

Abstract
Kisspeptins, the natural ligands of the G-protein-coupled receptor (GPR)-54, are the most potent stimulators of GnRH-1 secretion and as such are critical to reproductive function. However, the mechanism by which kisspeptins enhance calcium-regulated neuropeptide secretion is not clear. In the present study, we used GnRH-1 neurons maintained in mice nasal explants to examine the expression and signaling of GPR54. Under basal conditions, GnRH-1 cells exhibited spontaneous baseline oscillations in intracellular calcium concentration ([Ca2+]i), which were critically dependent on the operation of voltage-gated, tetrodotoxin (TTX)-sensitive sodium channels and were not coupled to calcium release from intracellular pools. Activation of native GPR54 by kisspeptin-10 initiated [Ca2+]i oscillations in quiescent GnRH-1 cells, increased the frequency of calcium spiking in oscillating cells that led to summation of individual spikes into plateau-bursting type of calcium signals in a subset of active cells. These changes predominantly reflected the stimulatory effect of GPR54 activation on the plasma membrane oscillator activity via coupling of this receptor to phospholipase C signaling pathways. Both components of this pathway, inositol 1,3,4-trisphosphate and protein kinase C, contributed to the receptor-mediated modulation of baseline [Ca2+]i oscillations. TTX and 2-aminoethyl diphenylborinate together abolished agonist-induced elevation in [Ca2+]i in almost all cells, whereas flufenamic acid was less effective. Together these results indicate that a plasma membrane calcium oscillator is spontaneously operative in the majority of prenatal GnRH-1 neurons and is facilitated by kisspeptin-10 through phosphatidyl inositol diphosphate hydrolysis and depolarization of neurons by activating TTX-sensitive sodium channels and nonselective cationic channels.
GnRH-1 neurons exhibit a spontaneously active calcium oscillator, dependent on tetrodotoxin-sensitive sodium conductance. Kisspeptin-10/GPR54, via phosphatidyl inositol diphosphate 2 hydrolysis, utilizes these channels and non-selective cationic channels.
Hypogonadal hypogonadism (HH) is a deficiency in pituitary secretion that underlies impairments in puberty and reproduction. One form of HH called idiopathic HH is due to inactivating mutations in the GPR54 gene (1,2). The human phenotype induced by these loss of function mutations is mimicked in gpr54 mutant mice (2,3); sexual immaturity. Kisspeptins (KPs) are the endogenous ligand for these receptors (4) and have been shown to stimulate gonadotropin release in vivo [mouse (5); rat, (6,7,8)] and in vitro [mouse (9)] but not in gpr54 mutant mice (10). It appears that this KP-induced gonadotropin release can occur through direct activation of GnRH-1 neurons because these cells express gpr54 mRNA transcripts (6) and functional receptors, as indicated by the ability of KPs to depolarize the cells (11) and activate c-Fos (6,8).
The downstream signals engaged after activation of G-protein-coupled receptor (GPR)-54 have been mainly studied in heterologous expression systems (4,12). These studies revealed coupling of GPR54 to the phospholipase C signaling pathway, leading to calcium mobilization, arachidonic acid release, and ERK1/2 and p38 MAP kinase phosphorylation. Consistent with these findings, it has been suggested that KP-induced GnRH-1 release predominantly reflects mobilization of intracellular Ca2+ and recruitment of ERK1/2 and p38 kinases but not voltage-gated Ca2+ influx (9). This conclusion, however, questioned the physiological relevance of KP-induced changes in electrical activity of GnRH-1 neurons (11,13,14) because depolarization of cells should facilitate voltage-gated Ca2+ influx and calcium is the main intracellular messenger controlling the exocytotic pathway. GnRH-1 neurons in hypothalamic slices exhibit intrinsic calcium oscillations that are dependent on inositol 1,4,5-trisphosphate (IP3)-induced calcium release from intracellular stores (15). In contrast, immortalized GnRH-1 neurons exhibit calcium transients dependent on voltage-gated calcium influx (16). Both tetrodotoxin (TTX)-dependent and voltage-gated calcium channel-dependent firing of action potentials were observed in GT1 cells (16,17). Thus, the nature of spontaneous calcium transients in GnRH-1-secreting neurons, channels involved in their generation, and the mechanism by which KPs modulate a calcium oscillator, remain unclear.
The studies reported here focus on the expression of gpr54 and the GPR54-induced calcium signaling in both individual GnRH-1 neurons and within the GnRH-1 neuronal population. Gpr54 transcript was identified in prenatal GnRH-1 neurons in vivo and in primary neurons maintained in nasal explants. This model, based on the extracentral nervous system origin of GnRH-1 neurons (18), has been successfully used to examine the regulation of GnRH-1 neurons (19,20,21,22). GnRH-1 neurons maintained in such explants exhibit spontaneous oscillations in intracellular calcium ([Ca2+]i) (23,24,25) and pulsatile calcium-controlled GnRH-1 release (26,27,28), suggesting operation of an endogenous calcium oscillator. Here we show that: 1) kisspeptin-10 (kp-10) increased the frequency of spontaneous calcium spiking in the majority of GnRH-1 cells, with most of these cells showing a summation of individual spikes into plateau-bursting type calcium signals; 2) kp-10-stimulated calcium oscillations are critically dependent on voltage-gated sodium channels and 2-aminoethyl diphenylborinate (2-APB)-sensitive signaling pathways; and 3) protein kinase-dependent signaling contributes to the modulation of calcium oscillations in kp-10 stimulated GnRH-1 neurons.
Materials and Methods
Animals
All mice were killed in accordance with National Institutes of Health (NIH), National Institute of Neurological Stroke and Disorders guidelines. NIH Swiss embryos were harvested at embryonic day (E) 12.5, E13.5, E14.5, and E17.5 (plug day, E0.5) and immediately frozen and stored (80 C) until processing for in situ hybridization histochemistry. Adult brains from NIH Swiss nonpregnant mice were also harvested, frozen, and stored (80 C) until processing.
Nasal explants
Nasal regions were cultured as previously described (21). Briefly, embryos were obtained from timed pregnant animals in accordance with NIH guidelines. Nasal pits of E11.5 staged NIH Swiss mice were isolated under aseptic conditions and adhered onto coverslips by a plasma (Cocalico Biologicals, Reamstown, PA)/thrombin (Sigma, St. Louis, MO) clot. Nasal explants were maintained at 37 C in a defined serum-free medium (SFM) in a humidified atmosphere with 5% CO2. On culture d 3, fresh media containing fluorodeoxyuridine (8 × 10−5 m; Sigma) was given for 3 d to inhibit proliferation of dividing olfactory neurons and nonneuronal explant tissue. On culture d 6 and every 2 d afterward, the medium was changed for fresh SFM. Explants were used between 6 and 10 d in vitro (div) (Fig. 1A).
Figure 1.

Nasal explants as a model system for studies on calcium signaling in GnRH-1-secreting neurons. A, Nasal explant obtained from E11.5 mouse and maintained 9 div, with nasal pit epithelium (NPE) and nasal midline cartilage (NMC), surrounded by mesenchyme. GnRH-1 neurons, immunostained for GnRH-1 (brown), migrate from NPE and follow olfactory axons to the NMC and into the periphery. Boxed area delimits a typical field in which cells were selected for calcium recording. B, Calcium imaging recordings were performed on cells maintained in mouse nasal explants for 6–10 d in vitro (div). Cells, identified by their bipolar morphology (left panel), were loaded with a calcium-sensitive dye (middle panel), and after recording the phenotype of the cells was confirmed by immunocytochemistry (right panel, brown). Arrows indicate same cells in all fields. C, Representative 16-min calcium imaging recording in cells bathed in SFM, showing spontaneous baseline oscillations in intracellular calcium level. The Y-scale is arbitrary. Such a pattern of signaling was observed in the majority of identified GnRH-1 neurons.
In situ hybridization histochemistry
Frozen mouse adult brain and embryonic sections (16 μm thickness) were cut on a cryostat and mounted on subbed slides. Adult brains were serially cut from the olfactory bulbs to the caudal hypothalamus. Serial sections were placed onto 50 slides, with the 51st section returning to slide 1. In this manner, serial atlas slides were generated with 10–12 sections/slide. Therefore, each slide had representative sections throughout the forebrain and two consecutive slides had serial sections from each region. In contrast, embryos were cut in serial series, with three series for E12.5, four series for E13.5 and E14.5, and five series for E17.5. The sections were processed for in situ hybridization histochemistry as previously described (18). Briefly, sections were fixed in 4% formaldehyde, rinsed in PBS, permeabilized in 0.3% Triton X-100/0.05 m EDTA/0.1 m Tris buffer (pH 8.0), rinsed in 0.05 m EDTA/0.1 m Tris buffer (pH 8.0), washed in 0.25% acetic anhydride/0.1 m triethanolamine, rinsed in 2× sodium chloride-sodium citrate (SSC), dehydrated through ethanol, delipidated in chloroform, rinsed in ethanol, and air dried. A 48-nucleotide oligo probe (2.5 pmol), complementary to the coding region of mouse GPR54 (5′-GAGTGGCACATGTGGCTTGCA-CCGAGACCTGCTGGATGTAGTTGACGA-3′) or GnRH-1 (5′-TTCAGTGTTTCTCTTTCCCCCA-GGGCGCAACCCATAGGACCAGTGCTG-3′) (29) was 3′-end labeled with S35-labeled dATP (PerkinElmer, Boston, MA) using 400 U terminal transferase (Roche, Indianapolis, IN), 5× tailing buffer and CoCl2 1.5 mm (Roche). Labeled probe (0.5 × 106 to 1 × 106 cpm) was applied to each slide in 100 μl of hybridization buffer (18). Slides were hybridized overnight in humid chambers (37 C). The following day, slides were rinsed in 1× SSC/65 mm dithiothreitol, washed at high stringency in 2× SSC/50% formamide/20 mm dithiothreitol at 45 C, and washed in 1× SSC at room temperature. Slides were then rinsed in water, dehydrated in ethanol, dried, and placed against film. After x-ray film exposure for 5 d, the slides were dipped in NTB autoradiography emulsion (Kodak, Rochester, NY) and exposed for 3–5 wk for GnRH-1 and 3–7 wk for GPR54. Emulsion-covered slides were developed as previously described (18). Adjacent sections and S35-labeled probes were used to optimize the signal for gpr54 transcript during prenatal development.
