-tetra-EET-8ZE, n=5). In the presence of 0.5μM norepinephrine, control diameter averaged 16.0 ± 0.9μm and after administration of iberiotoxin averaged 16.2 ± 0.5μm. * Significant difference from control diameter. † Significant difference from the control diameter response to the 11,12-EET analog.
Figure 4.

Effect of apamin (1μM), charybdotoxin (10nM) and TRAM-34 (1μM) on the afferent arteriolar response to 11-nonyloxy-undec-8(Z)-enoic acid (11,12-ether-EET-8-ZE, apamin, n=5; charybdotoxin, n=4; and TRAM-34, n=5). In the presence of 0.5μM norepinephrine, control diameter averaged 18.6 ± 0.8μm and after administration of inhibitors averaged 18.5 ± 0.7μm. * Significant difference from control diameter.
Figure 5.

Effect of okadaic acid (10nM) on the afferent arteriolar response to the Nmethylsulfonimide analog of 11,12-EET (11,12-EET-SI, n=6), 11-nonyloxy-undec-8(Z)-enoic acid (11,12-ether-EET-8-ZE, n=5) and 11,12-trans-oxido-eicosa-8(Z)-enoic acid (11,12-tetra-EET-8-ZE, n=4). In the presence of 0.5μM norepinephrine, control diameter averaged 19.5 ± 1.1μm and after administration of okadaic acid averaged 21.1 ± 1.3μm. * Significant difference from control diameter. † Significant difference from the control diameter response to the 11,12-EET analog.
11,12-EET analog effects on renal myocyte KCa channels and cAMP, cGMP and PP2A levels
The ability of 11,12-EET analogs to activate KCa channels was assessed by conducting patch-clamp studies in renal microvascular smooth muscle cells. Renal myocyte cell membranes contain a large-conductance (191pS) KCa channel (Figure 6A). 11,12-EET analogs increased the open probability of the large-conductance KCa channel (Figure 6B). 11,12-EET-SI (1μM) increased KCa channel activity (NPo) from 0.001 ± 0.0006 to 0.067 ± 0.033 (n=6); 11,12-ether-EET-8-ZE (1μM) increased NPo from 0.0004 ± 0.0001 to 0.125 ± 0.11 (n=4); and 11,12-tetra-EET-8-ZE (1mM) increased NPo from 0.003 ± 0.003 to 0.065 ± 0.04 (n=6). This 11,12-EET analog-mediated increase in KCa channel activity was greatly attenuated in the presence of PP2A inhibition (Figure 6C). 11,12-EET analogs increased NPo from 0.006 ± 0.002 to 0.085 ± 0.034 and was attenuated to 0.023 ± 0.015 (n=7) after the addition of okadaic acid (10 nM). Additionally, 11,12-EET analogs increased PP2A activity by 658% in cultured renal microvascular smooth muscle cells (Figure 6D). These data suggest that KCa channel activation by 11,12-EET analogs involves PP2A.
Figure 6.

Effect of 11,12-EET analogs on the renal microvascular smooth muscle cell largeconductance calcium-activated potassium channels (large-conductance KCa). Panel A: Average current-voltage relationship for single-channel activity recorded from insideout patches with symmetrical (140mM) K+. Each point represents the mean ± SE current for 4 to 5 patches, with the line fit by linear regression. The conductance for the single channel was 191.1 ± 3pS. Panel B: Representative current traces from the same cell-attached patch (+50mV) before and 10 minutes after addition of 1 μM 11,12-EET analogs. Renal myocyte large-conductance KCa channel response to the Nmethylsulfonimide analog of 11,12-EET (11,12-EET-SI), 11-nonyloxy-undec-8(Z)-enoic acid (11,12-ether-EET-8-ZE) and 11,12-trans-oxido-eicosa-8(Z)-enoic acid (11,12-tetra-EET-8-ZE). Left side: I. control; Right side: II. 11,12-EET analog response. Panel C: Representative traces of the effect of okadaic acid on the renal microvascular smooth muscle cell large-conductance KCa channels to 11,12-EET-SI (n=3) and 11,12-tetra-EET-8-ZE (n=4). Left side: I. 11,12-EET analog response; Right side: II. 11,12-EET analog responses in the presence of okadaic acid (10nM). Panel D: Percentage change of PP2A activity in cells stimulated with the N-methylsulfonimide analog of 11,12-EET (11,12-EET-SI, n=4), 11-nonyloxy-undec-8(Z)-enoic acid (11,12-ether-EET-8-ZE, n=4) and 11,12-trans-oxido-eicosa-8(Z)-enoic acid (11,12-tetra-EET-8-ZE, n=4). Control PP2A activity averaged 3.55nmol/min. * Significant increase in PP2A activity in response to 11,12-EET analogs.
