

Abstract
A discussion of the literature concerning the synthesis, function, and activity of heme c-containing proteins is presented. Comparison of the properties of heme c, which is covalently bound to protein, is made to heme b, which is bound noncovalently. A question of interest is why nature uses biochemically expensive heme c in many proteins when its properties are expected to be similar to heme b. Considering the effects of covalent heme attachment on heme conformation and on the proximal histidine interaction with iron, it is proposed that heme attachment influences both heme reduction potential and ligand-iron interactions.
1 Introduction
Heme (iron-protoporphyrin IX) prosthetic groups perform a wide range of functions in nature including electron transfer, oxygen transport and storage, catalysis, gas sensing, and gene regulation. The functional versatility of heme is attributed to the ability of the protein environment to tune heme reactivity, with important factors including the number and nature of protein-donated axial ligands to iron, the extent of heme burial in the protein, the accessibility of the heme to exogenous ligands, the distribution of polar and charged groups around the heme, and other properties of the heme-binding site and of the protein.1-3 An additional variable that receives less attention is the type of heme group, defined by the nature of substituents on the porphyrin macrocycle (Fig. 1). The most common types of heme are heme b and heme c. Heme b is iron-protoporphyrin IX and binds noncovalently to protein (Fig. 1A), whereas heme c is characterized by the presence of two (rarely, one) covalent thioether bonds formed between Cys side chains and the heme vinyl groups at positions 2 and 4 (Fig. 1B). The stereochemistry of heme attachment is the same in all known examples of heme c, and the vinyl groups at positions 2 and 4 are attached to the N- and C-terminal Cys of the CXXCH motif, respectively. Other, less common, derivatives of heme include heme d1, present in cytochrome cd1 nitrite reductase (Fig. 1C), heme a, found in cytochrome c oxidase, and the related heme o, found in some bacterial oxidases.
Figure 1.
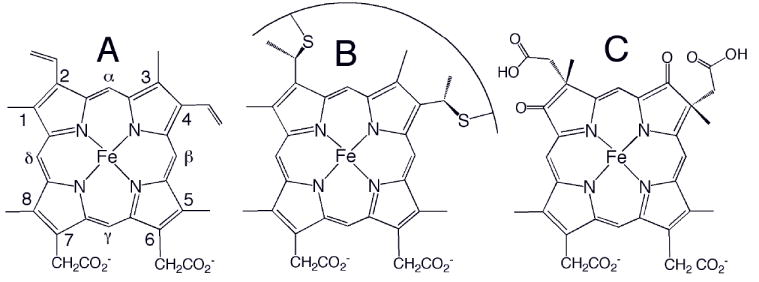
Chemical structures of (A) heme b, (B) heme c, and (C) heme d1. The Fisher numbering system for heme substituents is shown in (A). The curve in (B) represents a peptide segment; usually two residues separate the two Cys, and the His axial ligand follows the Cys attached to position 4. Heme o has a hydroxyfarnesyl group in place of vinyl 2 in (A), and heme a has a hydroxyfarnesyl group in place of vinyl 2 and a formyl group in place of methyl 8.
From a chemical perspective, hemes b and c are very similar to each other and thus are not expected to display significant inherent differences in electronic structure or reduction potential. From a biosynthetic perspective, however, these types of heme are quite different; because heme c is synthesized from heme b, the use of heme c requires a greater investment from the organism. The question of why covalently attached heme is sometimes used thus has garnered significant interest.4-6 The understanding that heme c biogenesis is not only complex but also achieved through diverse mechanisms underlines the importance of this type of heme in nature.6-10 The functional bases for the use of heme c, however, remain unclear. In this article, the synthesis, function and properties of heme c are discussed with an eye toward understanding the effects of covalent attachment on the properties of the heme iron.
2 Heme c occurrence in nature
Proteins containing heme c are usually referred to as cytochromes c, and often function as electron carriers, although heme c displays other functions as well. The heme attachment site in most cases is a CXXCH pentapeptide segment, where His serves as an axial ligand to iron, the Cys form the thioether bonds to the porphyrin, and the X represent variable residues (Fig. 2). Following conventions from the globin field, the side of the heme containing the CXXCH motif will be referred to as the proximal side, and the other side of the heme the distal side. Cytochromes c display a wide range of sizes and folds and examples of individual proteins with up to 16 heme c groups have been identified.11, 12 The sequence of the heme-binding motif is highly variable, and all amino acids have been found in the “XX” segment in nature (including Gly, Pro, and His), except for Cys.
Figure 2.
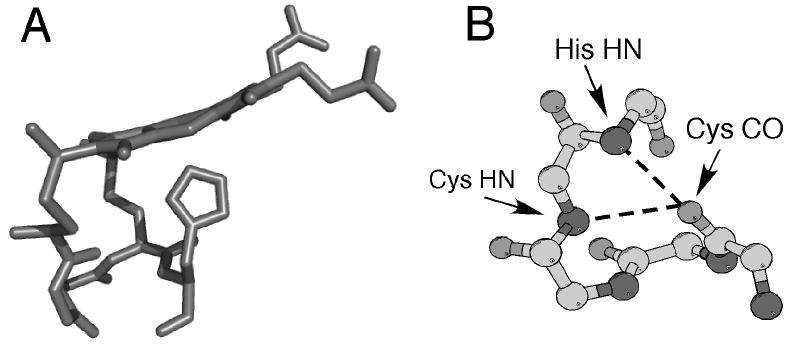
(A) Three-dimensional structure of heme c (side view) and the CXXCH (residues 14-18) pentapeptide from the structure of horse cytochrome c (PDB: 1HRC). The side chains of residues 15 and 16 are omitted for clarity. Note the nonplanar structure of the heme. (B) Ball-and-stick representation of backbone atoms of CXXCH motif. Hydrogen bonding interactions between the backbone HN of the second Cys and the His to the backbone CO of the first His are shown with dashed lines.
There is a small number of cytochromes c with heme attachment sites deviating from the signature CXXCH site with His as axial ligand. The photosynthetic protein complex cytochrome b6f contains a heme referred to as heme ci’ (also denoted x) attached to the cytochrome b component of the complex through a single thioether bond. Heme ci’ also is unusual for lacking protein-donated ligands to heme iron, being ligated axially only by a water or hydroxyl.13-15 Covalent binding to a single Cys is also observed for cytochromes c and c1 from the mitochondria of Euglena, Trypanosoma, and Leishmania species. These cytochromes c bind to a AXXCH or FXXCH motif with His as an iron ligand.16, 17 Another unconventional heme c binding motif is found at the active site of pentaheme nitrite reductases (NrfA) and consists of a CXXCK segment with heme c attached to the two Cys and the Lys serving as an axial ligand to iron.18 Longer forms of the heme attachment motif also have been identified. CXXXXCH motifs are found in tetraheme cytochromes c319 and CXXXCH motifs have been found in a diheme cytochrome c320 as well as a diheme cytochrome c552 from Pseudomonas stutzeri.21 Finally, a CX15CH heme attachment motif with both Cys forming covalent bonds to heme has been identified for one heme within the multiheme c-type cytochrome MccA from Wolinella succinogenes.22 As cytochromes c are identified by the presence of CXXCH in sequence data, the finding of this unusual attachment mode suggests that other variations are likely to exist that have not yet been identified.
Nomenclature has developed over many decades and there is not one universal system to name cytochromes c. One consistent feature of heme protein nomenclature is the use of a, b, c, d, d1, or o to refer to the composition of the heme group, although as mentioned above there is some variability in the mode of attachment for heme c. In addition, the term cytochrome f is used to describe some proteins that contain heme c and have the N-terminus as the distal ligand. There are four principal classes (also called types) of cytochromes c (Fig. 3). Class 1 is the largest group and consists of small (8-12 kDa) soluble proteins with His/Met heme axial ligation. They are usually low-spin (S = 0 in the ferrous and S = ½ in the ferric state) with a single high-potential heme bound near the N-terminus and the methionine ligand near the C-terminus. Most Class 1 cytochromes have one heme group, although a few have two domains, each of which binds one heme. Class 2 cytochromes c have a heme attachment site near the C-terminus of a four-helix bundle structure. Many are high-spin although there are examples that are low-spin with ligating methionine located near the N-terminus. The high-spin hemes c are typically denoted c’. Class 3 consists of multiheme cytochromes c; heme axial ligation typically is bis-His and reduction potentials of these proteins tend to be low. The Class 3 cytochromes c also have high heme:polypeptide ratios; the high molecular-weight examples of Class 3 cytochromes c have a large number of hemes. Finally, the high molecular weight (40 kDa) tetraheme reaction center cytochromes c constitute Class 4. Hemes in class 4 cytochromes c display His-Met or bis-His axial ligation. There are other cytochromes c that are not readily placed into these 4 classes. Individual cytochromes c are placed into subgroups named by one of two systems. In one system, each subgroup receives a subscript 1-8 which were assigned progressively (for example, cytochrome c2 and cytochrome c6 are Class 1 cytochromes c, and cytochrome c3 is a Class 3 cytochrome c). In a second system, a subscript is assigned that designates the wavelength of the low-energy absorbance band in the reduced protein (for example, cytochrome c551). For more details on subclasses of cytochromes c, the reader is referred to published descriptions of cytochrome c classification and sequences.23-25
Figure 3.
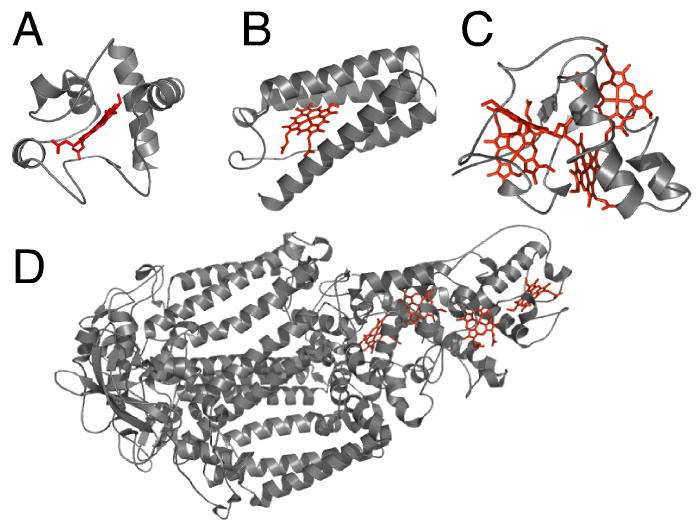
Three-dimensional structures of cytochromes c representative of each of the 4 structural classes. (A) Class 1: horse cytochrome c (PDB: 1HRC); (B) Class 2: Rhodopseudomonas capsulatus cytochrome c’ (1CPQ); (C) Class 3: Desulfovibrio vulgaris Hildenborough cytochrome c3 (1CTH); (D) Class 4: Photosynthetic reaction center from Rhodopseudomonas viridis (1PRC). In (D), cofactors other than the four hemes c are omitted for clarity. The polypeptides are shown in ribbon format and the hemes as sticks.