PCR on single GnRH-1 cells
For single cells, GnRH-1 like neurons, identified in vitro by their bipolar morphology, association with outgrowing axons, and location within the explant, were removed with pulled glass capillaries. For single cells and tissues, cDNAs were produced and PCR amplification performed as previously described (29,30). Based on the technique used to generate the cDNA pools, 3′ untranslated region biased primers are necessary. Primers were designed with the 5′-primer being less than 500 bases from the polyA site and the 3′-primer close to, but not into, the polyA region. All designed primers were screened using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to ensure specificity of binding. For each reaction, 30.5 μl H2O, 5 μl of 10× PCR buffer (Applied Biosystems, Foster City, CA), 4 μl of 25 mm MgCl2 (Applied Biosystems), 5 μl of deoxynucleotide triphosphate mix [25 μl of each 100 mm deoxynucleotide triphosphate, 900 μl H2O], 2 μl of 6.25 μm forward primer, 2 μl of 6.25 μm reverse primer, and 0.5 μl AmpliTaq Gold (Applied Biosystems) were added to 1 μl template cDNA. PCR was performed at 94 C (10 min), 94 C (30 sec), 55 or 65 C, depending on primers (30 sec), 72 C (2 min) for 40 cycles, with a postelongation at 72 C for 10 min. Amplified products were run on a 1.5% agarose gel. Specific bands of the predicted size were observed in the control total brain lane, whereas no bands were seen in water. cDNAs from single cells were initially screened by PCR for GnRH-1 (assuring the cell phenotype), III-tubulin, and L19 (two housekeeping genes, respectively, microtubule and ribosomal) (see primer sequences in Ref. 31). Only cells positive for all three transcripts (previously used in Refs. 29, 31, and 32) were used in this study. PCR was performed on single GnRH-1 cell cDNAs to determine the expression of gpr54 (3 div; n = 10, 7 div; n = 17, 14 div; n = 10).
Calcium imaging
Calcium imaging recordings were performed between 6 div to 10 div. The calcium Green-1 AM (Molecular Probes, Eugene, OR) was diluted to 2.7 mm concentration in 80% dimethylsulfoxide (DMSO) and 20% pluronic F-127 solution (Molecular Probes). This solution was diluted 1:200 with SFM to a final calcium Green-1 concentration of 13.5 μm. Nasal explants, maintained at 37 C in a 5% CO2 humidified incubator, were incubated with this loading solution for 20 min and then washed twice with fresh SFM (10 min each). Explants were mounted into a perfusion chamber and were continuously perfused with medium, at a rate of about 280 μl/min, using a peristaltic pump (Spectra Hardware, Inc., Westmoreland City, PA). Calcium Green-1 was visualized using an inverted Nikon microscope, through a ×20 fluorescence objective and an charge-coupled device camera (Retiga, Qimaging, Burnaby, Canada) connected to a Macintosh computer. Experiments were piloted by imaging software (IPLab Spectrum; Scanalytics Inc., Rockville, MD), controlling the shutter (Uniblitz; Vincent Associates, Rochester, NY) and acquiring pictures every second. Excitation wavelengths were provided through a medium-width excitation bandpass filter at 465–495 nm, and emission was monitored through a 40-nm bandpass centered on 535 nm. Fluctuations in [Ca2+]i were analyzed a posteriori with IPLab software (Scanalytics). Each cell, individually identified, was circled. Calcium Green-1 fluorescence intensity was plotted and analyzed with MATLAB (Mathworks, Natick, MA). For short recording experiments, subdivision into three to four periods was done for analysis [i.e. for four periods: SFM control period (5 min), pretreatment period (3 min), treatment period (3 min), and SFM washout period (5 min)]. All recordings were terminated by a 40 mm KCl stimulation to ensure the viability of the recorded cells.
Statistical analysis
When neuronal activity was estimated in term of calcium oscillations, a calcium fluctuation was first identified when a value was greater than the five previous and five subsequent points. Then that calcium fluctuation had to be greater than the mean of the five previous and five next points plus a minimal value (which represented small fluctuations in baseline) to be considered as a calcium oscillation (peak). The frequency of calcium oscillations was calculated as the number of detected calcium peaks per time unit (minutes). When the responsiveness of cells was evaluated, all cells were aligned on their baseline (determined during the control period) at the same OD value. The area under the curve was then measured for each experimental period and normalized to 1 min duration. A cell was considered as a responding cell to kp-10 when it showed an increase of the area under the curve greater than 2 times the sd of the previous period. The delay of peak was evaluated by determining the time corresponding to the maximal response minus the time of kp-10 application, and the slope of the response was evaluated from the time of application (baseline calcium level) to the time of the maximal response (maximal calcium level). The Student’s t test was used to compare both parameters (P < 0.05). Responding cells were than expressed in a percentage of the population recorded. Fischer’s exact test was used to compare percentages (P < 0.05), and these results are summarized in Tables 1 and 2. In Results, n and N represent the number of cells and explants recorded, respectively, with each explant derived from a different animal. To circumvent the heterogeneity of the cells, at least 30 cells (obtained from at least two different explants) were analyzed for each pharmacological group.
Table 1.
Summary of responses observed in GnRH-1 neurons
| Pretreatment | BIC | TTX | Cadmium | NIF | GVIA | SNX | TG | |
|---|---|---|---|---|---|---|---|---|
| Treatment | kp-10 | |||||||
| Responders % (cell number) | 85% (44/52) | 60%a (36/60) | 21%a (33/160) | 44%a (37/84) | 41%a,b (18/44) | 83% (39/47) | 72% (42/58) | 72% (36/50) |
NIF, Nifedipine; GVIA, ω-conotoxin GVIA; SNX, SNX-482.
Responders % different from responders % with kp-10 alone (Fisher’s exact test, P < 0.05).
Responders % similar to responders % after cadmium pretreatment.
Table 2.
Summary of responses observed in GnRH-1 neurons
| Pretreatment | 2-APB | 2-APB + TTX | PTX | CTX | DDA16 | BSM | DDA16 + BSM | DDA3 + BSM |
|---|---|---|---|---|---|---|---|---|
| Treatment | kp-10 | |||||||
| Responders % (cell number) | 50%a (45/90) | 4%a,b (3/67) | 80% (49/61) | 50%a (31/62) | 53%a,c,d (40/75) | 56%a,d (52/93) | 46%a (31/67) | 72%a (28/39) |
NIF, Nifedipine; GVIA, ω-conotoxin GVIA; SNX, SNX-482.
Responders % different from responders % with kp-10 alone (Fisher’s exact test, P < 0.05).
Responder percent different from responder percent after TTX pretreatment.
Responder percent similar to responder percent after CTX pretreatment.
Responder percent similar to responder percent after DDA16 + BSM pretreatment.
Drugs
kp-10 was a gift from Dr. U. Kaiser (Division of Endocrinology, Diabetes, and Hypertension, Brigham and Women’s Hospital, Boston, MA), (−)-bicuculline chloride [BIC; a γ-aminobutyric acid (GABA)A receptor antagonist], dideoxyadenosine (DDA; adenylyl cyclase inhibitor), cholera toxin (CTX; Gαs protein inhibitor), pertussis toxin (PTX; Gαi protein inhibitor), thapsigargin [sarcoplasmic/endoplasmic reticulum (Ca2+)ATPase inhibitor], phorbol 12-myristate 13-acetate (PMA), bisindolylmaleimide X hydrochloride (BSM; protein kinase C inhibitor), TTX (a voltage gated sodium channel blocker), nifedipine (an L-type calcium channel blocker), SNX-482 (an R-type calcium channel blocker), ionomycin (calcium ionophore), and flufenamic acid [FFA; transient receptor potential (TRP) inhibitor] were obtained from Sigma. Dantrolene (RyR inhibitor) and 2-APB [TRP and IP3 receptor (IP3R] inhibitor) were obtained from Tocris (Avonmouth, UK). All stock solutions (ethanol, DMSO, or SFM) were stored at −20 C, and solutions were prepared before each experiment by diluting stock solutions (1:500 to 1:2000) into SFM [used for growing the nasal explants (21)].
kp-10 as well as all drugs, except thapsigargin (TG), CTX, and PTX, were applied by superfusion to the explants. For TG, CTX, and PTX, explants were incubated for longer than 30 min (TG) or longer than 2 h (CTX and PTX) before recording and then superfused during the recordings.
Immunocytochemistry
To confirm the phenotype of all recorded cells (Fig. 1B), nasal explants were systematically immunocytochemically stained for GnRH-1 as previously described (21). Briefly, after calcium imaging recordings, each explant was fixed (4% formaldehyde, 1 h), rinsed in PBS, and placed in cryoprotectant until staining. Explants were washed in PBS, blocked in 10% normal goat serum/0.3% Triton X-100 for 1 h, washed several times in PBS, and then incubated in GnRH-1 antibody (1:3000, SW-1) overnight at 4 C. The next day, explants were washed in PBS, incubated for 1 h in biotinylated secondary antibody (1:500 in PBS/0.3% Triton X-100; Vector Laboratories, Inc., Burlingame, CA), washed in PBS, and processed for avidin-biotin-horseradish peroxidase/3′3-diaminobenzidine cytochemistry. Once stained, the recorded field was compared with the stained explant and only GnRH-1 positive cells analyzed [the majority loaded with calcium green are in fact GnRH-1 positive (33)].
Results
Endogenous GnRH-1 neuronal activity
Earlier studies revealed that GnRH-1 neurons maintained in SFM exhibit spontaneous baseline oscillations in [Ca2+]i, which reflect endogenous neuronal activity (24). In our preparations, about 85% of GnRH-1 identified cells exhibited such oscillations, with an averaged frequency of 1.30 ± 0.14 peaks/min (n = 60, N = 4), consistent with previous data (24,25). Figure 1C shows an example of a typical trace of spontaneous calcium oscillations in a single cell. In general, such oscillations can arise from calcium entry through calcium channels in the plasma membrane or from calcium discharge from internal stores such as release of calcium from the endoplasmic reticulum (ER) through IP3Rs or through ryanodine receptor-channels (RyRs), or a combination of the above. The calcium source(s) for the oscillations observed in GnRH-1 neurons was unclear. Thus, the contribution of external and internal calcium to the endogenous activity of GnRH-1 neurons was investigated.
All GnRH-1 neurons exhibiting spontaneous [Ca2+]i oscillations showed a decrease in the peak frequency followed by complete arrest of oscillations when treated with 1 μm TTX, a blocker of voltage-gated sodium channels (n = 164, N = 7; Fig. 2A). Consistent with the role of depolarization-driven calcium influx through plasma membrane channels on spontaneous [Ca2+]i oscillations, cadmium (200 μm for 3 min), a nonselective blocker of calcium channels, decreased the frequency of calcium peaks in about 40% of the GnRH-1 cells and completely stopped oscillations in about 15% of cells (n = 86, N = 4; Fig. 2B). Both TTX and cadmium blockades were reversible (data not shown). The fact that cadmium did not block oscillations in all GnRH-1 cells suggests that TTX-sensitive electrical activity is coupled to calcium release from ER stores and/or that cadmium-insensitive cation-conducting channels also contribute to calcium influx.