The involvement of signal transduction pathways in response to EET analogs was assessed in cultured renal myocytes. Measurement of cGMP and cAMP levels revealed that 11,12-EET analogs increased cAMP but not cGMP levels (Figure 7). The increase in cAMP observed in response to 11,12-EET analogs was eliminated by inhibition of adenylyl cyclase. 11,12-EET analogs increased cAMP levels by 203% and the 11,12-EET analog induced increases were greatly attenuated in the presence of SQ22536 averaging 115%. In contrast, 11,12-EET analogs had no effect on cultured renal myocyte cGMP levels. These findings are in agreement with previously published data that the afferent arteriolar dilation in response to 11,12-EET-SI was PKA-dependent but was not altered by the guanylate cyclase inhibitor ODQ [18]. Interestingly, inhibition of PP2A did not alter the increase in cAMP levels in response to 11,12-EET-SI and cAMP levels increased to 151 ± 11% of control (n=4).
Figure 7.

Effect of 11,12-EET analogs on cultured renal microvascular smooth muscle cell cAMP and cGMP levels. Panel A: Percentage change of cAMP levels in cells stimulated with the N-methylsulfonimide analog of 11,12-EET (EET-SI, n=4), 11-nonyloxy-undec-8(Z)-enoic acid (Ether-EET, n=4) and 11,12-trans-oxido-eicosa-8(Z)-enoic acid (Trans-EET, n=4) or their combination with SQ22536 (SQ, 100 μmol/L). Control cAMP levels averaged 0.61 pmol/ml. Panel B: Percentage change of cGMP levels in cells stimulated with 11,12-EET-SI (EET-SI, n=4), 11-nonyloxy-undec-8(Z)-enoic acid (Ether-EET, n=4) and 11,12-trans-oxido-eicosa-8(Z)-enoic acid (Trans-EET, n=4) or their combination with ODQ (10 μmol/L). Control cGMP levels averaged 0.78 pmol/ml. * Significant increase in cAMP or cGMP levels in response to 11,12-EET analogs. † Significant difference from the cAMP or cGMP response to the same dose of the 11,12-EET analog.
Additional studies were conducted in inside-out patches to determine the direct effects of PP2A (250 U/ml) and PKA (400 U/ml) to renal myocyte KCa channel activity (Figure 8). PP2A significantly increased KCa channel activity and this effect was inhibited by the addition of okadaic acid. PKA also increased KCa channel activity but this effect was only one fifth of that observed with the addition of PP2A. Taken together, the data from renal microvessel functional studies, electrophysiological studies in freshly isolated renal myocytes and signal transduction studies in cultured renal myocytes provide initial evidence that afferent arteriolar dilation initiated by 11,12-EET analogs involves a PP2A-dependent pathway to activate KCa channels and dilate the afferent arteriole.
Figure 8.

Effect of protein phosphatase 2A (PP2A) and protein kinase A (PKA) on the renal microvascular smooth muscle cell large-conductance calcium-activated potassium channels (large-conductance KCa). Panel A: Renal myocyte large-conductance KCa channel response to the PP2A (250 U/ml) and effect of okadaic acid (OA, 10nM) on the channel response to PP2A. Panel B: Renal myocyte large-conductance KCa channel response to the PKA (400 U/ml). Panel C: Bar graph depicting increase in open probability (NPo) to PP2A (n=4) and PKA (n=4). * Significant increase in NPo in response to PP2A and PKA.