3 Heme c function
Although proteins containing heme c display diverse structures, heme c function is less variable than that of heme b, with most cytochromes c serving as electron carriers, cycling between the reduced (ferrous) and oxidized (ferric) oxidation states to perform single electron transfers.26, 27 Diversity among these electron transfer proteins is expressed in the wide range of energy transduction processes involving cytochromes c including photosynthesis, various pathways of respiration, and the cycling of nitrogen and sulfur. Another greatly variable factor related to the function of heme c is its large range of reduction potentials in nature, spanning well over 1 V. The reduction potentials of the heme c and of the cytochrome’s redox partner(s) determine the thermodynamics of the electron transfer reaction and also play a role in determining electron transfer kinetics.28 It is particularly intriguing that heme c spans a significantly larger range of reduction potentials than does heme b when proteins with the same heme axial ligation are compared (the identity of the axial ligands is an important determinant of potential).29 Considering only sites with bis-His axial ligation (the most common ligand set), heme b is found with potentials ranging from -150 to +380 mV, whereas heme c potentials span from -450 to +380 mV vs. NHE.30, 31
Heme c can serve as a redox site for intramolecular or intermolecular electron transfer reactions (or both). The cytochrome c may transfer electrons to partner proteins, as does mitochondrial cytochrome c, or heme c may be present within an enzyme in which it serves to mediate electron transfer from a donor to an active catalytic site. Examples of this latter role are the hemes c in bacterial alcohol dehydrogenases, the bacterial caa3 and cbb3 oxidases, the mitochondrial and bacterial bc1 complex, cytochrome cd1 nitrite reductase, and nitrate reductase.11 Genome sequencing efforts in recent years have led to the discovery of many multi-heme cytochromes c. In particular, the genomes of organisms that respire metal ions such as iron, uranium, chromium, and manganese, for example, Geobacter sulfurreducens and Shewanella oneidensis, have been found to be rich in cytochromes c.32-38 Some of these cytochromes contain multiple hemes c in a monodimensional arrangement suggesting that the proteins act in “electron harvesting” and/or electron transfer over very long distances (Fig. 4),39 and some of these cytochromes are excreted for reduction of extracellular substrates.40, 41
Figure 4.
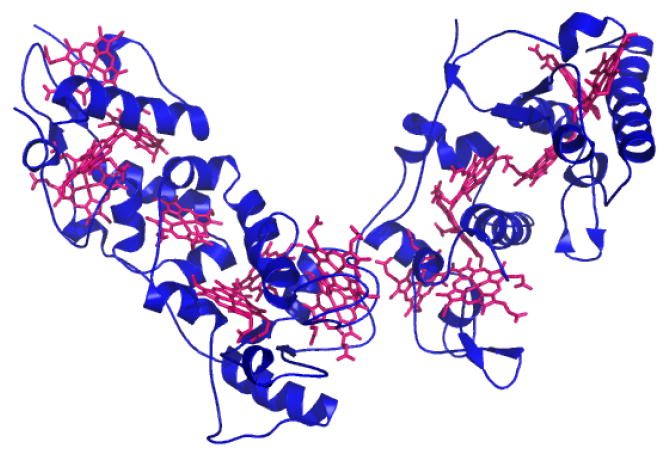
Three-dimensional structure of Desulfovibrio vulgaris Hildenborough HmcA (PDB: 1H29), a hexadecaheme cytochrome c, with hemes c shown in magenta.
Although cytochromes c most often function in electron transfer, a number of other functions are displayed by this versatile family of proteins. The finding that mitochondrial cytochrome c plays a key role in apoptosis was an unexpected development.42 While it is not clear that the heme plays a direct role in apoptosis triggering, the pro-apoptotic activity of mitochondrial cytochrome c has been linked to the heme redox state43, 44 and a conformational change involving the heme crevice.45 Mitochondrial cytochrome c also has been proposed to act in detoxification of reactive oxygen species.46 New functions have been proposed for other cytochromes c initially thought to function exclusively in electron transfer. For example, a Class 3 tetraheme cytochrome c3 has been found to bind CO specifically at heme 2 (replacing the distal histidine ligand) and is proposed to act as a CO sensor.47 This CO-binding activity was unexpected because each of the four hemes has two endogenous axial ligands. However, the displacement of an endogenous ligand by an exogenous ligand is seen for other heme proteins such as the hexacoordinate hemoglobins.48 Redox-state dependence of heme ligation also is observed in some cytochromes c, for example, cytochrome cd1 nitrite reductase, in which the heme c displays redox state-dependent ligation proposed to control electron transfer rates.49
Some hemes c have been identified that bind exogenous ligands to an open coordination site to act as a gas or redox sensor. The cytochromes c’ are 5-coordinate Class 2 cytochromes widely distributed among bacteria. They bind NO and are proposed to suppress toxic levels of NO.50, 51 Another ligand-binding high-spin heme c is found in Sphaeroides heme protein which binds oxygen but has an unknown function.52 Ten c-heme-containing sensor proteins have been identified in G. sulfurreducens, two of which have a PAS-type fold (which usually binds heme b) and are part of chemotaxis proteins.53 Interestingly, an internal redox-driven change in heme coordination also is observed for these PAS-type sensor proteins. The physiological effector molecule for these high-spin hemes c is not known but CO and NO are candidates. A similar redox-driven switching phenomenon was observed for another sensor protein, DcrA, a c-heme chemotaxis signal transducer that has two endogenous axial ligands in the reduced state, but has an open coordination site when oxidized; CO binds to the reduced protein, displacing the distal ligand.54
Although the active site of most heme enzymes is a heme b, heme c also can serve as a site for catalysis. The tetraheme cytochrome Nitrosomonas europaea cytochrome c554 has been reported to display NO reductase activity; like CO binding to cytochrome c3, this result is surprising because all hemes are 6-coordinate in the resting state.55 The bacterial di-heme cytochrome c peroxidases (CCPs) also have 6-coordinate c-type hemes in the resting (oxidized) state. Activation of the enzyme by calcium binding and reduction allows the low-potential peroxidatic heme to become accessible to substrate. The other, high-potential, heme is proposed to serve as an electron conduit.56, 57 A related enzyme is MauG which also contains two hemes c and plays a role in tryptophan tryptophylquinone biogenesis.58 The bifunctional catalase-peroxidases also contain two c-type hemes and share some similarity with the bacterial CCPs.59 Other enzymes with a heme c at the active site include cytochrome P46060 and hydroxylamine oxidoreductase.61 Both enzymes oxidize hydroxylamine to nitrite and also have an additional covalent cross link to heme along with the two thioether bonds. Two recently discovered enzymes with c-heme active sites are the SoxAX complex that is involved in thiosulfate oxidation,62, 63 and the rubber oxygenase RoxA that cleaves poly(cis-1,4-isoprene).64, 65
In summary, heme c displays a range of functions in nature, although it most frequently serves as an electron transfer site. In comparison with heme b, heme c sites show greater versatility with respect to reduction potential tuning, with low-spin hemes c reaching lower potentials than do low-spin hemes b. Comparing the functions of heme c and heme b provides some initial hints as to the functional basis for the covalent attachments. For one, covalent bonding may have effects that allow heme c potentials to be tuned over a larger range than heme b; this may be mediated by effects on the proximal histidine-iron (His-Fe) bond or on the heme conformation. These hypotheses will be discussed in subsequent sections.
4 Cytochrome c biogenesis and expression
The assembly of cytochromes c in biological systems displays surprising complexity and diversity, while yielding identical heme orientation relative to the CXXCH residues and identical stereochemistry at the thioether sulfurs (Fig. 1B). The considerable biosynthetic effort expended to convert heme b to heme c indicates that utilizing heme c in selected proteins confers a significant advantage to the organism.66 A number of heme c biogenesis systems exist that show varying degrees of complexity and specificity.9 The three most prevalent are denoted System I, II, and III.8 Systems I and II, also called Ccm (cytochrome c maturation) and Ccs (cytochrome c synthesis), respectively, are found in prokaryotes, plant mitochondria and chloroplasts, whereas System III, also called CCHL (cytochrome c heme lyase) is found in mitochondria of fungi, invertebrates, and vertebrates. In the case of Class 1 cytochromes c, it has been established experimentally for representative mitochondrial and bacterial proteins that covalent attachment of heme is necessary to achieve proper folding and stability.67-69 Indeed, as cytochromes c, and multiheme forms in particular, typically have low residue-to-heme ratios in comparison with other heme proteins, it is not surprising that the heme and its attachment play a critical role in folding.
Of the known systems for cytochrome c biogenesis, System I, found in some protozoan mitochondria and in α and γ proteobacteria such as the molecular biology workhorse Escherichia coli, is the most well studied.70, 71 System I for cytochrome c maturation consists of up to nine proteins (CcmABCDEFGHI) in addition to a number of accessory proteins. The cytochrome c structural gene is expressed as a pre-apoprotein with an N-terminal signal peptide that is recognized by the secretory (Sec) pathway transporting the polypeptide to the periplasm.72 Leader peptidases cleave the signal peptide to leave the apoprotein. Heme, which is synthesized in the cytoplasm, is transferred to the periplasm though an unknown mechanism. Formation of holocytochrome c requires heme delivery and ligation to the polypeptide in a manner that results in the correct orientation of the heme within the protein and the correct stereochemistry at the thioether sulfurs. Functions of some of the components of the ccm cassette in this process have been identified. CcmAB is similar to an ATP-dependent transporter, and CcmA hydrolyzes ATP.73 However, the substrate transported by CcmAB has not been identified, and it not thought to be heme. CcmC is required for delivery of heme to CcmE,74 which has been identified as a heme chaperone requiring the ATP hydrolysis activity of CcmAB.75, 76 CcmE forms a transient covalent bond between a histidine side chain and a heme vinyl group,75 and heme in holo-CcmE displays axial coordination by a Tyr side chain in one form and an unidentified weak-field ligand in a second form.77, 78 The details of the heme transfer mechanism remain unclear although ongoing studies continue to provide intriguing details. CcmD is a small polypeptide that is a component of a complex with CcmABC and is required for the release for holo-CcmE from CcmABCD.79 Holo-CcmE is the putative substrate for CcmF, which transfers heme to apocytochrome and along with CcmH is proposed to facilitate thioether bond formation.80 CcmG is a thioredoxin-like protein,81 likely involved in disulfide bond reduction and/or isomerization. DsbD also is proposed to pay this role.