Figure 2.
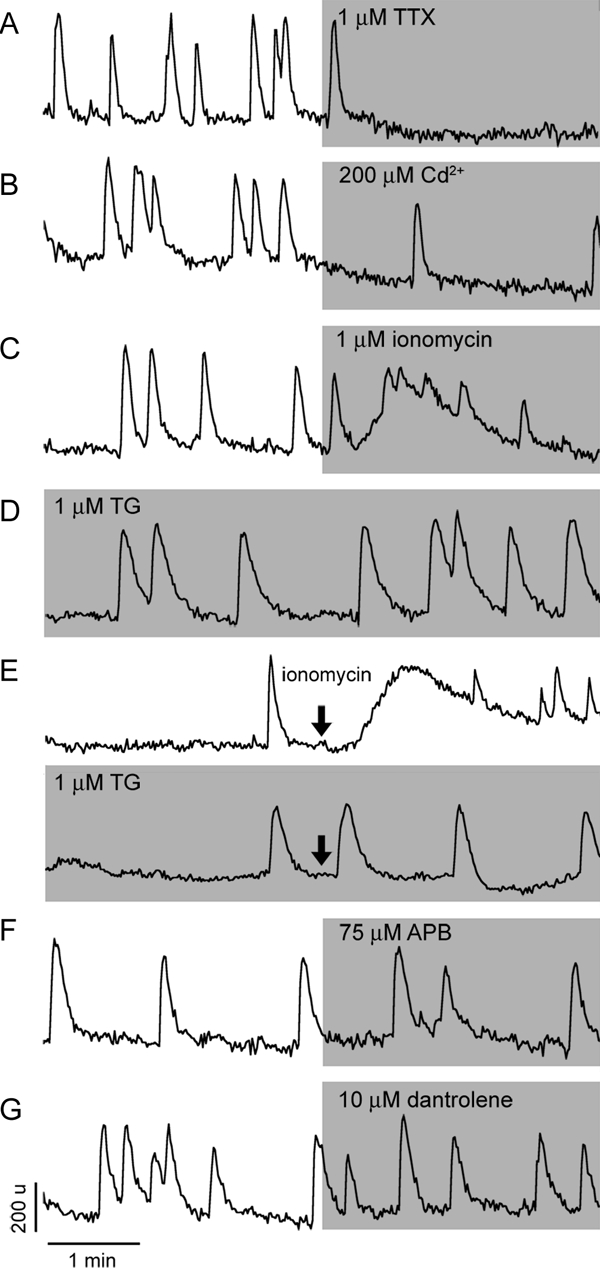
Characterization of spontaneous calcium oscillations in GnRH-1 neurons. A, Effects of TTX, a specific sodium channel blocker, on baseline calcium oscillations. Inhibition of calcium oscillations was observed in all spontaneously active GnRH-1 neurons. B, Effects of cadmium, a nonselective calcium channel blocker, on the frequency of calcium spiking. In a fraction of cells, cadmium abolished baseline calcium oscillations. C, Effects of a calcium ionophore ionomycin, which preferentially releases calcium from the ER, on spontaneous calcium oscillations. Note the low amplitude of calcium response and the persistence of fluctuations in [Ca2+]i in the presence of ionomycin, indicating the independence of calcium oscillations from the status of the ER calcium pool. D, Spontaneous calcium oscillations persist after depletion of the ER calcium pool by inhibition of (Ca2+)ATPases with TG. Note changes in the intraspike areas in TG-treated cells. E, Evaluation of the efficiency of TG treatment, example records showing the ability of ionomycin to elevate [Ca2+]i in controls (top panel) but not in TG-treated cells (bottom panel). The lack of effects of 2-APB, a blocker of IP3Rs (F), and dantrolene, an inhibitor of RyRs (G), on baseline calcium oscillations are shown. In this and following figures, vertical and horizontal bars represent respectively units of OD and time, and gray areas indicate duration of treatments.
Ionomycin, a calcium ionophore that preferentially releases calcium from the ER, was used to evaluate the status of the ER calcium pool to spontaneously oscillating GnRH-1 neurons. As shown in Fig. 2C, ionomycin induced a prolonged elevation in [Ca2+]i, on top of which additional fluctuations occurred, the latter probably reflecting the endogenous activity of the oscillator. The ratio of total area under the curve for spontaneous and ionomycin-induced responses was 1:15 (measured in 26 peaks from six cells in SFM vs. 14 ionomycin-induced calcium elevations), indicating that the size of the ER calcium pool might be sufficient to contribute to baseline [Ca2+]i oscillations.
To study the contribution of the ER calcium pool to spontaneous calcium oscillations, three types of experiments were performed. First, the ER was depleted by treating the cells with TG, an inhibitor of the sarcoendoplasmic reticulum type (Ca2+)ATPase in the ER membrane, with 1 μm for 30 min. Eighty-three of 84 TG-treated cells still exhibited spontaneous [Ca2+]i oscillations (n = 83, N = 4; Fig. 2D) without obvious changes in frequency and amplitude of calcium transients. However, the duration of the calcium spikes was larger in TG-treated cells, as indicated by an increase in the area under the curve (TG to SFM = 3:2, ratio measured in 14 peaks from six cells in TG vs. 26 peaks from six cells in SFM), suggesting the role of sarcoendoplasmic reticulum type (Ca2+)ATPase in the calcium reuptake after baseline [Ca2+]i oscillations. To ensure that the TG treatment was efficient and that the ER was effectively depleted, controls and TG-treated cells were exposed to 1 μm ionomycin. Figure 2E (bottom trace) shows no ionomycin-induced elevation in [Ca2+]i in TG-treated cells (n = 55, N = 2), in contrast to control cells in SFM (top trace; n = 36, N = 2).
Next, the contribution of IP3Rs to spontaneous [Ca2+]i oscillations was investigated by application of 2-APB (75 μm), a blocker of these channels. This treatment did not stop spontaneous oscillations in any of the cells studied (n = 91, N = 4; Fig. 2F). In about 10% of cells, a small decrease in the peak frequency was observed, which could reflect a nonspecific effect of this compound, also known as a blocker of TRP, subfamily C channels (34). Furthermore, the coupling of TTX-sensitive electrical activity to RyRs was tested by application of dantrolene (10 μm), a blocker of these channels. The pattern of spontaneous calcium oscillations was not affected in 90% of the GnRH-1 cells treated with dantrolene (n = 50/56, N = 3; Fig. 2G), strongly arguing against this mechanism, at least for the majority of cells. In the remaining cells (10%), dantrolene decreased the frequency of [Ca2+]i oscillations. However, this could be a side effect of this compound because none of these cells showed complete arrest of oscillations.
To ensure that the effects observed during these various paradigms were due to the drugs rather than to their diluents, controls were run in which explants were exposed to the diluents alone (1:500, ethanol and DMSO). [Ca2+]i oscillations in GnRH-1 cells were similar to those detected in SFM alone (1.50 ± 0.20 peaks/min in SFM, 1.68 ± 0.19 peaks/min in ethanol, 1.84 ± 0.17 peaks/min in DMSO; paired t test, P > 0.05).
A subpopulation of GABAergic neurons is known to be present in nasal explants and regulate GnRH-1 neuronal activity via an excitatory input through GABAA receptor-channels (33,35,36). To clarify whether the plasma membrane-driven calcium oscillator is present in GnRH-1 neurons independent of GABA signaling, explants were treated with the GABAA receptor antagonist BIC (20 μm). In the majority of cells, baseline calcium oscillations were preserved, with a frequency of about 0.6 peaks/min (1.30 ± 0.14 peaks/min in SFM vs. 0.57 ± 0.07 peaks/min in BIC, n = 60, N = 4; paired t test, P < 0.05). These data are consistent with previous data (24,25) and clearly indicate that baseline [Ca2+]i oscillations are an intrinsic property of GnRH-1 neurons but that GABA influences the oscillator activity.
Together these studies indicate that a plasma membrane calcium oscillator drives spontaneous baseline [Ca2+]i oscillations in GnRH-1 neurons and that this calcium influx is sufficient to sustain oscillations in the majority of cells. The maintenance of cytosolic calcium homeostasis occurs partially through activation of the ER (Ca2+) ATPase, but there is no coupling of calcium influx with calcium release from the ER calcium store.
Modulators of GnRH-1 neuronal activity: KPs
With this new insight into the mechanism driving basal endogenous GnRH-1 neuronal activity, one could now begin to examine how modulators of this system work. To date, KPs are the most potent stimulators of GnRH-1 neurons, however the mechanisms by which KPs regulate GnRH-1 calcium signaling are unclear. GPR54, the KP receptor, is expressed postnatally in GnRH-1 neurons (6,10,11). We found that several receptors expressed postnatally on GnRH-1 neurons are expressed prenatally in vivo and expressed in GnRH-1 neurons maintained in nasal explants (35,37,38). Thus, we determined whether gpr54 was expressed during embryonic development.
Expression of GPR54 mRNA during embryonic development
In situ hybridization histochemistry for gpr54 transcript and GnRH-1 transcript was performed on adult mouse brain sections (Fig. 3A, top panel) and embryonic sections from E12.5 to E17.5 mice (Fig. 3A, middle and bottom panel). As previously reported, a subset of GnRH-1 neurons in the forebrain express gpr54 transcript (Fig. 3A, white arrows). Although GnRH-1 transcript is abundant in cells in the nasal region at E12.5 [data not shown, (18)], no gpr54-positive cells were detected. However, 1 d, later (E13.5) cells expressing gpr54 transcript were detected in the nasal region (Fig. 3A, middle panel) and nasal forebrain junction (Fig. 3A, bottom panel). GnRH-1-positive cells were detected in these same regions on adjacent sections. A similar gpr54 expression pattern was found at E14.5 (data not shown). By E17.5, the majority of GnRH-1 cells have migrated into the forebrain (18). gpr54-positive cells were found in adjacent forebrain sections, but transcript levels had decreased further, with only a few cells detected above background (data not shown). Thus, during prenatal development, GnRH-1 cells expressing gpr54 transcript can be found, but many cells show low or undetectable levels. Next, single GnRH-1 cell cDNAs obtained from GnRH-1 cells in nasal explants were evaluated for the presence of gpr54 transcript by PCR. This method is extremely sensitive, detecting 10 copies of mRNA per cell (29). The gpr54 mRNA transcript was detected in about 70% of GnRH-1 neurons at three ages tested (n = 8/10 at 3 div, n = 9/17 at 7–8 div, and n = 9/10 at 14 div; Fig. 3B). These results are consistent with previous data obtained from GnRH-1 cells in adult mouse and rat (mRNA [(6,11), galactosidase expression under gpr54 promoter (10)]. Thus, both in vivo and in vitro prenatal GnRH-1 neurons express gpr54 transcript. This is the earliest documentation of gpr54 expression in GnRH-1 neurons and allowed us to use nasal explants as a model to investigate KP effects on GnRH-1 cellular behavior.