Discussion
Numerous studies have implicated epoxygenase metabolites of arachidonic acid as an EDHF [1,2,12,19,48]. We have previously reported that 11,12-EET is the primary vasoactive epoxide in the kidney and the 11,12-EET dilatory responses are dependent on PKA activation [18,19]. There is still a dearth of information concerning the cell signaling pathways activated by 11,12-EET due to difficulties encountered when studying this epoxide. The development of 11,12-EET analogs that are designed to resist esterification, β-oxidation, and metabolism by epoxide hydrolase will facilitate the vascular smooth muscle cell signaling studies and identify structural requirements. The present study evaluated the ability of 11,12-EET analogs to activate large-conductance KCa channels and dilate the afferent arteriole. In addition, the cell signaling pathways involved in renal microvascular smooth muscle cell relaxation and large-conductance KCa channel activation was investigated. The structural features of 11,12-EET for afferent arteriolar relaxation were also examined. Another aspect of this study was to evaluate the diameter responses of the pre- and postglomerular microvessels to 11,12-EET-SI. Overall, the data suggest that 11,12-EET is most active on the preglomerular vasculature and utilizes PP2A cell signaling to activate large-conductance KCa channels and dilate the afferent arteriole.
Although it has been suggested that epoxygenase metabolites could affect the pre- and post-glomerular vasculatures, there is no direct evidence that epoxides dilate the post-glomerular vessels of the kidney. We found that 11,12-EET-SI increased diameters of the pre-glomerular afferent arteriole and post-glomerular efferent arteriole; however, the diameters of the post-glomerular descending vasa recta vessels were unaffected by 11,12-EET-SI. The response of the afferent arteriole was also much greater than that of the efferent arteriole. As evidence for an effect of epoxides on the post-glomerular vasculature, a recent study demonstrated that release of EETs by the glomerulus contributed to bradykinin dilation of the rabbit efferent arteriole [33]. In contrast, acetylcholine dilation of the efferent arteriole does not depend on an EDHF [42]. Although there is a paucity of information concerning dilator influences on the descending vasa recta, studies conducted thus far would suggest that nitric oxide is the major endothelial-derived modulator of vascular resistance in this post-glomerular vascular segment [32]. We were unable to detect a change in the descending vasa recta diameter in response to 11,12-EET-SI. One possibility for these segmental differences could be that large-conductance KCa channels are primarily located in the pre-glomerular vessels whereas ATP-sensitive K+ channels play a more prominent role in controlling post-glomerular vascular resistance [42,43,48]. Since 11,12-EET had a greater effect on the pre-glomerular vasculature, additional studies were conducted on the afferent arteriole to evaluate structural requirements for 11,12-EET activities.
Evaluation of various 11,12-EET analogs revealed structural aspects required for afferent arteriolar dilator activity. As previously demonstrated [18,21], the substitution of a sulfonimide group for the EET carboxyl group to resist esterification and β-oxidation did not alter afferent arteriolar dilation. The 5,6 and 14,15 olefins do not appear essential for dilator activity and alterations of the cis-epoxide to trans-epoxide or ether did not significantly change afferent arteriolar dilator activity. Alteration of the cis-epoxide to an ether will be useful tool for future studies because it will resist epoxide hydrolase enzymes that are known to regulate vascular epoxide levels [6,20,38]. Hydroxylation of C(20) on 11,12-ether-EET-8-ZE resulted in a loss of afferent arteriolar dilator activity. This loss of activity with the hydroxylation of C(20) is different than a previous report that demonstrated a 25% decrease in the inhibition of TNF-α-induced endothelial VCAM-1 expression by 11,12-EET [11]. Besides this difference the structure activity requirements are similar to those described for the coronary dilator actions of 14,15-EET and the decrease in endothelial VCAM-1 expression by 11,12-EET [10,11,13]. Overall, 11,12-EET requires an Δ8,9-olefin to occupy a shallow pocket that conforms to a bent configuration, an oxygen or sulfur positioned between C(11) and C(12) and a terminal lipophilic pocket (Table 1).
Next, we utilized the chemically easier to synthesize and more stable 11,12-EET analogs and assessed K+ channel contribution to the 11,12-EET-mediated afferent arteriolar dilation. 11,12-EET analog dilation of the afferent arteriole was greatly attenuated by TEA and iberiotoxin but was not altered by charybdotoxin or apamin. TEA and iberiotoxin were used at a dose that would inhibit large-conductance KCa channels and apamin, charybdotoxin, and TRAM-34 were used at doses that inhibit intermediate- and small-conductance KCa channels, respectively [27,37,40,42,44]. This would suggest that 11,12-EET analogs dilate the afferent arteriole by activating large-conductance KCa channels. Further support for 11,12-EET-mediated activation of large-conductance KCa channels was obtained in patch-clamp studies. 11,12-EET analogs increased the open probability of a 191 pS KCa channel in isolated renal microvascular smooth muscle cells. Although these findings strongly suggest that the large-conductance KCa channels are the primary channels activated by 11,12-EET analogs, we cannot clearly exclude potential effects of 11,12-EET analogs on other ion channels. The finding that 11,12-EET activates renal vascular smooth muscle cells to elicit dilation of the afferent arteriole is consistent with the finding of previous studies [19,48]. A recent study demonstrated that 11,12-EET dilates the human mammary artery and rat mesenteric artery by activating large-conductance KCa channels [1,7,8]. 11,12-EET also dilates isolated perfused small renal arteries and increases the open probability of large-conductance KCa channels [48]. Interestingly, the ability to activate KCa channels may extend beyond smooth muscle cells because 11,12-EET has been shown to hyperpolarize platelets by activating KCa channels [23].