System II for cytochrome c biogenesis is found in Gram-positive bacteria, cyanobacteria, chloroplasts of plans and algae, and some β-, δ-, and ε-proteobacteria.8 As in System I, apocytochrome delivery is by the Sec system. However, System II has neither an ABC transporter complex nor a CcmE-like heme chaperone. Rather, CcsAB is proposed to act in heme attachment to apocytochrome; CcsA contains a WWD domain as a putative heme binding site. DsbD also serves as a component.82, 83 The simplest of the three major systems, System III, is found in the most complex organisms. System III makes use of CCHL enzymes with attachment occurring to apoprotein after its transport from cytoplasm into the mitochondrial intermembrane space.10 A conserved CPV motif has been established to be essential for CCHL function and is a possible heme binding site,84 and an inner membrane bound flavoprotein, Cyc2p, is proposed to play a role in reduction of heme iron and/or the apocytochrome cysteines to prepare for covalent ligation.85 The mechanism for apocytochrome transport from the cytoplasm into the intermembrane space depends upon the organism and the cytochrome; the means by which heme is transported into the intermembrane space is not understood.10 While System I is capable of maturing a wide range of cytochromes c, System III is rather specific, and distinct heme lyases are present that process mitochondrial cytochrome c and cytochrome c1, even within one organism.10 System III has been demonstrated not to mature bacterial cytochromes c.86
In addition to the prevailing maturation Systems I-III, a number of variants have been discovered. Cytochrome c biogenesis in archaea has been shown to occur through a variant of System I with a novel CcmE component.87 System I is not compatible with substitutions for the His ligand in the CXXCH motif,88 thus alternative systems are required for unusual cytochromes c lacking a proximal His. A set of proteins, denoted system CCB, have been identified that perform the biogenesis of the high-spin ci’ heme of cytochrome b6f, which is attached by a single Cys.89 In addition, unique maturation proteins are involved in attachment of the heme group to the CXXCK motif in NrfA.90 MccA has a heme lyase, CcsA1, proposed to be dedicated to attachment of heme to the two Cys of its unusual CX15CH motif.22 A/FXXCH sequences are not processed by System I, although they are processed by System II.91-93 An as-yet undiscovered maturation pathway is expected for these cytochromes because the available genomes of organisms containing cytochromes c with a single covalent attachment do not encode proteins for any of the known systems.91, 92
The identification and cloning of critical components of Systems I and III has allowed the development of heterologous expression systems for properly folded cytochromes c (Fig. 5). Early attempts to express holocytochromes c in non-native hosts met with mixed success (for a summary, see Pollock et al.94 and Keightley et al.73) Thus, studies of recombinant cytochrome c for many years were confined to functional mutants of Saccharomyces cerevisiae iso-1-cytochrome c which could be expressed in its native host.95 A major breakthrough in heterologous cytochrome c expression was the development of a system for expressing mitochondrial cytochromes c in E. coli. By co-expressing the cytochrome c structural gene and the yeast CCHL gene, Mauk and coworkers achieved the expression of properly folded and matured S. cerevisiae iso-1-cytochrome c in E. coli.94 Notably, the holoprotein was formed in the cytoplasm where the structural gene and lyase were expressed and heme is synthesized. This system has been applied to expressing related mitochondrial cytochromes c,96 but failed to achieve maturation of bacterial cytochromes c,86 reflecting the specificity of CCHL. An alternative method utilizes maturation System I by making use of Thöny-Meyer’s pEC86 plasmid containing the E. coli ccm cassette ccmABCDEFGH.97 Fee and coworkers incorporated a modified E. coli-compatible leader sequence from Thiobacillus versutus cytochrome c55098 to direct translocation of an apocytochrome c polypeptide to the periplasm for maturation;69 this system has been employed to express a range of properly folded bacterial and mitochondrial cytochromes c.99-102 Indeed, maturation of a broad range of cytochromes c has been achieved using System I including multiheme cytochromes c,103 a variant of cytochrome b562 with a c-type heme,104 tetraheme cytochrome c3 containing two CXXXXCH motifs along with two CXXCH motifs,105 a cytochrome c2 mutant with one “X” residue inserted into the motif (CXXXCH) by mutagenesis,106 and peptides designed with a CXXCH site for heme attachment to prepare custom microperoxidases (heme c-binding peptide fragments).107 System I also has been shown to be compatible with sequences containing an additional Cys within or adjacent to the heme attachment motif (CCXXCH, CCXCH, CXCCH, and CXXCHC), despite the fact that these sequences are not found in nature.108 The promiscuity of System I makes the pEC86 plasmid a versatile tool for preparing and studying a wide range of cytochromes c and heme c model compounds.
Figure 5.
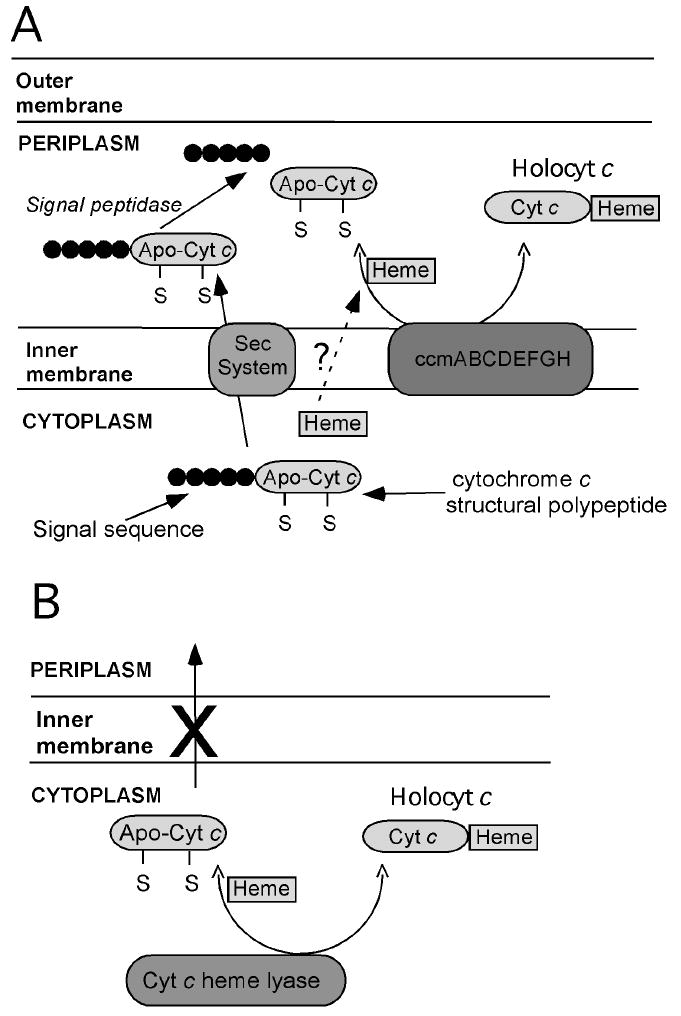
Schematic representation of systems for heterologous expression of cytochromes c in E. coli. (A) System based on E. coli Ccm machinery. A pre-apocytochrome c is expressed with an E. coli-compatible signal sequence directing translocation into the periplasm, where the Ccm apparatus assists with heme attachment. (B) System based on yeast CCHL. A cytochrome c structural gene is expressed along with yeast CCHL, which catalyzes heme attachment. As no signal sequence is used, heme attachment takes place in the cytoplasm.
5 Heme c covalent attachment chemistry
The stereospecific formation of thioether bonds by adding the Cys thiols to heme vinyl groups is chemically nontrivial. Nevertheless, a number of cases of spontaneous (not assisted by cytochrome c maturation factors) attachment of heme to Cys residues in a CXXCH motif (and related motifs) has been reported. The ability of this process to occur in the absence of maturation factors in selected cases raises questions about the roles of the maturation factors. At the same time, studies of cytochromes c undergoing anomalous maturation provide information relevant to understanding the roles of the maturation factors.
The first report of heme attachment without the assistance of maturation factors was for Hydrogenobacter thermophilus (Ht) cytochrome c552.109 It was found that upon expression of the cytochrome c552 structural gene in E. coli, holoprotein could be purified from the cytoplasm. The lack of an E. coli-compatible signal sequence resulted in the polypeptide remaining in the cytoplasm, whereas the maturation factors function on periplasmic apoprotein. Heme, however, is synthesized in the cytoplasm, and the apocytochrome and heme are able to form a productive complex allowing spontaneous thioether bond formation. NMR analysis of the polypeptide and heme chemical shifts of Ht holocytochrome c552 expressed and matured in the E. coli cytoplasm confirmed that the structure is indistinguishable from native protein once the retained N-terminal Met residue is cleaved.99 The hypothesis that heme and apoprotein spontaneously form a productive complex to drive thioether bond formation is supported by the observation that noncovalent heme binding with native-like tertiary structure is achieved in a Ht cytochrome c552 variant in which both Cys within the CXXCH motif are mutated to Ala.68, 110 In addition, single Cys-to-Ala mutants (AXXCH and CXXAH) were prepared and matured in the E. coli cytoplasm with single thioether attachments.111 This result demonstrates that disulfide bond formation in this motif is not necessary for unassisted thioether bond formation in the E. coli cytoplasm.
The ability to form properly folded holo-protein without maturation factors in good yield appears thus far to be unique to Ht cytochrome c552. In contrast, unassisted maturation of Thermus thermophilus (Tt) cytochrome c552 was demonstrated to yield a mixture of products.69, 73 Upon expressing the structural Tt cytochrome C552 gene in E. coli without a leader sequence so that it remains in the cytoplasm, three products were formed that were subjected to detailed structural and spectroscopic characterization. One is a monomeric product that resembles native Tt cytochrome C552, denoted Tt rC552.73 Despite its similarities to authentic holo-protein, detailed characterization of Tt rC552 reveals structural defects in the heme and a poorly defined tertiary structure.112 Nevertheless, Tt rC552 is amenable to crystallographic characterization and the 1.41-Å structure reveals an overall native-like fold for the protein, but an abnormal thioether linkage between Cys11 (the first in the CXXCH motif) and the heme 2-vinyl.113 The crystal structure, complemented by NMR, resonance Raman, and mass spectrometry is consistent with a – C(O)-CH2-S-CH2-Cys linkage between Cys 11 and heme (Fig. 6A) rather than the expected – CH(CH3)-S-CH2-Cys linkage. Gentle heating of Tt rC552 causes its conversion to a green product denoted p572, indicating its red-shifted α absorption band at 572 nm in the reduced state. The hyperfine-shifted resonances of the NMR spectrum also shift upon conversion to p572 to take on values that are unusual for a low-spin heme b or c, but consistent with a heme to which an electron-withdrawing group has been added.113 The crystal structure of p572 reveals a modified heme with electron density consistent with a formyl group coplanar with the porphyrin at position 2. The p572 product may be formed by base-assisted cleavage of the Cα-Cβ bond of the original 2-vinyl group, leaving a heme formyl (Fig. 6B).113 Finally, a third product, also with a red-shifted absorption spectrum, was denoted rC557. This product is an extremely stable dimer, and the crystal structure reveals that the heme is bound the cytochrome polypeptide in an incorrect orientation, rotated about the α,γ-meso axis. As a result, only one vinyl group is positioned to react with a Cys within each protomer; the other Cys forms an inter-protomer disulfide bond (Fig. 7).114
Figure 6.
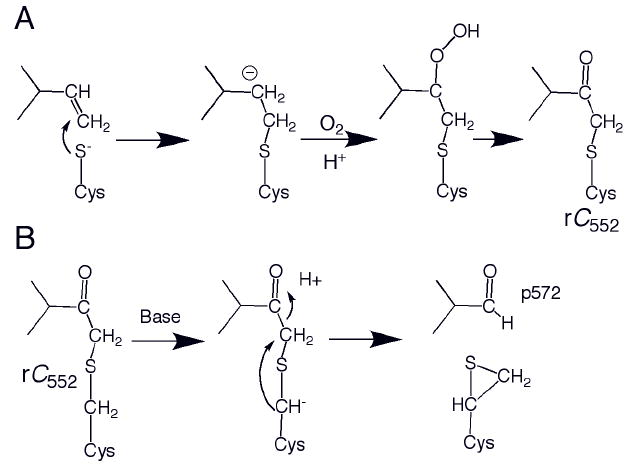
(A) Proposed nucleophilic mechanism for formation of Tt rC552. An analogous radial mechanism also is plausible.113 (B) Proposed mechanism for formation of p572 from Tt rC552. 113 The proposed thiirane product shown has not been characterized.
Figure 7.