Figure 3.

GnRH-1 neurons express a functional receptor to kp-10. A, In situ hybridization histochemistry for GnRH-1 mRNA and gpr54 mRNA performed on alternate sections from an adult mouse brain (upper panel) and E13.5 mouse sections (middle and lower panels). In agreement with the literature, in the adult brain preoptic area (POA; top), a subset of GnRH-1 neurons express gpr54 transcript (white arrows, boxed area is shown on the right for GnRH-1 and gpr54, and arrows point to cells shown in higher magnification). Gpr54-expressing cells, like GnRH-1-expressing cells, were detected at E13.5 in the nasal area (middle panel) and at the nasal/forebrain junction (bottom panel). oe, Olfactory epithelium; ob, olfactory bulb; fb, forebrain; III, third ventricle. B, Gel of PCR products from single-cell RT-PCR performed on GnRH-1 cells extracted from nasal explants after 7 or 8 d in vitro (div) using specific primers for gpr54. Seventy percent of the tested cells (n = 37) exhibited a band with gpr54-specific primers. Adult brain (Br) showed the expected band, whereas water (W) is negative. C, Effect of kp-10 on intracellular calcium level in 30 cells simultaneously recorded. Each row represents color-coded changes in intracellular calcium in a single cell. D, Average value of intracellular calcium level over time in 30 cells shown in C. E, Two representative traces of responding cells (85% of the cells), which either exhibited summation of single calcium spikes, termed plateau-bursting type of calcium signal (upper panel), or an increase in the frequency of calcium oscillations (lower panel). Note the long-lasting effect of kp-10 on calcium signaling after washout of agonist.
Modulation of GnRH-1 neuronal activity by kp-10
Application of kp-10 (0.1 nm) induced a slight elevation of the [Ca2+]i baseline (subthreshold response) and/or a reversible increase in the frequency of [Ca2+]i oscillations [subthreshold response: 1.29 ± 0.11 peaks/min in SFM vs. 1.52 ± 0.13 peaks/min in kp-10 (0.1 nm) vs. 1.21 + 0.11 peaks/min in washout SFM; paired t test P < 0.05] in about 57% of the GnRH-1 neurons (n = 50/87, N = 4). A more robust response to kp-10 was obtained with 10 nm, one tenth of the dose used in adult brain slice preparations (13,39). At this concentration, application of kp-10 induced elevation in [Ca2+]i in about 85% of GnRH-1 neurons (n = 44/52, N = 3), which is consistent with recent data obtained in slices from adult mice (39). Figure 3C illustrates the time course of changes in [Ca2+]i in one experiment using a pseudocolor representation, and Fig. 3D shows the data averaged across all GnRH-1 cells. Single-cell tracings revealed two types of responses in GnRH-1 cells. In the majority of cells (29 of 44), the baseline [Ca2+]i oscillations became a plateau-bursting type of signals (Fig. 3E, top trace) and in the remaining cells (15 of 44), the baseline oscillations were preserved or recovered rapidly but showed a significant increase in the spike frequency (SFM = 0.64 ± 0.12 peaks/min vs. treated = 2.58 ± 0.23 peaks/min; n = 15, N = 3; paired t test, P < 0.01; Fig. 3E, bottom trace). Note also the sustained receptor activity, which persisted in all responding cells after the washout of the agonist, in accordance with previous data (11,13,14).
To ensure that the endogenous oscillator activity of GnRH-1 cells was not artificially accelerated during recording, control recordings were performed in SFM without addition of kp-10. During 16-min recordings, none of control cells exhibited similar changes in their calcium oscillations (n = 64, N = 2). To clarify whether kp-10-induced responses recorded in GnRH-1 neurons could result from stimulation of GABAergic neurons rather than a direct action on GnRH-1 neurons, explants were treated with the GABAA receptor antagonist BIC (20 μm) before application of kp-10. When GABAergic inputs were disrupted, about 60% of GnRH-1 neurons still responded to kp-10 (n = 36 of 60, N = 4; responder percent lower than with kp-10 alone (Fisher’s exact test, P < 0.05; Table 1). Both patterns of calcium responses, plateau bursting and single spiking, were preserved, but the latter dominated (23 of 36), in contrast to SFM+kp-10 recordings.
These results indicate that functional GPR54 receptors are expressed in the majority of GnRH-1 neurons in nasal explants allowing integration of kp-10 signals directly but that tonic GABAergic input facilitates the action of kp-10. The lack of calcium response in a portion of the GnRH-1 neuronal population could indicate that the levels of GPR54 receptor expression in these cells are not sufficient to significantly alter the pattern of spontaneous [Ca2+]i oscillations and/or that some cells do not express gpr54, which is consistent with the RT-PCR analysis of single cells.
Dependence of GPR54 actions on extracellular and [Ca2+]i
When the spontaneous plasma membrane pacemaking activity was stopped by TTX, kp-10-induced a rise in [Ca2+]i in only 21% of cells (n = 33 of 160, N = 7; responder percent lower than with kp-10 alone). Note that the pattern of calcium signal in TTX-treated responders (Fig. 4A) was comparable with the plateau-bursting type of response observed in most GnRH-1 cells during SFM+kp-10 recordings. However, the delay in response was longer (133.7 ± 6.5 sec in TTX+kp-10 vs. 102.2 ± 7.2 sec in kp-10; Student’s t test, P < 0.05), and the slope of the response was reduced (2.1 ± 0.2 OD units/s in TTX+kp-10 vs. 4.1 ± 0.5 OD units/s in kp-10; Student t test, P < 0.05), suggesting that the fast component of the response is TTX sensitive (Fig. 3E).
Figure 4.
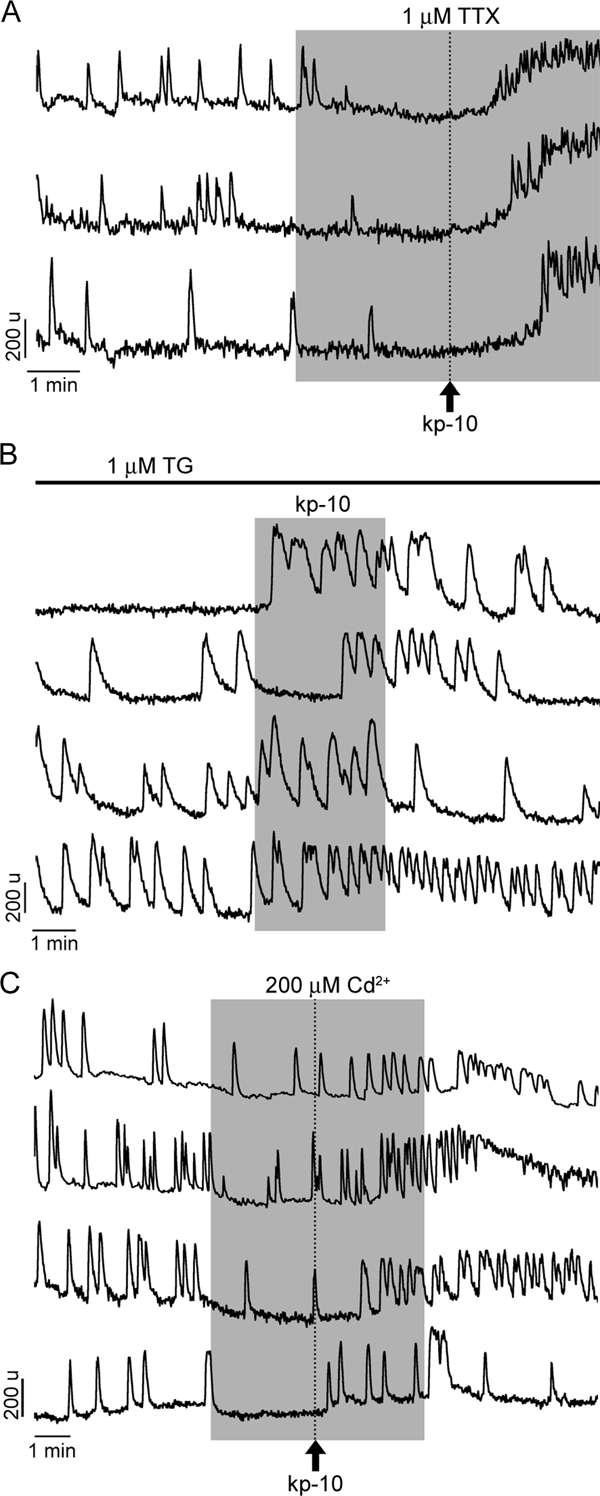
Identification of the source of calcium for kp-10-induced calcium signaling. A, A small fraction of cells (21%) with blocked spontaneous calcium oscillations by TTX responded to kp-10 with increase in [Ca2+]I, suggesting a role of intracellular calcium pools in response. Note the pattern of calcium signals in three representative cells. In the residual cells (79%) bathed in TTX-containing medium, kp-10 was completely ineffective, suggesting dependence of agonist action on the extracellular calcium pool. B, Consistent with the major role of calcium influx in kp-10 action, in 72% of cells with depleted ER calcium pool with TG, kp-10 also induced rise in [Ca2+]i. Four representative traces are shown. C, Only in a fraction of cells (44%, four representative traces are shown) bathed in medium contain cadmium, a kp-10-induced rise in [Ca2+]i occurred, further indicating the relevance of intracellular calcium pools for kp-10 action for a subpopulation of cells. In this and following figures, vertical dotted lines indicate the moment of kp-10 application, which was removed at the same time as cadmium.