Cell signaling events that could possibly contribute to the 11,12-EET mediated activation of renal microvascular smooth muscle cells were investigated. We previously demonstrated that the afferent arteriolar dilation to 11,12-EET-SI was substantially reduced by PKA inhibition [18]. We provide further biochemical evidence that 11,12-EET increases renal vascular smooth muscle cell cAMP levels. In fact, every 11,12-EET analog increased cultured renal vascular smooth muscle cell cAMP levels but did not alter cGMP levels. Likewise, 11,12-EET induction of cultured rat aortic smooth muscle cell tissue-type plasminogen activator (t-PA) gene transcription requires activation of adenylyl cyclase and PKA [31]. Thus, afferent arteriolar dilation to 11,12-EET involves increasing cAMP, PKA activation and opening renal vascular smooth muscle cell KCa channels.
The contribution of PP2A to the 11,12-EET mediated afferent arteriolar dilation is a controversial finding of the present study. We initially observed that the PP2A inhibitor, okadaic acid greatly attenuated the afferent arteriolar dilation in response to 11,12-EET analogs. 11,12-EET also increased PP2A activity in cultured renal microvascular smooth muscle cells and PP2A inhibition blocked 11,12-EET mediated activation of the large-conductance KCa channel. We also demonstrated that PP2A activates large-conductance KCa channels to a much greater extent than PKA. Taken together, these findings would suggest that 11,12-EET activates PP2A resulting in the opening of large-conductance KCa channels and afferent arteriolar dilation.
The finding of a phosphatase contributing to K+ channel activation goes against the current dogma that phosphorylation activates and dephosphorylation deactivates K+ channels [22,36]. A number of studies have demonstrated that stimulation of protein kinases and K+ channel phosphorylation increases the activity of the K+ channels including the KCa channels [22,36]. Studies have also shown that phosphatase inhibitors will amplify the activation of K+ channels by protein kinases [22,36]. PP2A inhibition has also been shown to increase whereas exogenous PP2A decreases large-conductance KCa channel activity in human mesangial cells [35,39]. Thus, the current finding of PP2A contributing to activation of renal microvascular smooth muscle cell large-conductance KCa channels is in direct opposition to this body of literature. On the other hand, there is evidence that PP2A dephosphorylation of large-conductance KCa channels can increase channel activity and protein kinase C phosphorylation can inhibit smooth muscle cell large-conductance KCa channels [24,44-46]. KCa dephosphorylation by PP2A has been demonstrated to be responsible for increasing channel activity in response to somatostatin and atrial natriuretic peptide in pituitary tumor cells [44,45]. The relaxation of human airway smooth muscle in response to 14,15-EET is dependent on activation of KCa channels and hyperpolarization [28]. These intracellular effects on human bronchi are related to the dephosphorylation of the myosin phosphatase inhibitor protein (CPI-17) [28]. These would initially appear at odds with our previous finding that afferent arteriolar dilation to 11,12-EET-SI was substantially reduced by PKA inhibition [18]. Interestingly, protein kinase activation of PP2A and subsequent activation of KCa channels has been demonstrated in pituitary tumor cells, myometrial cells and tracheal smooth muscle cells [44-47]. Previous studies have also demonstrated that PP2A activation contributes to the mesenteric resistance artery relaxation to 11,12-EET and 11,12-EET analogs [7,8]. Thus, there is evidence to support the notion that 11,12-EET activates renal microvascular smooth muscle cell PP2A that opens KCa channels leading to afferent arteriolar dilation.