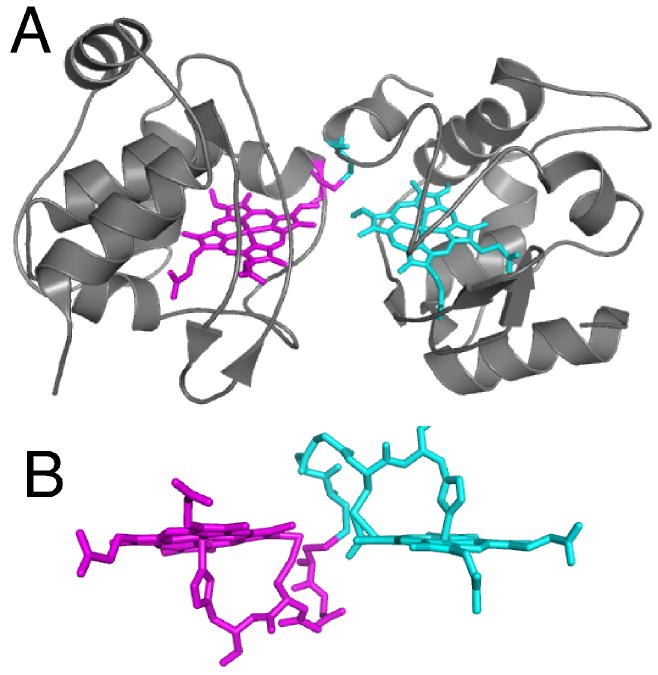
(A) Structure of T. thermophilus rC557 (PDB: 1FOC). Each heme is rotated about the α,γ-meso axis relative to the orientation in correctly matured cytochromes c, and each heme is attached via one thioether bond between 2-vinyl and Cys14. The Cys11 residues form an inter-protomer disulfide bond rather than binding to heme. (B) Detailed view of heme and residues 11-15 (CXXCH) in rC557. The atoms from each protomer are depicted in different colors. Note that the heme groups and CXXCH units from the two protomers are linked to each other via the interprotomer disulfide.
The analysis of the products of faulty Tt cytochrome c552 maturation provides some insight into incorrect pathways of heme attachment that are suppressed by the biological maturation process. The incorrect mode of linkage to the 2-vinyl Cβ seen in Tt rC552 represents an anti-Markovnikov addition of the Cys RSH to the vinyl group, which would suggest a free-radical mechanism. A nucleophilic mechanism also is possible, in particular if improper protein folding holds the Cys Sγ away from heme vinyl Cα; the carbanion on Cα could be stabilized through conjugation with the porphyrin π system.113 Both Markovnikov and anti-Markovnikov adducts have been observed in photochemical and in acid-catalyzed addition of thiol to a vinyl group of bilirubin, thus, it is not clear that one pathway is favored over another.115 These results indicate that the Ccm factors assist with directing attack of thiol at the Cα. Secondly, the rC557 product results from the heme adopting the incorrect rotational isomer. Rotational isomerism is routinely observed upon reconstitution of globins,116, 117 but not otherwise in cytochromes c. Thus, the Ccm factors apparently aid in stabilizing or selecting the proper heme rotational isomer prior to covalent attachment.
In addition to the studies of anomalous cytochrome c maturation in vivo, covalent heme attachment in vitro also has been demonstrated. Ht holocytochrome c552 formation was shown to occur by combining apoprotein and heme under reducing conditions, confirming that indeed the process can occur in the absence of biological maturation factors.118 Likewise, in vitro reactions of apocytochromes c from horse heart and from Paracoccus denitrificans with heme to form covalent thioether bonds have been observed, but a mixture of products results and the reaction is slow.119 As Ht holocytochrome c552 is particularly amenable to formation in vitro, it has proven to be a useful tool for understanding the chemistry of cytochrome c synthesis. One study considered metal dependence by examining the reaction of Ht apocytochrome c552 with Co(III), Zn(II), Mn(II), Mn(III), and metal-free derivatives of protoporphyrin IX. It was found that thioether bond formation occurred only for divalent M(II) derivatives,120 consistent with the finding that this reaction occurred for Fe(II) but not Fe(III) heme.118 A mechanistic basis for this observation was not given, but it is possible that the higher hydrophobicity of the divalent metalloporphyrin facilitates formation of a productive metalloporphyrin-apocytochrome complex. Similarly, in semisynthesis of horse cytochrome c, which requires complex formation between two peptides, one of which has bound heme, the iron must be reduced for productive complex formation.121 Other studies examined the dependence on the Cys residues. During maturation in vitro, heme was found to form a covalent bond and bind in the correct orientation in single Cys-Ala mutants (AXXCH or CXXAH) of Ht cytochrome c552,122 despite the fact that these single-Cys variants are not processed by the maturation apparatus of E. coli.91, 92 This result suggests that although disulfide bond formation is necessary in the biological maturation pathway, it is not necessary for the chemistry of thioether bond formation. The single Cys-to-Ala mutants formed in vitro were shown to bind heme in the correct orientation. Thus, the heme-polypeptide interaction in itself is sufficient for defining the correct heme orientation in Ht cytochrome c552. However, this was not the case for unassisted maturation of Tt cytochrome c552, suggesting that the maturation factors do assist in orienting heme and polypeptide.114 The ability of System I to mature a range of cytochromes c and even microperoxidases suggests that the necessary interactions that yield the correct orientation of heme relative to the CXXCH motif occurs right at the motif itself. In the case of System III, however, its specificity for particular mitochondrial cytochromes c suggests such recognition may involve regions of the polypeptide beyond the CXXCH motif.
6 Heme c conformation
The substitution of peripheral heme vinyl groups for thioethers in itself is not expected to significantly perturb heme electronic structure or reactivity. Nevertheless, analysis of heme protein reduction potentials reveals that heme c accesses a larger range than heme b, with heme c being found with substantially lower potentials when comparing groups with the same axial ligation.30, 31 This observation suggests that heme c displays greater “tunability” in terms of its reduction potential relative to heme b, particularly to lower potentials. While having thioether rather than vinyl substituents alone cannot account for this difference, consideration of the effects of the tightly bound CXXCH motif (Fig. 2) yields some clues. Indeed, the structure of the heme-CXXCH unit is defined primarily by local bonding, as demonstrated by the close similarity of the structure of this segment in the heme peptide microperoxidase-8 (containing the heme bound to the CXXCH and three additional residues) and in folded cytochrome c.123 The local stability of this segment suggests that the pentapeptide heme-binding segment may have an influence on the His-Fe bond (discussed in the next section) and on the conformation of the heme itself. These factors in turn are expected to influence the heme reduction potential.
Shelnutt and coworkers developed a normal-coordinate structural decomposition (NSD) method for analyzing the distortion of porphyrins from planarity. In this analysis, deviations from planarity are described as displacements along lowest-frequency normal coordinates and are described as saddling, ruffling, doming, waving, and propellering (Fig. 8).124 NSD analysis of a range of heme proteins reveals that proteins of a common functional class show strong similarities in heme conformation. Interestingly, hemes c are typically highly nonplanar, with ruffling usually being the primary mode of distortion.125 Resonance Raman studies of microperoxidase-8 reveal that the CXXCH pentapeptide is primarily responsible for inducing the heme ruffling,126 in agreement with molecular mechanics studies of the heme-CXXCH unit.127 The functional significance is that heme conformation plays a role in determining reduction potential; as the heme becomes more distorted, potential is lowered.128, 129
Figure 8.
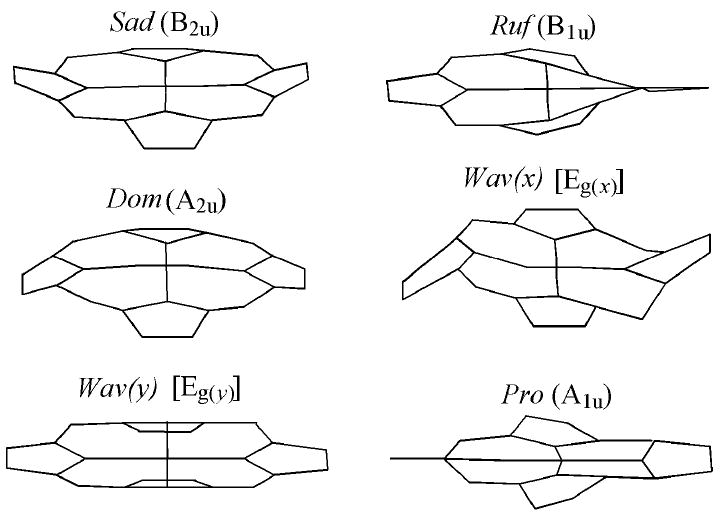
Out-of-plane modes representing nonplanar deformations of the porphyrin macrocycle, from work of Shelnutt and coworkers.124
Although the heme-attachment peptide is known to play a key role in determining conformation of heme c, the extent and type of distortion varies significantly among different cytochromes c, and even among different hemes within a multi-heme cytochrome c.19 For example, analysis of the varied conformations of the hemes in tetraheme cytochromes c3 reveals that within the protein the composition of the heme attachment peptide (which can be CXXCH or CXXXXCH) exerts a strong influence on the heme conformation, although other factors such as the distal ligand, interactions with heme propionates, and other interactions with heme also contribute.130, 131 In another example, mitochondrial cytochromes c display highly ruffled hemes, whereas the heme in cytochromes c2 is only slightly distorted from planarity (Fig. 9).19 Indeed, factors other than the fingerprint peptide itself influence heme conformation; even hemes b display high amounts of ruffling in the nitrophorins as a result of interactions between heme pocket residues and the porphyrin.132 Elucidating the role of heme distortion in determining reduction potential is complicated, and there is not a clear correlation between reduction potential and extent of heme distortion among cytochromes c. This lack of a clear correlation is not surprising as many factors contribute to determining reduction potential such that any one factor alone rarely shows a clear correlation with potential. Nevertheless, altering the conformation of hemes c via the attachment peptide properties does provide a means by which potential of hemes c can be tuned.
Figure 9.
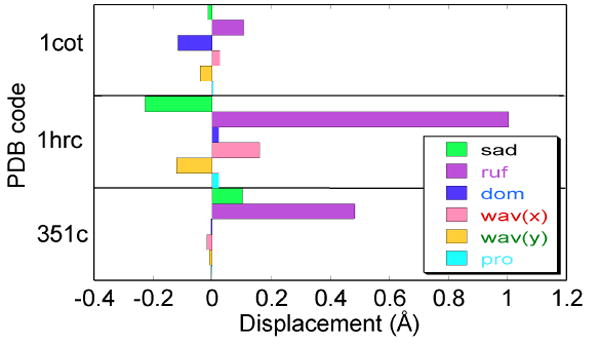
NSD results for horse heart cytochrome c (1HRC), P. aeruginosa cytochrome c551 (351C), and Paracoccus denitrificans cytochrome c2 (1COT). The primary mode of distortion of heme c usually is ruffling. However, the amount of ruffling varies substantially among cytochromes c, and the heme of cytochrome c2 is nearly planar.
The large variation seen in heme c conformation among cytochromes c is somewhat surprising given the evidence that its being attached to a CXXCH peptide plays a key role in determining heme conformation. Examination of the structures of this segment in a range of cytochromes c reveals that the backbone structure of the CXXCH motif shows little variation, even among proteins in which the variable “XX” residues have very different properties (charge and size; see Fig. 10). Despite the similar structures seen for different proteins, there is evidence that this peptide displays varying extents of hydrogen bonding within the motif depending on the identity of the “XX” residues and/or the peptide’s environment. Weaker hydrogen bonding within this motif has been proposed to allow the heme to relax into a more planar conformation.126, 127 In a study of microperoxidase-8, the extent of ruffling was shown to be greater in a hydrophobic environment; this was proposed to be a result of enhanced hydrogen bonding from the amide NH of Cys 17 and His18 to the backbone carbonyl of Cys14 tighten the pentapeptide structure and distort the heme.126 Variability in the stability of these hydrogen bonds also is observed in studies of full-length cytochromes c. This is seen in hydrogen-deuterium exchange rates of the backbone HN of the second Cys and the His in this motif. Given the strong conservation of the CXXCH backbone structure (Fig. 10), slow exchange is expected for the amide protons second Cys and the His in cytochromes c as they hydrogen bond with the first Cys carbonyl. Nevertheless, hydrogen-deuterium exchange rates of the second Cys and His of this motif are quite variable among cytochromes c.100, 133-138 The means by which such variations impact heme conformation remains to be examined in a systematic manner, but represents an intriguing direction of future study. Comparison between different protein species presents difficulties because of the multiple factors influencing heme conformation. Recently, point mutations that alter the hydrogen exchange rates within this motif have been reported,135 and analysis of heme conformation in these and related mutants may provide some insight into this interesting question.