Consistent with the involvement of a voltage-dependent pathway activated by kp-10 when the ER calcium pool was depleted by TG, kp-10 still induced calcium responses in 72% of GnRH-1 neurons (n = 36 of 50, N = 4; Fig. 4B; responder percent similar to kp-10 alone), indicating the major action of GPR54 is on the plasma membrane calcium oscillator. The extracellular calcium dependency of the kp-10-induced response was then investigated using cadmium (200 μm, nonselective blocker of voltage-gated calcium channels). When the cells were exposed to kp-10 in the presence of cadmium, the rise in [Ca2+]i was observed in only 44% of GnRH-1 neurons (n = 37 of 84; N = 4; Fig. 4C; responder percent lower than with kp-10 alone). The identity of the calcium channels involved in the kp-10-induced response was then investigated with application of specific calcium blockers before kp-10 exposure: nifedipine (L-type specific: 1 μm), conotoxin cleaved peptide (VIA) product GVIA (N-type specific: 1 μm), and SNX-482 (R-type specific: 100 nm). Blockade of L-type channels was as effective as cadmium application (41% of cells responded; n = 18 of 44, N = 2; responder percent lower than with kp-10 alone but similar to the response with cadmium), whereas blockade of N-type and R-type channels was ineffective: 83% (n = 39 of 47, N = 2) and 72% (n = 42 of 58, N = 2) of cells responded, respectively, both similar to responder percent with kp-10 alone.
To further evaluate the contribution of extracellular and intracellular calcium on kp-10-induced calcium signals, cells were perfused with 2-APB (75 μm) before application of kp-10. Under these conditions, 60% of GnRH-1 neurons were still able to exhibit calcium responses (n = 54 of 90, N = 3; Fig. 5A; responder percent lower than with kp-10 alone). Finally, in cells treated with both TTX and 2-APB, only 4% of the recorded cells showed a calcium response to kp-10 (n = 3 of 67, N = 4; Fig. 5B; responder percent lower than with kp-10 alone or combined with TTX). These results indicate an agonist-induced facilitation of the plasma membrane calcium oscillator activity, supplemented in a fraction of the cells by IP3 induced calcium release from the ER and/or stimulation of TRPC channels. To further investigate the involvement of TRPC channels in the kp-10-induced response, FFA (100 μm), another inhibitor of these channels, was applied before kp-10 exposure. In the presence of FFA, 69% of GnRH-1 neurons still responded (n = 42 of 61, N = 2; responder percent higher than with 2-APB but lower than with kp-10), suggesting that FFA was less effective than 2-APB in inhibiting TRP channels or 2-APB also affected other channels. The involvement of RyRs in the kp-10 response was not evaluated because neither TG depletion of ER nor 2-APB treatment alone was effective on the kp-10-induced response, but 2-APB and TTX together abolished it.
Figure 5.
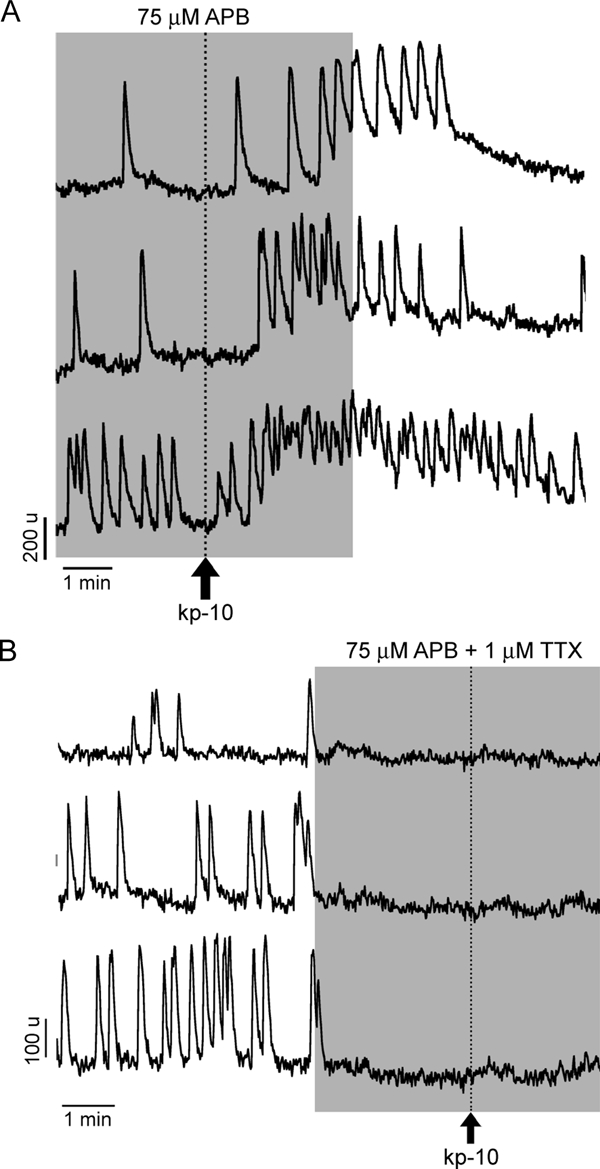
Contribution of IP3R-sensitive calcium pool on kp-10-induced calcium signaling. A, The majority of cells (60%) responded to kp-10 in the presence of 2-APB. In the residual cells, kp-10 stimulatory action was abolished. B, In the presence of TTX and 2-APB, the stimulatory action of kp-10 on calcium signaling was abolished in all cells.
Pathways involved in GPR54-mediated modulation of the oscillator activity
Three G protein-dependent signaling pathways are operative in GnRH-1-secreting neurons Gαs (40), Gαq/11 (41), and GαI (42). In general, the Gαi/o signaling pathway is associated with inhibition of adenylyl cyclase (AC) via binding of their α-subunits to the enzyme and, in some cases, stimulation of phospholipase C (PLC) through β/γ-dimers, which dissociate phosphatidyl inositol diphosphate (PIP2) into diacylglycerol and IP3 and then operate IP3R. The Gαq/11 signaling pathway also stimulates PLC but via their α-subunits. To determine which of the two calcium-mobilizing pathways account for the kp-10-induced calcium response, cells were treated with 250 ng/ml PTX, a blocker of the Gαi/o coupling, for at least 2 h. Subsequent application of 10 nm kp-10 still resulted in elevation in calcium levels in 80% of GnRH-1 neurons (n = 49 of 61, N = 2; Fig. 6A; responder percent similar to kp-10 alone), suggesting that the Gαq/11 coupling represents the main signaling pathway for the calcium-signaling action of kp-10.
Figure 6.
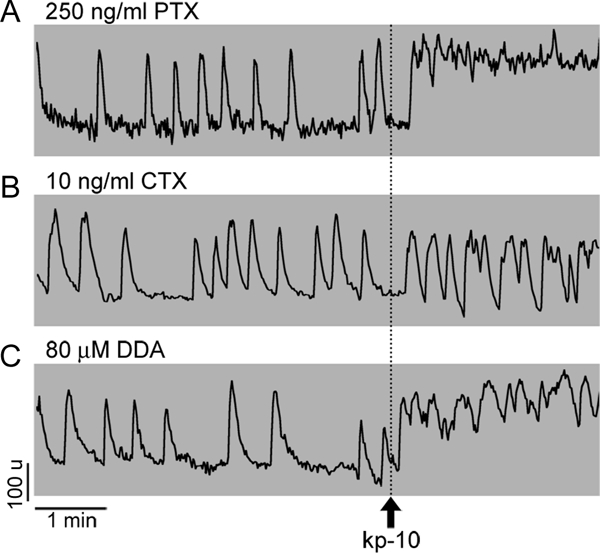
Identification of the G protein signaling pathways involved in kp-10 action. A, Uncoupling the inhibitory G protein (Gαi/o) with PTX did not alter the kp-10-induced calcium response, suggesting that the calcium mobilizing action of kp-10 in a fraction of cells was probably mediated by Gαq/11 signaling pathway. B and C, Uncoupling the stimulatory G protein (Gαs) with CTX (B) or its downstream effector AC by DDA (C) was accompanied with a decrease in the percentage of responding cells to 50 and 53%, respectively), suggesting a facilitating role of the Gαs/AC couple in the kp-10 response in a fraction of cells.
Activation of PLC leads to stimulation of protein kinase C (PKC). To test the relevance of GPR54-induced signaling on PKC, two sets of experiments were performed. In the first set, the enzyme was activated by the addition of phorbol ester, PMA. In these experiments, GABAergic input was blocked to ensure the observed action was on GnRH-1 neurons rather than GABAergic neurons. In presence of BIC, 79% of GnRH-1 neurons exhibited an increase in neuronal activity after PMA application (n = 58 of 73, N = 2; Fig. 7A). The PMA-induced increase of GnRH-1 neuronal activity was attenuated by an 8 min treatment with 100 nm BSM, a specific blocker of PKC (n = 12 of 77, N = 3; Fig. 7B). In the second set, PKC was blocked by BSM before kp-10 application, kp-10 was still effective in 55% of GnRH-1 neurons (n = 52 of 93, N = 4; Fig. 7C; responder percent lower than with kp-10 alone). These results indicate that the endogenous calcium oscillator activity can be facilitated by PLC signaling pathway through PKC-dependent and independent mechanisms.
Figure 7.
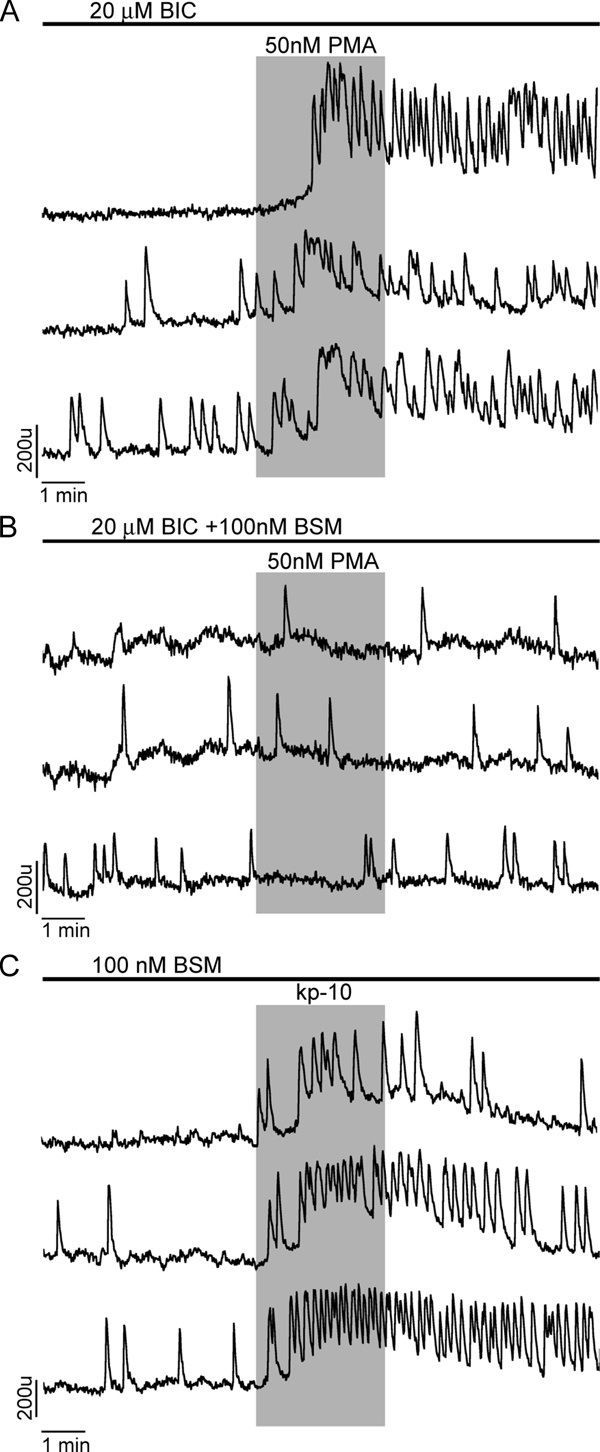
Spontaneous calcium oscillations in GnRH-1 neurons are modulated by PKC. Three representative traces per treatment are shown. A, PMA, an activator of PKC, stimulated calcium spiking in all cells with inhibited GABAergic input by BIC. A similar effect was observed in control cells. B, The stimulatory action of PMA was abolished in all cells bathed in medium containing BSM, a PKC inhibitor. C, BSM treatment lowered the percentage of cells responding to kp-10 to 55%, suggesting a facilitating role of PKC in the kp-10 response.