In conclusion, our study demonstrates that 11,12-EET-SI preferentially dilates the renal preglomerular vasculature. We determined the structural requirements for 11,12-EET activity by comparing afferent arteriolar dilation to various 11,12-EET analogs. In addition, the 11,12-EET analogs were utilized to determine the renal microvascular smooth muscle cell signaling mechanisms responsible for afferent arteriolar relaxation. We present data from perfused afferent arterioles, freshly isolated and cultured renal microvascular smooth muscle cells that implicate PP2A and KCa channels in the dilator response to 11,12-EET. Overall, these findings provide initial evidence that afferent arteriolar dilation in response to 11,12-EET involves renal microvascular smooth muscle cell PP2A activation and large-conductance KCa channel opening.
Acknowledgements
This work was supported by the National Institutes of Health (NIH) grants HL-59699, DK-38226, HL-074167 and an American Heart Association Established Investigator Award to J.D. Imig. NIH grant GM31278 and the Robert A. Welch Foundation to J.R. Falck. The authors thank Roger Williams, Sachie Hase and Connie Snead for technical assistance.
References
- 1.Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR, Michelakis ED. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation. 2003;107:769–776. doi: 10.1161/01.cir.0000047278.28407.c2. [DOI] [PubMed] [Google Scholar]
- 2.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:15–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 3.Capdevila JH, Falck JR. The CYP P450 arachidonic acid monooxygenases: from cell signaling to blood pressure regulation. Biochem Biophys Res Commun. 2001;285:571–576. doi: 10.1006/bbrc.2001.5167. [DOI] [PubMed] [Google Scholar]
- 4.Chen JK, Falck JR, Reddy KM, Capdevila J, Harris RC. Epoxyeicosatrienoic acids and their sulfonimide derivatives stimulate tyrosine phosphorylation and induce mitogenesis in renal epithelial cells. J Biol Chem. 1998;273:29254–29261. doi: 10.1074/jbc.273.44.29254. [DOI] [PubMed] [Google Scholar]
- 5.Cheng MK, Doumad AB, Jiang H, Falck JR, McGiff JC, Carroll MA. Epoxyeicosatrienoic acids mediate adenosine-induced vasodilation in rat preglomerular microvessels (PGMV) via A2A receptors. Br J Pharmacol. 2004;141:441–448. doi: 10.1038/sj.bjp.0705640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH. Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A. 2002;99:2222–2227. doi: 10.1073/pnas.261710799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimitropoulou C, Han G, Miller AW, Molero M, Fuchs LC, White RE, Carrier GO. Potassium (BK(Ca)) currents are reduced in microvascular smooth muscle cells from insulin-resistant rats. Am J Physiol Heart Circ Physiol. 2002;282:H908–917. doi: 10.1152/ajpheart.00382.2001. [DOI] [PubMed] [Google Scholar]
- 8.Dimitropoulou C, West L, Field MB, White RE, Reddy LM, Falck JR, Imig JD. Protein phosphatase 2A and Ca(2+)-activated K(+) channels contribute to 11,12-epoxyeicosatrienoic acid analog mediated mesenteric arterial relaxation. Prostaglandins Other Lipid Mediat. 2007;83:50–61. doi: 10.1016/j.prostaglandins.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 10.Falck JR, Krishna UM, Reddy YK, Kumar PS, Reddy KM, Hittner SB, Deeter C, Sharma KK, Gauthier KM, Campbell WB. Comparison of vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am J Physiol Heart Circ Physiol. 2003;284:H337–349. doi: 10.1152/ajpheart.00831.2001. [DOI] [PubMed] [Google Scholar]
- 11.Falck JR, Reddy LM, Reddy YK, Bondlela M, Krishna UM, Ji Y, Sun J, Liao JK. 11,12-epoxyeicosatrienoic acid (11,12-EET): structural determinants for inhibition of TNF-alpha-induced VCAM-1 expression. Bioorg Med Chem Lett. 2003;13:4011–4014. doi: 10.1016/j.bmcl.2003.08.060. [DOI] [PubMed] [Google Scholar]