Figure 10.
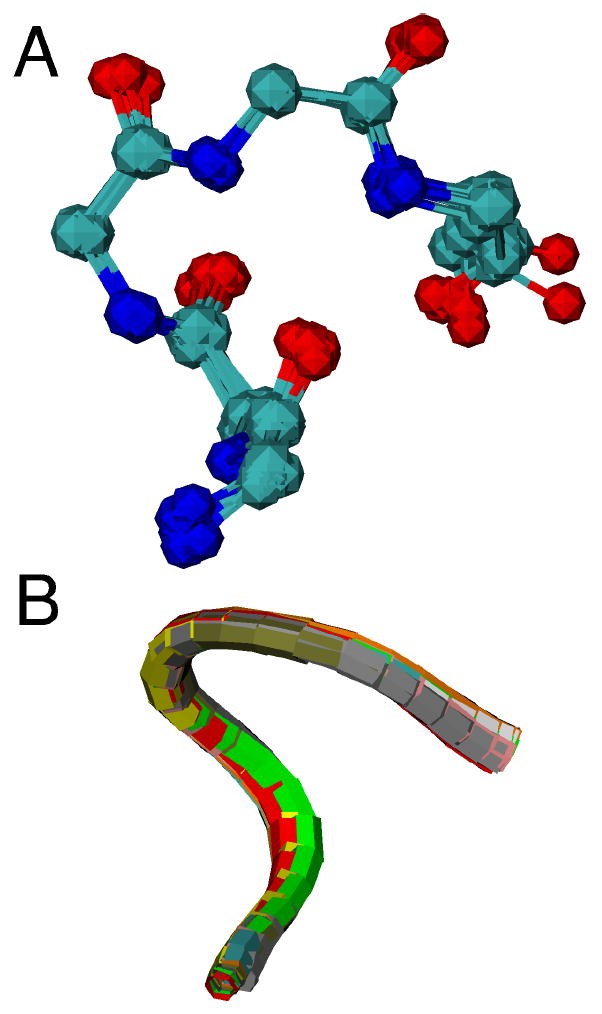
Overlay of CXXCH pentapeptide backbone for ten cytochromes c with diverse residues in the variable “XX” positions shown in (A) ball-and-stick and (B) ribbon format. Each protein is represented with a different color in (B). Proteins with crystal structures with 1.5-Å or better resolution were selected. The PDB accession codes and the identity of the variable “XX” resides in the structures are: 1B7V (IS), 1GU6 (AD), 1IQC (NS), 1YNR (MA), 1CTJ (AA), 2CTH (GD), 351C (VA), 1HRC (AQ), 1C2R (KT), 2YCC (LQ).
7 The His-Fe interaction in heme c
The nature of His-Fe coordination plays an important role in modulating reactivity of heme proteins.31, 139-144 In terms of electron transfer, the most common function of heme c, it is important to consider reduction potential and iron spin state which influence thermodynamics and kinetics of electron transfer. Factors that differentially impact the strength of the His-Fe interaction in the oxidized and reduced states of heme have corresponding effects on reduction potential, and factors that affect the ligand-field strength of the His influence iron spin state and reactivity. One method by which these factors can be tuned is by modulating the extent of histidine (neutral imidazole) versus histidinate (anionic imidazolate) character of the axial His side chain through the His and heme environment and the heme structure itself. A more anionic His favors a lower reduction potential by stabilizing the higher iron oxidation state, and also favors a low-spin heme. Intriguingly, a number of studies, discussed subsequently, suggest that the properties of the peptide segment attaching the heme c prosthetic group to polypeptide modulate the His binding affinity for ferric vs. ferrous iron and thus heme c reduction potential.
The effect of covalent attachment on His-Fe bonding has been examined using both model systems and cytochromes c. One type of model system used is synthetic peptide-sandwiched mesoheme (PSM), an effective mimic of proteins with covalently bound heme with bis-His axial coordination (Fig. 11A, B).145, 146 Tailed porphyrin covalent linkers are used to attach the porphyrin ring to a peptide unit, lowering the energy required for intramolecular His ligation to iron. Modifications of PSMs have been introduced that stabilize the His-Fe(III) state, and it is proposed that the diminished peptide mobility arising from the presence of covalent peptide-porphyrin bonding results in stabilization of His-Fe(III) coordination relative to His-Fe(II) coordination and therefore a negative shift in reduction potential.146
Figure 11.
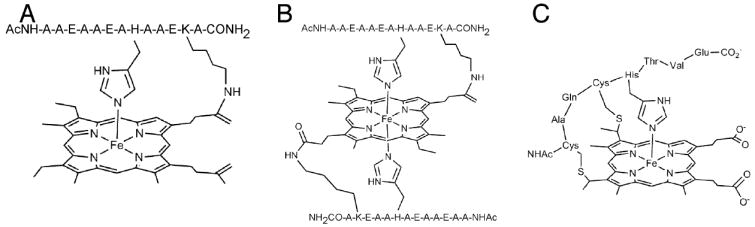
Examples of model systems utilized to study the effects of covalent attachment on the His-Fe interaction. (A, B) Peptide-sandwiched mesohemes (PSMs),145, 146 (C) Microperoxidase-8 (MP-8).
Another important class of models for studying metal-ligand interactions in heme c has been the microperoxidases.147, 148 A comparison of Fe(III) microperoxidase-8 (MP-8, Fig. 11C), which has two covalent peptide-heme linkages, with a synthetic heme peptide containing only one covalent linkage to the heme prosthetic group reveals greater thermodynamic stability of metal-ligand interactions in MP-8.149, 150 In addition, heme affinity for thioether ligands is increased as His-Fe(III) binding is enhanced; thioether interactions with ferric iron are inherently weak.151 The two heme-peptide covalent bonds provided by the CXXCH motif were demonstrated to enhance the His ligand field strength such that a ferric heme with His/Met axial ligation is low-spin; this results in stronger thioether bonding. In contrast, in a heme with only one such covalent bond, His/Met axial ligation does not yield a pure low-spin state for the ferric state.150 Notably, ferric cytochrome b562, which has His/Met axial ligation, also exists as a spin-state mixture.152 Maintaining a low-spin state in both the oxidized and reduced forms of the protein has implications for electron transfer function and heme reactivity more broadly.
Mutation of residues in and near the CXXCH motif in cytochromes c has been a valuable tool for assessing the effect of the heme-binding motif on the His-Fe interaction. In one study, the Cys14Ser mutant of S. cerevisiae iso-1-cytochrome c was prepared and a fraction of the ferric protein was found to be high-spin.153 Similarly, variants of Ht cytochrome c552 with one and no Cys linkages to heme have been prepared; the b-type variant displays a decrease in reduction potential of 75 mV, but no characterization of electronic structure has been reported.68 Mutants of two homologous bacterial cytochromes c, Pseudomonas aeruginosa cytochrome c551 and Ht cytochrome c552, have been examined to gain insight into how properties of amino acid residues in and near the heme binding motif influence the conformational flexibility of the heme attachment peptide as well as the His-Fe interaction.135 Specifically, mutating one of the variable “X” residues, at position 13, along with nearby residue 22, which lies across the CXXCH loop, was found to cause significant changes in the local flexibility of the CXXCH motif.135 Decreased conformational fluctuations of CXXCH were correlated with decreased reduction potential in these variants. In the cytochromes c from Pa and Ht, the HNδ1 of the axially ligated His participates in a hydrogen bonding interaction with the carbonyl oxygen of Pro25. 1H and 15N NMR studies revealed that this hydrogen bond is strengthened in the ferric but not the ferrous state of lower potential mutants, in support of the proposal that the properties of the CXXCH motif influence the anionic character of His.135
Although there is compelling evidence that covalent heme attachment influences the His-Fe interaction, the number of systematic studies of such effects is limited. One challenge is the difficulty of preparing appropriate model complexes; the report that a range of microperoxidases can be prepared by expression may provide a route to more systematic studies of this effect.107 Another challenge comes with characterizing subtle effects on the His-Fe bond within a complex molecule such as a protein. Further application of methods such as NMR of paramagnetic species154 and nuclear resonance vibrational spectroscopy155 is expected to aid in these studies in the future.
8 Functional bases for heme c attachment
In the literature there are a number of discussions that address the question of why covalent attachment is important for the function of cytochromes c. Reasons put forth include enhancing protein stability, stabilizing a particular orientation of partially solvent-exposed heme, minimizing the residue-to-heme ratio, and facilitating the assembly and folding of multi-heme proteins and/or structures with closely packed hemes.4-6, 66 In this review, we discuss the question of the functional significance of heme c from the bioinorganic chemistry perspective, that is, we consider the influence of heme covalent attachment on the properties of the heme itself and on iron-ligand interactions. In addition, we remark on the specific importance of covalent heme attachment in mitochondrial cytochromes c.
The discovery that release of cytochrome c from mitochondria is an important apoptosis trigger revealed a previously undiscovered function of cytochrome c in cell death.42 Notably, apocytochrome c does not display pro-apoptotic activity.156 Subsequently, the pro-apoptotic activity of cytochrome c has been linked to a conformational change involving the heme crevice45 and to heme redox state.43, 44 A biophysical study of mitochondrial cytochrome c binding to apoptotic protease activating factor 1 (APAF-1) revealed an extremely high affinity interaction, such that presence of only a few molecules of holocytochrome c in the cytoplasm is sufficient to induce apoptosis.157 Thus, the presence of any holocytochrome c in the cytoplasm in the absence of pro-apoptotic stimuli needs to be prevented, and the assembly of holocytochrome c within the mitochondrial intermembrane space assures that holocytochrome c is not present in the cytoplasm upon synthesis. In fact, cytoplasmic apocytochrome c blocks Bax-induced apoptosis in cells.158 A specialized basis for covalent attachment of heme to mitochondrial cytochrome c thus is assuring that newly synthesized holocytochrome c is not in the cytoplasm. Notably, many proteins in the intermembrane space are small, with folding stabilized by binding cofactors or forming disulfide bridges. Such “folding-traps” have been proposed more generally to ensure unidirectional transport of proteins into the intermembrane space.159
Looking more broadly beyond the special role of mitochondrial cytochrome c in apoptosis, there are a number of ways in which heme c covalent attachment influences heme reactivity; these have been addressed in sections above but will be summarized here. One is the finding that heme c attachment enhances the bonding of the His within the CXXCH motif with iron. A result is an increase in the ligand-field strength such that hemes c with axial His/Met ligation are typically purely low-spin in both oxidation states. A further effect is enhancing distal Met-iron bonding, an interaction particularly weak in the ferric state. The greater ligand field also prevents redox-linked changes in spin state which are expected to increase reorganization energy associated with electron transfer.