To examine kp-10 action via the Gαs signaling pathway, cells were treated with CTX (10 ng/ml) for at least 2 h. In these treated cells, subsequent application of 10 nm kp-10 resulted in calcium responses in 50% of the GnRH-1 neurons (n = 31 of 62, N = 4; Fig. 6B), in contrast to 75–80% of GnRH-1 neurons detected in SFM+kp-10. These results suggest that if GPR54 can couple to the Gαs signaling pathway, it occurs in a small group of GnRH-1 cells, in contrast to GPR54 coupling to the PLC signaling pathway. However, even this possibility should be taken with reservation because accumulation of cAMP occurs during a 2-h treatment with CTX (43), and the plasma membrane oscillator of GnRH-1 neurons has been shown to be sensitive to forskolin-induced cAMP production (24,25).
The relevance of the AC signaling pathway in kp-10 action was further examined by treating the cells with DDA (80 μm), an AC inhibitor for 16 min. As in CTX-treated cells, subsequent application of kp-10 was effective in 53% of GnRH-1 neurons (n = 40 of 75, N = 6; Fig. 6C; responder percent lower than with kp-10 alone but similar to the response with CTX). When cells were treated with DDA and BSM simultaneously, the percent of cells inhibited was not augmented, 46% of the cells still responded to kp-10 (n = 31 of 67, N = 2; responder percent similar to DDA or BSM alone). To reduce the impact of DDA on basal AC activity, in another set of experiments, cells were treated with DDA for only 3 min before kp-10. Under these conditions, 72% of the cells responded to kp-10 application (n = 28 of 39, N = 2). These results indicate that basal AC activity is relevant for determining the responsiveness of the GnRH-1 cells to kp-10 rather than contributing in the response itself. To evaluate whether basal AC activity was altering the responsiveness of GnRH-1 neurons through cAMP-dependent phosphorylation, the cells were treated for 8 min with H89, an inhibitor of the cAMP-dependent protein kinase (PKA), prior to the application of kp-10. Application of kp-10 elicited a response in 65% of the cells (n = 71 of 109, N = 4; responder percent lower than with kp-10 alone but similar to the response with DDA), Together these results favor an AC signaling pathway via PKA-dependent phosphorylation determining the responsiveness of GnRH-1 cells to kp-10 but not an essential pathway.
Discussion
GnRH-1 neurons are essential for reproduction, and KPs, via its receptor GPR54, play a major role in regulating these neurons and thus the reproductive axis. This report examines spontaneous baseline oscillations in [Ca2+]i in GnRH-1 neurons, individual and population dynamics, and the mechanism(s) by which kp-10/GPR54 activation alters GnRH-1 cellular behavior. When stimulated by kp-10, quiescent GnRH-1 cells responded by generating [Ca2+]i oscillations and spontaneously oscillatory cells responded by an increase in the frequency of calcium spikes or summation of individual spikes in plateau-bursting type of calcium signals. These changes predominantly reflected the stimulatory effect of GPR54 activation on the plasma membrane oscillator activity via the coupling of this receptor to PLC signaling pathway.
Postnatally GnRH-1 neurons are diffusely distributed from the olfactory bulbs to the caudal hypothalamus (44). As such, sampling in even a GnRH-1 cell-rich region, such as the preoptic area, is difficult. Thus, an in vitro model of postmitotic primary GnRH-1 cells obtained from nasal explants (21) was used in this study. Similar to in vivo, GnRH-1 neurons in nasal explants exhibit spontaneous electrical activity (35,45), pulsatile secretion (26), and depolarization-induced secretion (46). As in vivo, the levels of GnRH-1 released in vitro are difficult to quantify due to the small amount of releasable GnRH-1; thus, changes in secretion require pooling multiple mouse nasal explants per group (46). In contrast, calcium imaging techniques allow one to monitor the dynamics of the GnRH-1 neuronal population (23,31,33,47), and calcium oscillations have been shown to reflect electrical events in individual GnRH-1 neurons in nasal explants (24).
Here we demonstrate that GnRH-1 neurons exhibit spontaneous baseline calcium oscillations with a frequency of about one peak per minute. These baseline [Ca2+]I oscillations are driven by a plasma membrane oscillator that does not require the ER calcium pool. Oscillatory activity is often driven by a balance between voltage-dependent membrane conductances. For example, in thalamocortical relay cells, the pacemaking activity is mediated by alternative activation and inactivation of low-threshold calcium channels, hyperpolarization-gated cyclic nucleotide-modulated (HCN) channels, high-threshold calcium channels, and potassium channels (48). Two models have also been proposed for the oscillatory activity of GnRH-1 neurons, focusing on the role of cyclic nucleotides alone (49) or in combination with nonselective leaky cationic channels (50). A recent study demonstrated that HCN channels are not required for the rhythmic activity of prenatal GnRH-1 neurons and suggested that cAMP exhibits its stimulatory action on the plasma membrane oscillator through other effector(s) in a PKA-dependent manner (25).
Consistent with earlier work on prenatal GnRH-1 cells (33,38) and adult GnRH-1 neurons (15), here we also show that TTX blocked the oscillatory activity in GnRH-1 neurons, suggesting that the mechanism triggering [Ca2+]i oscillations in GnRH-1 cells in mouse is dependent on periodic activity of voltage-gated sodium channels. In GnRH-1 cells maintained in nasal explants derived from monkeys, a significant blockade of [Ca2+]i oscillations was not observed after application of TTX (51), although TTX-sensitive action currents were present (45). In GT1 cells, TTX-sensitive sodium channels have been shown to participate in the firing of action potentials with the effect dependent on the resting membrane potential (17). The efficiency of TTX to block [Ca2+]i oscillations in our experiments suggested a subsequent activation of voltage-gated calcium channels. However, incomplete inhibition of [Ca2+]I oscillations with cadmium and nifedipine, which is in agreement with data from acute brain slices (15) and GT1 cells showing sensitivity to nimodipine but not nifedipine (16), implies that other Ca2+-conducting cationic channels also contribute to the Ca2+ influx.
In general, sodium-driven depolarization leads to facilitation of calcium influx alone or in combination with Ca2+ release from the ER store. Release from the ER store can be mediated by activation of IP3Rs and/or RyRs by Ca2+ or direct coupling of voltage-gated calcium channels to the RyRs. IP3Rs have been shown to be a major contributor in the genesis of [Ca2+]i oscillations in GnRH-1 neurons in adult mice (15). In contrast, GnRH-1 neurons in nasal explants have a small releasable ER calcium pool and did not exhibit sensitivity to 2-APB. The difference in 2-APB sensitivity in these two preparations may be due to intrinsic developmental properties of calcium signaling in neurons, as shown for IP3R in vivo (52) and in vitro (53). Furthermore, because only a fraction of GnRH-1 neurons exhibited sensitivity to dantrolene, it is reasonable to conclude that depolarization-induced activation of the RyRs is not required for generation of [Ca2+]i oscillations in GnRH-1 neurons. Consistent with this conclusion, [Ca2+]i oscillations persisted in GnRH-1 neurons in which the ER (Ca2+)ATPase was inhibited with thapsigargin.
The expression of gpr54 and mechanism(s) by which activation of these receptors alters spontaneous GnRH-1 calcium oscillations was next examined. The expression of the gpr54 transcript was determined to be present in GnRH-1 neurons in vivo, as early as E13.5 in GnRH-1 cells still within the nasal region. To date, these data provide the earliest time point for gpr54 expression in GnRH-1 neurons and subsequently the ability of kp-10 to directly stimulate GnRH-1 neurons. Whether the activation of GPR54 occurs physiologically at such early developmental stage remains unknown, especially because the transcript appeared to be down-regulated as GnRH-1 cells entered the central nervous system. However, consistent with early expression of GPR54 in GnRH-1 cells and its extrinsic down-regulation, GnRH-1 neurons obtained from nasal explants and maintained in vitro for 1 wk were found to express gpr54. Using calcium imaging recordings to demonstrate the functionality of the GPR54, we found that the majority of GnRH-1 cells responded in a dose-dependent manner. The responsiveness of GnRH-1 neurons in vitro provides a model to study, in a large population of cells, the cellular mechanisms by which kp-10 can regulate GnRH-1 neurons. Three kinds of responses were detected in GnRH-1 neurons when stimulated by kp-10: quiescent GnRH-1 cells responded by generating [Ca2+]i oscillations and spontaneously oscillatory cells responded by an increase in the frequency of calcium spikes or summation of individual spikes in plateau-bursting type of calcium signals.
Excitatory GABAergic drives modulate GnRH-1 neuronal activity in nasal explants (33,35). Inhibition of GABAergic input decreased the percentage of GnRH-1 cells responding to kp-10 from 85 to 60%, with both patterns of calcium responses, plateau bursting, and single spiking, still present. No detectable levels of gpr54 mRNA were found in the nasal pit during development, the known location of the GABAergic nasal neurons (36). In the adult mouse, β-galactosidase expression driven by the gpr54 promoter was also limited to GnRH-1 neurons (10). Thus, it seems likely that the tonic GABAergic input determines the level of responsiveness of GnRH-1 neurons. In vivo, GABAergic input has been showed to be excitatory to GnRH-1 neurons before puberty (54,55). Whether a switch to inhibition of GnRH-1 neurons occurs after puberty remains controversial (54,55). However, the excitatory GABAergic input observed in explants may mimic a prepubertal status and thus GABAergic modulation of the kp-10 response by GnRH-1 neurons physiologically relevant.