- 12.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 13.Gauthier KM, Falck JR, Reddy LM, Campbell WB. 14,15-EET analogs: characterization of structural requirements for agonist and antagonist activity in bovine coronary arteries. Pharmacol Res. 2004;49:515–524. doi: 10.1016/j.phrs.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 14.Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol. 1992;263:H519–525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- 15.Honkanen RE, Zwiller J, Moore RE, Daily SL, Khatra BS, Dukelow M, Boynton AL. Characterization of microcystin-LR, a potent inhibitor of type 1 and type 2A protein phosphatases. J Biol Chem. 1990;265:19401–19404. [PubMed] [Google Scholar]
- 16.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289:F496–503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 17.Imig JD, Falck JR, Wei S, Capdevila JH. Epoxygenase metabolites contribute to nitric oxide-independent afferent arteriolar vasodilation in response to bradykinin. J Vasc Res. 2001;38:247–255. doi: 10.1159/000051053. [DOI] [PubMed] [Google Scholar]
- 18.Imig JD, Inscho EW, Deichmann PC, Reddy KM, Falck JR. Afferent arteriolar vasodilation to the sulfonimide analog of 11,12-epoxyeicosatrienoic acid involves protein kinase A. Hypertension. 1999;33:408–413. doi: 10.1161/01.hyp.33.1.408. [DOI] [PubMed] [Google Scholar]
- 19.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol. 1996;7:2364–2370. doi: 10.1681/ASN.V7112364. [DOI] [PubMed] [Google Scholar]
- 20.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 21.Imig JD, Zhao X, Falck JR, Wei S, Capdevila JH. Enhanced renal microvascular reactivity to angiotensin II in hypertension is ameliorated by the sulfonimide analog of 11,12-epoxyeicosatrienoic acid. J Hypertens. 2001;19:983–992. doi: 10.1097/00004872-200105000-00020. [DOI] [PubMed] [Google Scholar]
- 22.Korovkina VP, England SK. Molecular diversity of vascular potassium channel isoforms. Clin Exp Pharmacol Physiol. 2002;29:317–323. doi: 10.1046/j.1440-1681.2002.03651.x. [DOI] [PubMed] [Google Scholar]
- 23.Krotz F, Riexinger T, Buerkle MA, Nithipatikom K, Gloe T, Sohn HY, Campbell WB, Pohl U. Membrane-potential-dependent inhibition of platelet adhesion to endothelial cells by epoxyeicosatrienoic acids. Arterioscler Thromb Vasc Biol. 2004;24:595–600. doi: 10.1161/01.ATV.0000116219.09040.8c. [DOI] [PubMed] [Google Scholar]
- 24.Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 2006;21:69–78. doi: 10.1152/physiol.00040.2005. [DOI] [PubMed] [Google Scholar]
- 25.Li PL, Chen CL, Bortell R, Campbell WB. 11,12-Epoxyeicosatrienoic acid stimulates endogenous mono-ADP-ribosylation in bovine coronary arterial smooth muscle. Circ Res. 1999;85:349–356. doi: 10.1161/01.res.85.4.349. [DOI] [PubMed] [Google Scholar]
- 26.Li PL, Zhang DX, Ge ZD, Campbell WB. Role of ADP-ribose in 11,12-EET-induced activation of K(Ca) channels in coronary arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. 2002;282:H1229–1236. doi: 10.1152/ajpheart.00736.2001. [DOI] [PubMed] [Google Scholar]
- 27.McSherry IN, Sandow SL, Campbell WB, Falck JR, Hill MA, Dora KA. A role for hetrocellular coupling and EETs in dilation of rat cremaster arteries. Microcirculation. 2006;13:119–130. doi: 10.1080/10739680500466400. [DOI] [PubMed] [Google Scholar]
- 28.Morin C, Sirois M, Echave V, Gomes MM, Rousseau E. EET Relaxing Effects Involve BKCa Channel Activation and CPI-17 Dephosphorylation in Human Bronchi. Am J Respir Cell Mol Biol. 2007 doi: 10.1165/rcmb.2006-0281OC. PMID:17237191. [DOI] [PubMed] [Google Scholar]
- 29.Namboodiripad AN, Jennings ML. Permeability characteristics of erythrocyte membrane to okadaic acid and calyculin A. Am J Physiol. 1996;270:C449–456. doi: 10.1152/ajpcell.1996.270.2.C449. [DOI] [PubMed] [Google Scholar]