The enhancement of ligand field strength by heme covalent attachment also has implications for functionally important conformational changes. As has been noted previously,5 there is a number of examples of heme c-containing proteins that undergo conformational changes linked with changes in heme redox state. Examples include cytochrome cd1 nitrite reductase, in which the heme c displays redox state-dependent ligation proposed to control electron transfer rates,49 and DcrA, a c-heme chemotaxis signal transducer that has two endogenous axial ligands in the reduced state, but has an open coordination site when oxidized.54 In addition, ten c-heme-containing sensor proteins that undergo an internal redox-driven change in heme coordination have been identified in G. sulfurreducens.53 Covalent heme attachment has two possible roles in these proteins. One is enhancement of metal-ligand bonding will promote rearrangements of the protein fold to be driven by or coupled to binding of the distal ligand to heme, in particular when that ligand is Met. Secondly, covalent attachment of heme is expected to make the protein structure more robust in multiple conformations, and to prevent dissociation of heme in the course of large conformational changes. However, it is important to note that redox-linked changes in structure and heme ligation are not limited to heme c-containing systems.
Another major influence covalent attachment has on heme is to alter its conformation. Hemes c tend to have a ruffled conformation, and heme ruffling lowers reduction potential. Not all hemes c are highly ruffled, and this variation points to heme c conformation being highly tunable based on the properties of the attachment peptide and other heme pocket residues. Changing heme conformation changes heme electronic structure and reactivity, and affects other functions such as ligand binding.160 Our understanding of the effects of heme conformation on protein function is at an early stage, although analyses in particular by NMR of paramagnetic heme proteins have been informative.132, 161 In addition, femtosecond correlation spectroscopy, which allows observation of low-frequency modes associated with heme distortions from planarity, is a recently developed tool that will be valuable in studies of heme conformation.162 In summary, analyses of the influence of heme covalent attachment on heme conformation and ligand interactions provide compelling evidence that enhancement of ruffling and of the His-Fe(III) interaction, both of which lower reduction potential, may account for the greater range of reduction potentials seen for hemes c compared to hemes b.
Acknowledgments
The authors acknowledge support from NIH grant GM63170 and an Elon Huntington Hooker Graduate Fellowship to S. E. J. B.
References
- 1.Chapman SK, Daff S, Munro AW. Metal Sites in Proteins and Models. 1997:39–70. [Google Scholar]
- 2.Schneider S, Marles-Wright J, Sharp KH, Paoli M. Nat Prod Rep. 2007;24:621–630. doi: 10.1039/b604186h. [DOI] [PubMed] [Google Scholar]
- 3.Paoli M, Marles-Wright J, Smith A. DNA Cell Biol. 2002;21:271–280. doi: 10.1089/104454902753759690. [DOI] [PubMed] [Google Scholar]
- 4.Stevens JM, Daltrop O, Allen JWA, Ferguson SJ. Acc Chem Res. 2004;37:999–1007. doi: 10.1021/ar030266l. [DOI] [PubMed] [Google Scholar]
- 5.Barker PD, Ferguson SJ. Structure. 1999;7:R281–R290. doi: 10.1016/s0969-2126(00)88334-3. [DOI] [PubMed] [Google Scholar]
- 6.Allen JWA, Daltrop O, Stevens JM, Ferguson SJ. Philos Trans R Soc Lond Ser B-Biol Sci. 2003;358:255–266. doi: 10.1098/rstb.2002.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Page MD, Sambongi Y, Ferguson SJ. Trends Biochem Sci. 1998;23:103–108. doi: 10.1016/s0968-0004(98)01173-6. [DOI] [PubMed] [Google Scholar]
- 8.Kranz R, Lill R, Goldman B, Bonnard G, Merchant S. Mol Microbiol. 1998;29:383–396. doi: 10.1046/j.1365-2958.1998.00869.x. [DOI] [PubMed] [Google Scholar]
- 9.Allen JWA, Jackson AP, Rigden DJ, Willis AC, Ferguson SJ, Ginger ML. Febs J. 2008;275:2385–2402. doi: 10.1111/j.1742-4658.2008.06380.x. [DOI] [PubMed] [Google Scholar]
- 10.Giegé P, Grienenberger JM, Bonnard G. Mitochondrion. 2008;8:61–73. doi: 10.1016/j.mito.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 11.Bertini I, Cavallaro G, Rosato A. Chem Rev. 2006;106:90–115. doi: 10.1021/cr050241v. [DOI] [PubMed] [Google Scholar]
- 12.Mowat CG, Chapman SK. Dalton Trans. 2005:3381–3389. doi: 10.1039/b505184c. [DOI] [PubMed] [Google Scholar]
- 13.Stroebel D, Choquet Y, Popot JL, Picot D. Nature. 2003;426:413–418. doi: 10.1038/nature02155. [DOI] [PubMed] [Google Scholar]
- 14.Kurisu G, Zhang HM, Smith JL, Cramer WA. Science. 2003;302:1009–1014. doi: 10.1126/science.1090165. [DOI] [PubMed] [Google Scholar]
- 15.de Vitry C, Desbois A, Redeker V, Zito F, Wollman FA. Biochemistry. 2004;43:3956–3968. doi: 10.1021/bi036093p. [DOI] [PubMed] [Google Scholar]
- 16.Ikegami I, Katoh S, Takamiya A. Biochim Biophys Acta. 1968;162:604–606. doi: 10.1016/0005-2728(68)90066-2. [DOI] [PubMed] [Google Scholar]
- 17.Pettigrew GW, Leaver JL, Meyer TE, Ryle AP. Biochem J. 1975;147:291–302. doi: 10.1042/bj1470291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Einsle O, Messerschmidt A, Stach P, Bourenkov GP, Bartunik HD, Huber R, Kroneck PMH. Nature. 1999;400:476–480. doi: 10.1038/22802. [DOI] [PubMed] [Google Scholar]
- 19.Jentzen W, Ma JG, Shelnutt JA. Biophys J. 1998;74:753–763. doi: 10.1016/S0006-3495(98)74000-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aragao D, Frazao C, Sieker L, Sheldrick GM, LeGall J, Carrondo MA. Acta Crystallogr Sect D-Biol Crystallogr. 2003;59:644–653. doi: 10.1107/s090744490300194x. [DOI] [PubMed] [Google Scholar]
- 21.Jüngst A, Wakabayashi S, Matsubara H, Zumft WG. FEBS Lett. 1991;279:205–209. doi: 10.1016/0014-5793(91)80150-2. [DOI] [PubMed] [Google Scholar]
- 22.Hartshorne RS, Kern M, Meyer B, Clarke TA, Karas M, Richardson DJ, Simon J. Mol Microbiol. 2007;64:1049–1060. doi: 10.1111/j.1365-2958.2007.05712.x. [DOI] [PubMed] [Google Scholar]
- 23.Ambler RP. Biochim Biophys Acta. 1991;1058:42–47. doi: 10.1016/s0005-2728(05)80266-x. [DOI] [PubMed] [Google Scholar]
- 24.Meyer TE. In: Cytochrome c: A Multidisciplinary Approach. Scott RA, Mauk AG, editors. University Science Books; Sausalito, CA: 1996. pp. 33–99. [Google Scholar]
- 25.Moore GR, Pettigrew GW. Cytochormes c; Evolutionary, Structural, and Physicochemical Aspects. Springer-Verlag; Berlin: 1990. [Google Scholar]
- 26.Scott RA, Mauk AG. Cytochrome c: A Multidisciplinary Approach. University Science Books; Sausalito, CA: 1996. [Google Scholar]
- 27.Pettigrew GW, Moore GR. Cytochromes c: Biological Aspects. Springer-Verlag; Berlin: 1987. [Google Scholar]
- 28.Marcus RA, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 29.Raphael AL, Gray HB. J Am Chem Soc. 1991;113:1038–1040. [Google Scholar]
- 30.Reedy CJ, Elvekrog MM, Gibney BR. Nucleic Acids Res. 2008;36:D307–D313. doi: 10.1093/nar/gkm814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker FA. Chem Rev. 2004;104:589–615. doi: 10.1021/cr020634j. [DOI] [PubMed] [Google Scholar]
- 32.Butler JE, Kaufmann F, Coppi MV, Nunez C, Lovley DR. J Bacteriol. 2004;186:4042–4045. doi: 10.1128/JB.186.12.4042-4045.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi L, Squier TC, Zachara JM, Fredrickson JK. Mol Microbiol. 2007;65:12–20. doi: 10.1111/j.1365-2958.2007.05783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lovley DR, Holmes DE, Nevin KP. Advances in Microbial Physiology. 2004;49:219–286. doi: 10.1016/S0065-2911(04)49005-5. [DOI] [PubMed] [Google Scholar]
- 35.Marshall MJ, Beliaev AS, Dohnalkova AC, Kennedy DW, Shi L, Wang ZM, Boyanov MI, Lai B, Kemner KM, McLean JS, Reed SB, Culley DE, Bailey VL, Simonson CJ, Saffarini DA, Romine MF, Zachara JM, Fredrickson JK. PLoS Biol. 2006;4:1324–1333. doi: 10.1371/journal.pbio.0040268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Assfalg M, Bertini I, Bruschi M, Michel C, Turano P. Proc Natl Acad Sci U S A. 2002;99:9750–9754. doi: 10.1073/pnas.152290999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Methe BA, Nelson KE, Eisen JA, Paulsen IT, Nelson W, Heidelberg JF, Wu D, Wu M, Ward N, Beanan MJ, Dodson RJ, Madupu R, Brinkac LM, Daugherty SC, DeBoy RT, Durkin AS, Gwinn M, Kolonay JF, Sullivan SA, Haft DH, Selengut J, Davidsen TM, Zafar N, White O, Tran B, Romero C, Forberger HA, Weidman J, Khouri H, Feldblyum TV, Utterback TR, Van Aken SE, Lovley DR, Fraser CM. Science. 2003;302:1967–1969. doi: 10.1126/science.1088727. [DOI] [PubMed] [Google Scholar]
- 38.Heidelberg JF, Paulsen IT, Nelson KE, Gaidos EJ, Nelson WC, Read TD, Eisen JA, Seshadri R, Ward N, Methe B, Clayton RA, Meyer T, Tsapin A, Scott J, Beanan M, Brinkac L, Daugherty S, DeBoy RT, Dodson RJ, Durkin AS, Haft DH, Kolonay JF, Madupu R, Peterson JD, Umayam LA, White O, Wolf AM, Vamathevan J, Weidman J, Impraim M, Lee K, Berry K, Lee C, Mueller J, Khouri H, Gill J, Utterback TR, McDonald LA, Feldblyum TV, Smith HO, Venter JC, Nealson KH, Fraser CM. Nat Biotechnol. 2002;20:1118–1123. doi: 10.1038/nbt749. [DOI] [PubMed] [Google Scholar]