Since their discovery, only a few studies have been focused on intracellular signaling pathways triggered by KPs binding to its receptor. Two studies were done with recombinant receptors, one using CHO cells (4) and the other using HEK293 cells (12) as expression systems. In both of these studies, the KPs induced calcium mobilization, presumably through the Gαq/11 coupling of these receptors and PIP2 hydrolysis. A third study performed in hypothalamic explants and assaying GnRH-1 secretion showed kp-10 activation required PLC, mobilization of intracellular calcium, and recruitment of ERK1/2 and p38 kinases but not voltage-gated calcium influx (9). In contrast, a recent study showed that kp-10-induced depolarization of GnRH-1 neurons is dependent on activation of cationic TRPC channels (13), which should facilitate calcium influx and calcium-dependent secretion.
Our results with native GPR54 in prenatal GnRH-1 neurons are consistent with both views and suggest that coupling of GPR54 to the PLC-dependent signaling pathway in most GnRH-1 cells accounts for the action of kp-10. In accordance with data from heterologous expressing systems, CHO (4) and HEK293 (12), we confirmed that Gαi/o coupling of GPR54 was not necessary for kp-10 to stimulate GnRH-1 neurons, suggesting that GPR54 stimulates PLC through Gq/11 signaling pathways. Furthermore, it appears that both PLC-dependent signaling molecules, IP3 and PKC, are involved in this regulation because: 1) the stimulatory action of kp-10 was abolished in about 25% of the GnRH-1 cells by PKC inhibition; 2) phorbol esters mimicked the action of kp-10 on calcium signaling in a PKC-dependent manner, consistent with the observation that phorbol esters stimulate GnRH-1 secretion in rat hypothalamus (56); and 3) 2-APB application in combination with TTX abolished the kp-10-induced calcium signaling in all cells.
These results fit with data obtained in GnRH-1 neurons in brain slices from the preoptic area that showed a TTX-resistant depolarization (11,13,14), mediated by nonselective cationic channels and sensitive to 2-APB presumably by inhibiting TRPC channels (13,39). TRPC transcripts are found in adult GnRH-1 neurons (13) as well as in GnRH-1 neurons maintained in nasal explants (our unpublished data). In our experiments, the number of GnRH-1 cells responding to kp-10 was attenuated by 2-APB to a greater extent than by FFA, suggesting that IP3R could also be inhibited in addition to TRPC channels. The mechanism by which Gαq/11-coupled receptors activate TRPC channels is not completely characterized and may include PKC-independent diacylglycerol action and ER calcium store depletion-induced stimulation (57). In accordance with this, the ER store depletion-induced membrane depolarization was observed in GT1 cells (58). In addition, a TTX-independent mechanism for stimulating GnRH-1 secretion at the level of GnRH-1 nerve terminals has been recently shown (59), data in agreement with work by Castellano et al. (9) that highlighted a mechanism dependent on ER calcium store.
However, in the present experiments, 75% of the GnRH-1 neuronal population failed to respond to kp-10 in the presence of TTX alone. This indicates that kp-10-induced PIP2 hydrolysis leads to calcium responses in the majority of prenatal GnRH-1 neurons by facilitating the plasma membrane oscillator through activation of TTX-sensitive sodium channels. In contrast, Liu et al. (39) observed negligible changes in the calcium response of GnRH-1 neurons to kp-10 in the presence of TTX, whereas electrical responsiveness to kp-10 was almost completely lost with 2-APB. It is important to note that TTX also partially inhibits [Ca2+]i oscillations in adult acute brain slices (15), whereas a complete blockade is observed in prenatal GnRH-1 neurons in the present study. Whether these are intrinsic or extrinsic induced, developmental changes in GnRH-1 cells remains to be examined. However, the delay of the response to kp-10 observed in the presence of TTX in the responding GnRH-1 cells in nasal explants, suggests a chronological activation of the cells. These data are in agreement with the alternative pathway described in adult preoptic area GnRH-1 cells involving kp-10 evoked inhibition of potassium channels (13,14,39). The prevalence of one vs. the other in the two models remains an interesting developmental and/or anatomical question. In the case of the secretory response, differences might also arise from the subcellular compartment studied because the relationship between calcium oscillations at the cell body and secretion at the nerve terminal remains difficult to assay.
In summary, the data in this report demonstrate that, in prenatal GnRH-1 neurons, a calcium oscillator is spontaneously active and is critically dependent on the TTX-sensitive sodium conductance. In a subset of kp-10-responsive GnRH-1 neurons, stimulation of GPR54 by kp-10 facilitates oscillator activity by stimulating calcium mobilization. However, in the majority of the kp-10-responsive GnRH-1 neurons, facilitation of the activity of this oscillator occurs via depolarization through TTX-sensitive sodium channels, resulting in increased frequency of [Ca2+]i oscillations via PKC. In addition, in a subset of kp-10-responsive GnRH-1 cells, stimulation of GPR54 leads to integration of calcium influx across the plasma membrane via depolarization of the cells through 2-APB-sensitive nonselective cationic channels.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorder and Stroke.
Disclosure Statement: The authors have nothing to disclose.
First Published Online October 23, 2008
Abbreviations: AC, Adenylyl cyclase; 2-APB, 2-aminoethyl diphenylborinate; BIC, bicuculline chloride; BSM, bisindolylmaleimide X hydrochloride; [Ca2+]i, intracellular calcium concentration; CTX, cholera toxin; DDA, dideoxyadenosine; div, days in vitro; DMSO, dimethylsulfoxide; E, embryonic day; ER, endoplasmic reticulum; FFA, flufenamic acid; GABA, γ-aminobutyric acid; GPR, G-protein-coupled receptor; HH, hypogonadal hypogonadism; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; KP, kisspeptins; kp-10, kisspeptin-10; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; PIP2, phosphatidyl inositol diphosphate; PMA, phorbol 12-myristate 13-acetate; PTX, pertussis toxin; RyR, ryanodine receptor channel; SFM, serum-free medium; SSC, sodium chloride-sodium citrate; TG, thapsigargin; TRP, transient receptor potential; TTX, tetrodotoxin.
References
- de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E 2003 Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA 100:10972–10976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno Jr JS, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O'Rahilly S, Carlton MB, Crowley Jr WF, Aparicio SA, Colledge WH 2003 The GPR54 gene as a regulator of puberty. N Engl J Med 349:1614–1627 [DOI] [PubMed] [Google Scholar]
- Funes S, Hedrick JA, Vassileva G, Markowitz L, Abbondanzo S, Golovko A, Yang S, Monsma FJ, Gustafson EL 2003 The KiSS-1 receptor GPR54 is essential for the development of the murine reproductive system. Biochem Biophys Res Commun 312:1357–1363 [DOI] [PubMed] [Google Scholar]
- Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, Brezillon S, Tyldesley R, Suarez-Huerta N, Vandeput F, Blanpain C, Schiffmann SN, Vassart G, Parmentier M 2001 The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem 276:34631–34636 [DOI] [PubMed] [Google Scholar]
- Gottsch ML, Cunningham MJ, Smith JT, Popa SM, Acohido BV, Crowley WF, Seminara S, Clifton DK, Steiner RA 2004 A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology 145:4073–4077 [DOI] [PubMed] [Google Scholar]
- Irwig MS, Fraley GS, Smith JT, Acohido BV, Popa SM, Cunningham MJ, Gottsch ML, Clifton DK, Steiner RA 2004 Kisspeptin activation of gonadotropin releasing hormone neurons and regulation of KiSS-1 mRNA in the male rat. Neuroendocrinology 80:264–272 [DOI] [PubMed] [Google Scholar]
- Navarro VM, Castellano JM, Fernandez-Fernandez R, Barreiro ML, Roa J, Sanchez-Criado JE, Aguilar E, Dieguez C, Pinilla L, Tena-Sempere M 2004 Developmental and hormonally regulated messenger ribonucleic acid expression of KiSS-1 and its putative receptor, GPR54, in rat hypothalamus and potent luteinizing hormone-releasing activity of KiSS-1 peptide. Endocrinology 145:4565–4574 [DOI] [PubMed] [Google Scholar]
- Matsui H, Takatsu Y, Kumano S, Matsumoto H, Ohtaki T 2004 Peripheral administration of metastin induces marked gonadotropin release and ovulation in the rat. Biochem Biophys Res Commun 320:383–388 [DOI] [PubMed] [Google Scholar]
- Castellano JM, Navarro VM, Fernandez-Fernandez R, Castano JP, Malagon MM, Aguilar E, Dieguez C, Magni P, Pinilla L, Tena-Sempere M 2006 Ontogeny and mechanisms of action for the stimulatory effect of kisspeptin on gonadotropin-releasing hormone system of the rat. Mol Cell Endocrinol 257–258:75–83 [DOI] [PubMed] [Google Scholar]
- Messager S, Chatzidaki EE, Ma D, Hendrick AG, Zahn D, Dixon J, Thresher RR, Malinge I, Lomet D, Carlton MB, Colledge WH, Caraty A, Aparicio SA 2005 Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc Natl Acad Sci USA 102:1761–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Gottsch ML, Lee KJ, Popa SM, Smith JT, Jakawich SK, Clifton DK, Steiner RA, Herbison AE 2005 Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci 25:11349–11356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir AI, Chamberlain L, Elshourbagy NA, Michalovich D, Moore DJ, Calamari A, Szekeres PG, Sarau HM, Chambers JK, Murdock P, Steplewski K, Shabon U, Miller JE, Middleton SE, Darker JG, Larminie CG, Wilson S, Bergsma DJ, Emson P, Faull R, Philpott KL, Harrison DC 2001 AXOR12, a novel human G protein-coupled receptor, activated by the peptide KiSS-1. J Biol Chem 276:28969–28975 [DOI] [PubMed] [Google Scholar]