- 30.Navar LG, Inscho EW, Majid SA, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiol Rev. 1996;76:425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- 31.Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, Liao JK. Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J Biol Chem. 2001;276:15983–15989. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 32.Pallone TL, Zhang Z, Rhinehart K. Physiology of the renal medullary microcirculation. Am J Physiol Renal Physiol. 2003;284:F253–266. doi: 10.1152/ajprenal.00304.2002. [DOI] [PubMed] [Google Scholar]
- 33.Ren Y, Garvin JL, Falck JR, Renduchintala KV, Carretero OA. Glomerular autacoids stimulated by bradykinin regulate efferent arteriole tone. Kidney Int. 2003;63:987–993. doi: 10.1046/j.1523-1755.2003.00810.x. [DOI] [PubMed] [Google Scholar]
- 34.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 35.Sansom SC, Stockand JD, Hall D, Williams B. Regulation of large calcium-activated potassium channels by protein phosphatase 2A. J Biol Chem. 1997;272:9902–9906. doi: 10.1074/jbc.272.15.9902. [DOI] [PubMed] [Google Scholar]
- 36.Schubert R, Nelson MT. Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol Sci. 2001;22:505–512. doi: 10.1016/s0165-6147(00)01775-2. [DOI] [PubMed] [Google Scholar]
- 37.Schweitz H, Stansfeld CE, Bidard JN, Fagni L, Maes P, Lazdunski M. Charybdotoxin blocks dendrotoxin-sensitive voltage-activated K+ channels. FEBS Lett. 1989;250:519–522. doi: 10.1016/0014-5793(89)80788-4. [DOI] [PubMed] [Google Scholar]
- 38.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 39.Stockand JD, Silverman M, Hall D, Derr T, Kubacak B, Sansom SC. Arachidonic acid potentiates the feedback response of mesangial BKCa channels to angiotensin II. Am J Physiol. 1998;274:F658–664. doi: 10.1152/ajprenal.1998.274.4.F658. [DOI] [PubMed] [Google Scholar]
- 40.Strong PN. Potassium channel toxins. Pharmacol Ther. 1990;46:137–162. doi: 10.1016/0163-7258(90)90040-9. [DOI] [PubMed] [Google Scholar]
- 41.Wang D, Borrego-Conde LJ, Falck JR, Sharma KK, Wilcox CS, Umans JG. Contributions of nitric oxide, EDHF, and EETs to endothelium-dependent relaxation in renal afferent arterioles. Kidney Int. 2003;63:2187–2193. doi: 10.1046/j.1523-1755.2003.00036.x. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Loutzenhiser R. Determinants of renal microvascular response to ACh: afferent and efferent arteriolar actions of EDHF. Am J Physiol Renal Physiol. 2002;282:F124–132. doi: 10.1152/ajprenal.0157.2001. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Trottier G, Loutzenhiser R. Determinants of renal afferent arteriolar actions of bradykinin: evidence that multiple pathways mediate responses attributed to EDHF. Am J Physiol Renal Physiol. 2003;285:F540–549. doi: 10.1152/ajprenal.00127.2003. [DOI] [PubMed] [Google Scholar]
- 44.White RE, Lee AB, Shcherbatko AD, Lincoln TM, Schonbrunn A, Armstrong DL. Potassium channel stimulation by natriuretic peptides through cGMP-dependent dephosphorylation. Nature. 1993;361:263–266. doi: 10.1038/361263a0. [DOI] [PubMed] [Google Scholar]
- 45.White RE, Schonbrunn A, Armstrong DL. Somatostatin stimulates Ca(2+)-activated K+ channels through protein dephosphorylation. Nature. 1991;351:570–573. doi: 10.1038/351570a0. [DOI] [PubMed] [Google Scholar]
- 46.Zhou XB, Ruth P, Schlossmann J, Hofmann F, Korth M. Protein phosphatase 2A is essential for the activation of Ca2+-activated K+ currents by cGMP-dependent protein kinase in tracheal smooth muscle and Chinese hamster ovary cells. J Biol Chem. 1996;271:19760–19767. doi: 10.1074/jbc.271.33.19760. [DOI] [PubMed] [Google Scholar]
- 47.Zhou XB, Schlossmann J, Hofmann F, Ruth P, Korth M. Regulation of stably expressed and native BK channels from human myometrium by cGMP- and cAMP-dependent protein kinase. Pflugers Arch. 1998;436:725–734. doi: 10.1007/s004240050695. [DOI] [PubMed] [Google Scholar]
- 48.Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, Roman RJ. Stereospecific effects of epoxyeicosatrienoic acids on renal vascular tone and K(+)-channel activity. Am J Physiol. 1996;270:F822–832. doi: 10.1152/ajprenal.1996.270.5.F822. [DOI] [PubMed] [Google Scholar]