- 39.Leys D, Scrutton NS. Curr Opin Struct Biol. 2004;14:642–647. doi: 10.1016/j.sbi.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 40.Gralnick JA, Newman DK. Mol Microbiol. 2007;65:1–11. doi: 10.1111/j.1365-2958.2007.05778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mehta T, Coppi MV, Childers SE, Lovley DR. Appl Environ Microbiol. 2005;71:8634–8641. doi: 10.1128/AEM.71.12.8634-8641.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu XS, Kim CN, Yang J, Jemmerson R, Wang XD. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 43.Pan ZH, Voehringer DW, Meyn RE. Cell Death Differ. 1999;6:683–688. doi: 10.1038/sj.cdd.4400544. [DOI] [PubMed] [Google Scholar]
- 44.Brown GC, Borutaite V. Biochim Biophys Acta. 2008;1777:877–881. doi: 10.1016/j.bbabio.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 45.Jemmerson R, Liu J, Hausauer D, Lam KP, Mondino A, Nelson RD. Biochemistry. 1999;38:3599–3609. doi: 10.1021/bi9809268. [DOI] [PubMed] [Google Scholar]
- 46.Zhao YG, Wang ZB, Xu JX. J Biol Chem. 2003;278:2356–2360. doi: 10.1074/jbc.M209681200. [DOI] [PubMed] [Google Scholar]
- 47.Takayama Y, Kobayashi Y, Yahata N, Saitoh T, Hori H, Ikegami T, Akutsu H. Biochemistry. 2006;45:3163–3169. doi: 10.1021/bi051867i. [DOI] [PubMed] [Google Scholar]
- 48.Hoy JA, Robinson H, Trent JT, Kakar S, Smagghe BJ, Hargrove MS. J Mol Biol. 2007;371:168–179. doi: 10.1016/j.jmb.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 49.Farver O, Kroneck PMH, Zumft WG, Pecht I. Proc Natl Acad Sci U S A. 2003;100:7622–7625. doi: 10.1073/pnas.0932693100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moir JWB. Biochim Biophys Acta-Protein Struct Molec Enzym. 1999;1430:65–72. doi: 10.1016/s0167-4838(98)00276-3. [DOI] [PubMed] [Google Scholar]
- 51.Cross R, Aish J, Paston SJ, Poole RK, Moir JWB. J Bacteriol. 2000;182:1442–1447. doi: 10.1128/jb.182.5.1442-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klarskov K, Van Driessche G, Backers K, Dumortier C, Meyer TE, Tollin G, Cusanovich MA, Van Beeumen JJ. Biochemistry. 1998;37:5995–6002. doi: 10.1021/bi972498w. [DOI] [PubMed] [Google Scholar]
- 53.Pokkuluri PR, Pessanha M, Londer YY, Wood SJ, Duke NEC, Wilton R, Catarino T, Saigueiro CA, Schiffer M. J Mol Biol. 2008;377:1498–1517. doi: 10.1016/j.jmb.2008.01.087. [DOI] [PubMed] [Google Scholar]
- 54.Yoshioka S, Kobayashi K, Yoshimura H, Uchida T, Kitagawa T, Aono S. Biochemistry. 2005;44:15406–15413. doi: 10.1021/bi0513352. [DOI] [PubMed] [Google Scholar]
- 55.Upadhyay AK, Hooper AB, Hendrich MP. J Am Chem Soc. 2006;128:4330–4337. doi: 10.1021/ja055183+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimizu H, Schuller DJ, Lanzilotta WN, Sundaramoorthy M, Arciero DM, Hooper AB, Poulos TL. Biochemistry. 2001;40:13483–13490. doi: 10.1021/bi011481h. [DOI] [PubMed] [Google Scholar]
- 57.Arciero DM, Hooper AB. J Biol Chem. 1994;269:11878–11886. [PubMed] [Google Scholar]
- 58.Wang YT, Graichen ME, Liu AM, Pearson AR, Wilmot CM, Davidson VL. Biochemistry. 2003;42:7318–7325. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 59.Smulevich G, Jakopitsch C, Droghetti E, Obinger C. J Inorg Biochem. 2006;100:568–585. doi: 10.1016/j.jinorgbio.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 60.Arciero DM, Hooper AB. FEBS Lett. 1997;410:457–460. doi: 10.1016/s0014-5793(97)00635-2. [DOI] [PubMed] [Google Scholar]
- 61.Igarashi N, Moriyama H, Fujiwara T, Fukumori Y, Tanaka N. Nat Struct Biol. 1997;4:276–284. doi: 10.1038/nsb0497-276. [DOI] [PubMed] [Google Scholar]
- 62.Friedrich CG, Quentmeier A, Bardischewsky F, Rother D, Kraft R, Kostka S, Prinz H. J Bacteriol. 2000;182:4677–4687. doi: 10.1128/jb.182.17.4677-4687.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheesman MR, Little PJ, Berks BC. Biochemistry. 2001;40:10562–10569. doi: 10.1021/bi0100081. [DOI] [PubMed] [Google Scholar]
- 64.Braaz R, Fischer P, Jendrossek D. Appl Environ Microbiol. 2004;70:7388–7395. doi: 10.1128/AEM.70.12.7388-7395.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Braaz R, Armbruster W, Jendrossek D. Appl Environ Microbiol. 2005;71:2473–2478. doi: 10.1128/AEM.71.5.2473-2478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allen JWA, Barker PD, Daltrop O, Stevens JM, Tomlinson EJ, Sinha N, Sambongi Y, Ferguson SJ. Dalton Trans. 2005:3410–3418. doi: 10.1039/b508139b. [DOI] [PubMed] [Google Scholar]
- 67.Dumont ME, Corin AF, Campbell GA. Biochemistry. 1994;33:7368–7378. doi: 10.1021/bi00189a043. [DOI] [PubMed] [Google Scholar]
- 68.Tomlinson EJ, Ferguson SJ. Proc Natl Acad Sci U S A. 2000;97:5156–5160. doi: 10.1073/pnas.090089397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fee JA, Chen Y, Todaro TR, Bren KL, Patel KM, Hill MG, Gomez-Moran E, Loehr TM, Ai JY, Thöny-Meyer L, Williams PA, Stura E, Sridhar V, McRee DE. Protein Sci. 2000;9:2074–2084. doi: 10.1110/ps.9.11.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thöny-Meyer L. Microbiol Mol Biol Rev. 1997;61:337–&. doi: 10.1128/mmbr.61.3.337-376.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thöny-Meyer L. Biochim Biophys Acta. 2000;1459:316–324. doi: 10.1016/s0005-2728(00)00167-5. [DOI] [PubMed] [Google Scholar]
- 72.Thöny-Meyer L, Kunzler P. Eur J Biochem. 1997;246:794–799. doi: 10.1111/j.1432-1033.1997.t01-1-00794.x. [DOI] [PubMed] [Google Scholar]
- 73.Keightley JA, Sanders D, Todaro TR, Pastuszyn A, Fee JA. J Biol Chem. 1998;273:12006–12016. doi: 10.1074/jbc.273.20.12006. [DOI] [PubMed] [Google Scholar]
- 74.Schulz H, Fabianek RA, Pellicioli EC, Hennecke H, Thöny-Meyer L. Proc Natl Acad Sci U S A. 1999;96:6462–6467. doi: 10.1073/pnas.96.11.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schulz H, Hennecke H, Thöny-Meyer L. Science. 1998;281:1197–1200. doi: 10.1126/science.281.5380.1197. [DOI] [PubMed] [Google Scholar]
- 76.Christensen O, Harvat EM, Thöny-Meyer L, Ferguson SJ, Stevens JM. Febs J. 2007;274:2322–2332. doi: 10.1111/j.1742-4658.2007.05769.x. [DOI] [PubMed] [Google Scholar]
- 77.Uchida T, Stevens JM, Daltrop O, Harvat EM, Hong L, Ferguson SJ, Kitagawa T. J Biol Chem. 2004;279:51981–51988. doi: 10.1074/jbc.M408963200. [DOI] [PubMed] [Google Scholar]
- 78.Garcia-Rubio I, Braun M, Gromov I, Thony-Meyer L, Schweiger A. Biophys J. 2007;92:1361–1373. doi: 10.1529/biophysj.106.098277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Richard-Fogal CL, Frawley ER, Kranz RG. J Bacteriol. 2008;190:3489–3493. doi: 10.1128/JB.00146-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ren Q, Ahuja U, Thöny-Meyer L. J Biol Chem. 2002;277:7657–7663. doi: 10.1074/jbc.M110979200. [DOI] [PubMed] [Google Scholar]
- 81.Edeling MA, Guddat LW, Fabianek RA, Thöny-Meyer L, Martin JL. Structure. 2002;10:973–979. doi: 10.1016/s0969-2126(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 82.Feissner RE, Richard-Fogal CL, Frawley ER, Loughman JA, Earley KW, Kranz RG. Mol Microbiol. 2006;60:563–577. doi: 10.1111/j.1365-2958.2006.05132.x. [DOI] [PubMed] [Google Scholar]
- 83.Goldman BS, Beck DL, Monika EM, Kranz RG. Proc Natl Acad Sci U S A. 1998;95:5003–5008. doi: 10.1073/pnas.95.9.5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Steiner H, Kispal G, Zollner A, Haid A, Neupert W, Lill R. J Biol Chem. 1996;271:32605–32611. doi: 10.1074/jbc.271.51.32605. [DOI] [PubMed] [Google Scholar]
- 85.Bernard DG, Quevillon-Cheruel S, Merchant S, Guiard B, Hamel PP. J Biol Chem. 2005;280:39852–39859. doi: 10.1074/jbc.M508574200. [DOI] [PubMed] [Google Scholar]
- 86.Sanders C, Lill H. Biochim Biophys Acta. 2000;1459:131–138. doi: 10.1016/s0005-2728(00)00122-5. [DOI] [PubMed] [Google Scholar]
- 87.Allen JWA, Harvat EM, Stevens JM, Ferguson SJ. FEBS Lett. 2006;580:4827–4834. doi: 10.1016/j.febslet.2006.07.073. [DOI] [PubMed] [Google Scholar]
- 88.Allen JWA, Leach N, Ferguson SJ. Biochem J. 2005;389:587–592. doi: 10.1042/BJ20041894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuras R, Saint-Marcoux D, Wollman FA, de Vitry C. Proc Natl Acad Sci U S A. 2007;104:9906–9910. doi: 10.1073/pnas.0702340104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eaves DJ, Grove J, Staudenmann W, James P, Poole RK, White SA, Griffiths I, Cole JA. Mol Microbiol. 1998;28:205–216. doi: 10.1046/j.1365-2958.1998.00792.x. [DOI] [PubMed] [Google Scholar]
- 91.Allen JWA, Ginger ML, Ferguson SJ. Biochem J. 2004;383:537–542. doi: 10.1042/BJ20040832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Allen JWA, Tomlinson EJ, Hong L, Ferguson SJ. J Biol Chem. 2002;277:33559–33563. doi: 10.1074/jbc.M204963200. [DOI] [PubMed] [Google Scholar]
- 93.Simon J, Eichler R, Pisa R, Biel S, Gross R. FEBS Lett. 2002;522:83–87. doi: 10.1016/s0014-5793(02)02885-5. [DOI] [PubMed] [Google Scholar]