- Zhang C, Roepke TA, Kelly MJ, Ronnekleiv OK 2008 Kisspeptin depolarizes gonadotropin-releasing hormone neurons through activation of TRPC-like cationic channels. J Neurosci 28:4423–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielecka-Fortuna J, Chu Z, Moenter SM 2008 Kisspeptin acts directly and indirectly to increase GnRH neuron activity and its effects are modulated by estradiol. Endocrinology 149:1979–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasoni CL, Todman MG, Strumia MM, Herbison AE 2007 Cell type-specific expression of a genetically encoded calcium indicator reveals intrinsic calcium oscillations in adult gonadotropin-releasing hormone neurons. J Neurosci 27:860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles AC, Hales TG 1995 Mechanisms of spontaneous calcium oscillations and action potentials in immortalized hypothalamic (GT1–7) neurons. J Neurophysiol 73:56–64 [DOI] [PubMed] [Google Scholar]
- Van Goor F, Krsmanovic LZ, Catt KJ, Stojilkovic SS 1999 Control of action potential-driven calcium influx in GT1 neurons by the activation status of sodium and calcium channels. Mol Endocrinol 13:587–603 [DOI] [PubMed] [Google Scholar]
- Wray S, Grant P, Gainer H 1989 Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci USA 86:8132–8136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasawa E, Quanbeck CD, Schulz CA, Burich AJ, Luchansky LL, Claude P 1993 A primary cell culture system of luteinizing hormone releasing hormone neurons derived from embryonic olfactory placode in the rhesus monkey. Endocrinology 133:2379–2390 [DOI] [PubMed] [Google Scholar]
- Daikoku S, Koide I, Chikamori-Aoyama M, Shimomura Y 1993 Migration of LHRH neurons derived from the olfactory placode in rats. Arch Histol Cytol 56:353–370 [DOI] [PubMed] [Google Scholar]
- Fueshko S, Wray S 1994 LHRH cells migrate on peripherin fibers in embryonic olfactory explant cultures: an in vitro model for neurophilic neuronal migration. Dev Biol 166:331–348 [DOI] [PubMed] [Google Scholar]
- Duittoz AH, Batailler M, Caldani M 1997 Primary cell culture of LHRH neurones from embryonic olfactory placode in the sheep (Ovis aries). J Neuroendocrinol 9:669–675 [DOI] [PubMed] [Google Scholar]
- Terasawa E, Schanhofer WK, Keen KL, Luchansky L 1999 Intracellular Ca(2+) oscillations in luteinizing hormone-releasing hormone neurons derived from the embryonic olfactory placode of the rhesus monkey. J Neurosci 19:5898–5909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin S, Wray S 2008 Gonadotropin-releasing hormone-1 neuronal activity is independent of cyclic nucleotide-gated channels. Endocrinology 149:279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin S, Wray S 2008 GnRH-1 neuronal activity is independent of HCN channels but is sensitive to PKA-dependent phosphorylation. Endocrinology 149:3500–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasawa E, Keen KL, Mogi K, Claude P 1999 Pulsatile release of luteinizing hormone-releasing hormone (LHRH) in cultured LHRH neurons derived from the embryonic olfactory placode of the rhesus monkey. Endocrinology 140:1432–1441 [DOI] [PubMed] [Google Scholar]
- Duittoz AH, Batailler M 2000 Pulsatile GnRH secretion from primary cultures of sheep olfactory placode explants. J Reprod Fertil 120:391–396 [PubMed] [Google Scholar]
- Funabashi T, Daikoku S, Shinohara K, Kimura F 2000 Pulsatile gonadotropin-releasing hormone (GnRH) secretion is an inherent function of GnRH neurons, as revealed by the culture of medial olfactory placode obtained from embryonic rats. Neuroendocrinology 71:138–144 [DOI] [PubMed] [Google Scholar]
- Kramer PR, Krishnamurthy R, Mitchell PJ, Wray S 2000 Transcription factor activator protein-2 is required for continued luteinizing hormone-releasing hormone expression in the forebrain of developing mice. Endocrinology 141:1823–1838 [DOI] [PubMed] [Google Scholar]
- Dulac C, Axel R 1995 A novel family of genes encoding putative pheromone receptors in mammals. Cell 83:195–206 [DOI] [PubMed] [Google Scholar]
- Giacobini P, Kopin AS, Beart PM, Mercer LD, Fasolo A, Wray S 2004 Cholecystokinin modulates migration of gonadotropin-releasing hormone-1 neurons. J Neurosci 24:4737–4748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple JL, Wray S 2005 Developmental changes in GABA receptor subunit composition within the gonadotrophin-releasing hormone-1 neuronal system. J Neuroendocrinol 17:591–599 [DOI] [PubMed] [Google Scholar]
- Moore Jr JP, Shang E, Wray S 2002 In situ GABAergic modulation of synchronous gonadotropin releasing hormone-1 neuronal activity. J Neurosci 22:8932–8941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE 2007 SnapShot: mammalian TRP channels. Cell 129:220 [DOI] [PubMed] [Google Scholar]
- Kusano K, Fueshko S, Gainer H, Wray S 1995 Electrical and synaptic properties of embryonic luteinizing hormone-releasing hormone neurons in explant cultures. Proc Natl Acad Sci USA 92:3918–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray S, Fueshko SM, Kusano K, Gainer H 1996 GABAergic neurons in the embryonic olfactory pit/vomeronasal organ: maintenance of functional GABAergic synapses in olfactory explants. Dev Biol 180:631–645 [DOI] [PubMed] [Google Scholar]
- Sharifi N, Reuss AE, Wray S 2002 Prenatal LHRH neurons in nasal explant cultures express estrogen receptor β transcript. Endocrinology 143: 2503–2507 [DOI] [PubMed] [Google Scholar]
- Giacobini P, Wray S 2007 Cholecystokinin directly inhibits neuronal activity of primary gonadotropin-releasing hormone cells through cholecystokinin-1 receptor. Endocrinology 148:63–71 [DOI] [PubMed] [Google Scholar]
- Liu X, Lee K, Herbison AE 2008 Kisspeptin excites gonadotropin-releasing hormone (GnRH) neurons through a phospholipase C/calcium-dependent pathway regulating multiple ion channels. Endocrinology 149:4605–4614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez de la Escalera G, Choi AL, Weiner RI 1992 β1-Adrenergic regulation of the GT1 gonadotropin-releasing hormone (GnRH) neuronal cell lines: stimulation of GnRH release via receptors positively coupled to adenylate cyclase. Endocrinology 131:1397–1402 [DOI] [PubMed] [Google Scholar]
- Krsmanovic LZ, Stojilkovic SS, Mertz LM, Tomic M, Catt KJ 1993 Expression of gonadotropin-releasing hormone receptors and autocrine regulation of neuropeptide release in immortalized hypothalamic neurons. Proc Natl Acad Sci USA 90:3908–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krsmanovic LZ, Mores N, Navarro CE, Saeed SA, Arora KK, Catt KJ 1998 Muscarinic regulation of intracellular signaling and neurosecretion in gonadotropin-releasing hormone neurons. Endocrinology 139:4037–4043 [DOI] [PubMed] [Google Scholar]
- Kostic TS, Tomic M, Andric SA, Stojilkovic SS 2002 Calcium-independent and cAMP-dependent modulation of soluble guanylyl cyclase activity by G protein-coupled receptors in pituitary cells. J Biol Chem 277:16412–16418 [DOI] [PubMed] [Google Scholar]
- Herbison AE 2006 Physiology of the gonadotropin-releasing hormone neuronal network. In: Knobil E, Neill J, eds. The physiology of reproduction. 3rd ed. San Diego: Academic Press; 1415–1482 [Google Scholar]
- Abe H, Terasawa E 2005 Firing pattern and rapid modulation of activity by estrogen in primate luteinizing hormone releasing hormone-1 neurons. Endocrinology 146:4312–4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore Jr JP, Wray S 2000 Luteinizing hormone-releasing hormone (LHRH) biosynthesis and secretion in embryonic LHRH. Endocrinology 141:4486–4495 [DOI] [PubMed] [Google Scholar]
- Temple JL, Laing E, Sunder A, Wray S 2004 Direct action of estradiol on gonadotropin-releasing hormone-1 neuronal activity via a transcription-dependent mechanism. J Neurosci 24:6326–6333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss R, Seifert R 2000 Pacemaker oscillations in heart and brain: a key role for hyperpolarization-activated cation channels. Chronobiol Int 17:453–469 [DOI] [PubMed] [Google Scholar]
- Khadra A, Li YX 2006 A model for the pulsatile secretion of gonadotropin-releasing hormone from synchronized hypothalamic neurons. Biophys J 91:74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBeau AP, Van Goor F, Stojilkovic SS, Sherman A 2000 Modeling of membrane excitability in gonadotropin-releasing hormone-secreting hypothalamic neurons regulated by Ca2+-mobilizing and adenylyl cyclase-coupled receptors. J Neurosci 20:9290–9297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H, Keen KL, Terasawa E 2008 Rapid action of estrogens on intracellular calcium oscillations in primate luteinizing hormone-releasing hormone-1 neurons. Endocrinology 149:1155–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure AV, Grunwald D, Moutin MJ, Hilly M, Mauger JP, Marty I, De Waard M, Villaz M, Albrieux M 2001 Developmental expression of the calcium release channels during early neurogenesis of the mouse cerebral cortex. Eur J Neurosci 14:1613–1622 [DOI] [PubMed] [Google Scholar]
- Genazzani AA, Carafoli E, Guerini D 1999 Calcineurin controls inositol 1,4,5-trisphosphate type 1 receptor expression in neurons. Proc Natl Acad Sci USA 96:5797–5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Abraham IM, Herbison AE 2002 Effect of GABA on GnRH neurons switches from depolarization to hyperpolarization at puberty in the female mouse. Endocrinology 143:1459–1466 [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Heger S, Ojeda SR, Moenter SM 2002 Activation of A-type γ-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol 16:2872–2891 [DOI] [PubMed] [Google Scholar]
- Rochdi L, Theraulaz L, Enjalbert A, Gautron JP 2000 Differential in vitro secretion of gonadotropin-releasing hormone (GnRH) and [hydroxyproline] GnRH from the rat hypothalamus during postnatal development. J Neuroendocrinol 12:919–926 [DOI] [PubMed] [Google Scholar]
- Ambudkar IS, Bandyopadhyay BC, Liu X, Lockwich TP, Paria B, Ong HL 2006 Functional organization of TRPC-Ca2+ channels and regulation of calcium microdomains. Cell Calcium 40:495–504 [DOI] [PubMed] [Google Scholar]
- Van Goor F, Krsmanovic LZ, Catt KJ, Stojilkovic SS 1999 Coordinate regulation of gonadotropin-releasing hormone neuronal firing patterns by cytosolic calcium and store depletion. Proc Natl Acad Sci USA 96:4101–4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Tassigny XD, Fagg LA, Carlton MB, Colledge WH 2008 Kisspeptin can stimulate GnRH release by a direct action at GnRH nerve terminals. Endocrinology 149:3926–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]