- 94.Pollock WBR, Rosell FI, Twitchett MB, Dumont ME, Mauk AG. Biochemistry. 1998;37:6124–6131. doi: 10.1021/bi972188d. [DOI] [PubMed] [Google Scholar]
- 95.Pielak GJ, Mauk AG, Smith M. Nature. 1985;313:152–154. doi: 10.1038/313152a0. [DOI] [PubMed] [Google Scholar]
- 96.Patel CN, Lind MC, Pielak GJ. Protein Expr Purif. 2001;22:220–224. doi: 10.1006/prep.2001.1438. [DOI] [PubMed] [Google Scholar]
- 97.Arslan E, Schulz H, Zufferey R, Kunzler P, Thöny-Meyer L. Biochem Biophys Res Commun. 1998;251:744–747. doi: 10.1006/bbrc.1998.9549. [DOI] [PubMed] [Google Scholar]
- 98.Ubbink M, Vanbeeumen J, Canters GW. J Bacteriol. 1992;174:3707–3714. doi: 10.1128/jb.174.11.3707-3714.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karan EF, Russell BS, Bren KL. J Biol Inorg Chem. 2002;7:260–272. doi: 10.1007/s007750100292. [DOI] [PubMed] [Google Scholar]
- 100.Russell BS, Zhong L, Bigotti MG, Cutruzzolà F, Bren KL. J Biol Inorg Chem. 2003;8:156–166. doi: 10.1007/s00775-002-0401-z. [DOI] [PubMed] [Google Scholar]
- 101.Bren KL, Kellogg JA, Kaur R, Wen X. Inorg Chem. 2004;43:7934–7944. doi: 10.1021/ic048925t. [DOI] [PubMed] [Google Scholar]
- 102.Kellogg JA, Bren KL. Biochim Biophys Acta. 2002;1601:215–221. doi: 10.1016/s1570-9639(02)00471-5. [DOI] [PubMed] [Google Scholar]
- 103.Londer YY, Pokkuluri PR, Erickson J, Orshonsky Y, Schiffer M. Protein Expr Purif. 2005;39:254–260. doi: 10.1016/j.pep.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 104.Allen JWA, Barker PD, Ferguson SJ. J Biol Chem. 2003;278:52075–52083. doi: 10.1074/jbc.M307196200. [DOI] [PubMed] [Google Scholar]
- 105.Herbaud ML, Aubert C, Durand MC, Guerlesquin F, Thony-Meyer L, Dolla A. Biochim Biophys Acta-Protein Struct Molec Enzym. 2000;1481:18–24. doi: 10.1016/s0167-4838(00)00117-5. [DOI] [PubMed] [Google Scholar]
- 106.Ríos-Velázquez C, Cox RL, Donohue TJ. Arch Biochem Biophys. 2001;389:234–244. doi: 10.1006/abbi.2001.2330. [DOI] [PubMed] [Google Scholar]
- 107.Braun M, Thöny-Meyer L. Proc Natl Acad Sci U S A. 2004;101:12830–12835. doi: 10.1073/pnas.0402435101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Allen JWA, Ferguson SJ. Biochem Soc Trans. 2006;34:91–93. doi: 10.1042/BST0340091. [DOI] [PubMed] [Google Scholar]
- 109.Sanbongi Y, Yang JH, Igarashi Y, Kodama T. Eur J Biochem. 1991;198:7–12. doi: 10.1111/j.1432-1033.1991.tb15979.x. [DOI] [PubMed] [Google Scholar]
- 110.Wain R, Redfield C, Ferguson SJ, Smith LJ. J Biol Chem. 2004;279:15177–15182. doi: 10.1074/jbc.M311869200. [DOI] [PubMed] [Google Scholar]
- 111.Tomlinson EJ, Ferguson SJ. J Biol Chem. 2000;275:32530–32534. doi: 10.1074/jbc.M004022200. [DOI] [PubMed] [Google Scholar]
- 112.Fee JA, Chen Y, Todaro TR, Bren KL, Patel KM, Hill MG, Gomez-Moran E, Loehr TM, Ai JY, Thony-Meyer L, Williams PA, Stura E, Sridhar V, McRee DE. Protein Sci. 2000;9:2074–2084. doi: 10.1110/ps.9.11.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fee JA, Todaro TR, Luna E, Sanders D, Hunsicker-Wang LM, Patel KM, Bren KL, Gomez-Moran E, Hill MG, Ai JY, Loehr TM, Oertling WA, Williams PA, Stout CD, McRee D, Pastuszyn A. Biochemistry. 2004;43:12162–12176. doi: 10.1021/bi048968l. [DOI] [PubMed] [Google Scholar]
- 114.McRee DE, Williams PA, Sridhar V, Pastuszyn A, Bren KL, Patel KM, Chen Y, Todaro TR, Sanders D, Luna E, Fee JA. J Biol Chem. 2001;276:6537–6544. doi: 10.1074/jbc.M008421200. [DOI] [PubMed] [Google Scholar]
- 115.Lightner DA, McDonagh AF, Wijekoon WMD, Reisinger M. Tetrahedron Lett. 1988;29:3507–3510. [Google Scholar]
- 116.La Mar GN, Budd DL, Viscio DB, Smith KM, Langry KC. Proc Natl Acad Sci U S A. 1978;75:5755–5759. doi: 10.1073/pnas.75.12.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.La Mar GN, Satterlee JD, de Ropp JS. In: The Porphyrin Handbook. Kadish KM, Smith KM, Ruilard R, editors. Academic Press; New York: 2000. pp. 185–298. [Google Scholar]
- 118.Daltrop O, Allen JWA, Willis AC, Ferguson SJ. Proc Natl Acad Sci U S A. 2002;99:7872–7876. doi: 10.1073/pnas.132259099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Daltrop O, Ferguson SJ. J Biol Chem. 2003;278:4404–4409. doi: 10.1074/jbc.M211124200. [DOI] [PubMed] [Google Scholar]
- 120.Daltrop O, Ferguson SJ. J Biol Chem. 2004;279:45347–45353. doi: 10.1074/jbc.M408637200. [DOI] [PubMed] [Google Scholar]
- 121.Corradin G, Harbury HA. Biochem Biophys Res Commun. 1974;61:1400–1406. doi: 10.1016/s0006-291x(74)80439-0. [DOI] [PubMed] [Google Scholar]
- 122.Daltrop O, Smith KM, Ferguson SJ. J Biol Chem. 2003;278:24308–24313. doi: 10.1074/jbc.M301967200. [DOI] [PubMed] [Google Scholar]
- 123.Low DW, Gray HB, Duus JØ. J Am Chem Soc. 1997;119:1–5. [Google Scholar]
- 124.Jentzen W, Song XZ, Shelnutt JA. J Phys Chem B. 1997;101:1684–1699. [Google Scholar]
- 125.Shelnutt JA, Song XZ, Ma JG, Jia SL, Jentzen W, Medforth CJ. Chem Soc Rev. 1998;27:31–41. [Google Scholar]
- 126.Ma JG, Vanderkooi JM, Zhang J, Jia SL, Shelnutt JA. Biochemistry. 1999;38:2787–2795. doi: 10.1021/bi982332a. [DOI] [PubMed] [Google Scholar]
- 127.Ma JG, Laberge M, Song XZ, Jentzen W, Jia SL, Zhang J, Vanderkooi JM, Shelnutt JA. Biochemistry. 1998;37:5118–5128. doi: 10.1021/bi972375b. [DOI] [PubMed] [Google Scholar]
- 128.Barkigia KM, Chantranupong L, Smith KM, Fajer J. J Am Chem Soc. 1988;110:7566–7567. [Google Scholar]
- 129.Ravikanth M, Chandrashekar TK. Coordination Chemistry. 1995:105–188. [Google Scholar]
- 130.Ma JG, Zhang J, Franco R, Jia SL, Moura I, Moura JJG, Kroneck PMH, Shelnutt JA. Biochemistry. 1998;37:12431–12442. doi: 10.1021/bi981189i. [DOI] [PubMed] [Google Scholar]
- 131.Othman S, Desbois A. Eur Biophys J Biophys Lett. 1998;28:12–25. [Google Scholar]
- 132.Shokhireva TK, Berry RE, Uno E, Balfour CA, Zhang HJ, Walker FA. Proc Natl Acad Sci U S A. 2003;100:3778–3783. doi: 10.1073/pnas.0536641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Milne JS, Mayne L, Roder H, Wand AJ, Englander SW. Protein Sci. 1998;7:739–745. doi: 10.1002/pro.5560070323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Marmorino JL, Auld DS, Betz SF, Doyle DF, Young GB, Pielak GJ. Protein Sci. 1993;2:1966–1974. doi: 10.1002/pro.5560021118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Michel LV, Ye T, Bowman SEJ, Levin BD, Hahn MA, Russell BS, Elliott SJ, Bren KL. Biochemistry. 2007;46:11753–11760. doi: 10.1021/bi701177j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Michel LV, Bren KL. J Biol Inorg Chem. 2008;13:837–845. doi: 10.1007/s00775-008-0370-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gooley PR, Caffrey MS, Cusanovich MA, Mackenzie NE. Biochemistry. 1992;31:443–450. doi: 10.1021/bi00117a020. [DOI] [PubMed] [Google Scholar]
- 138.Bartalesi I, Rosato A, Zhang W. Biochemistry. 2003;42:10923–10930. doi: 10.1021/bi0348258. [DOI] [PubMed] [Google Scholar]
- 139.Goodin DB. J Biol Inorg Chem. 1996;1:360–363. [Google Scholar]
- 140.Banci L, Rosato A, Turano P. J Biol Inorg Chem. 1996;1:364–367. [Google Scholar]
- 141.Capece L, Marti MA, Crespo A, Doctorovich F, Estrin DA. Journal of the American Chemical Society. 2006;128:12455–12461. doi: 10.1021/ja0620033. [DOI] [PubMed] [Google Scholar]
- 142.Monson EK, Ditta GS, Helinski DR. Journal of Biological Chemistry. 1995;270:5243–5250. doi: 10.1074/jbc.270.10.5243. [DOI] [PubMed] [Google Scholar]
- 143.Negrerie M, Bouzhir L, Martin JL, Liebl U. Journal of Biological Chemistry. 2001;276:46815–46821. doi: 10.1074/jbc.M102224200. [DOI] [PubMed] [Google Scholar]
- 144.Yamashita T, Hoashi Y, Watanabe K, Tomisugi Y, Ishikawa Y, Uno T. Journal of Biological Chemistry. 2004;279:21394–21400. doi: 10.1074/jbc.M400512200. [DOI] [PubMed] [Google Scholar]
- 145.Arnold PA, Benson DR, Brink DJ, Hendrich MP, Jas GS, Kennedy ML, Petasis DT, Wang MX. Inorg Chem. 1997;36:5306–5315. [Google Scholar]
- 146.Cowley AB, Kennedy ML, Silchenko S, Lukat-Rodgers GS, Rodgers KR, Benson DR. Inorg Chem. 2006;45:9985–10001. doi: 10.1021/ic052205k. [DOI] [PubMed] [Google Scholar]
- 147.Marques HM, Munro OQ, Munro T, de Wet M, Vashi PR. Inorg Chem. 1999;38:2312–2319. [Google Scholar]
- 148.Marques HM. Dalton Trans. 2007:4371–4385. doi: 10.1039/b710940g. [DOI] [PubMed] [Google Scholar]
- 149.Cowley AB, Benson DR. Inorg Chem. 2007;46:48–59. doi: 10.1021/ic060682c. [DOI] [PubMed] [Google Scholar]
- 150.Cowley AB, Lukat-Rodgers GS, Rodgers KR, Benson DR. Biochemistry. 2004;43:1656–1666. doi: 10.1021/bi035531p. [DOI] [PubMed] [Google Scholar]
- 151.Tezcan FA, Winkler JR, Gray HB. J Am Chem Soc. 1998;120:13383–13388. [Google Scholar]
- 152.Wu JZ, La Mar GN, Yu LP, Lee KB, Walker FA, Chiu ML, Sligar SG. Biochemistry. 1991;30:2156–2165. doi: 10.1021/bi00222a020. [DOI] [PubMed] [Google Scholar]
- 153.Rosell FI, Mauk AG. Biochemistry. 2002;41:7811–7818. doi: 10.1021/bi016060e. [DOI] [PubMed] [Google Scholar]
- 154.Bren KL. In: Application of Physical Methods to Inorganic and Bioinorganic Chemistry. Scott RA, Lukehart CM, editors. John Wiley & Sons, Ltd.; Chichester, UK: 2007. pp. 357–384. [Google Scholar]
- 155.Scheidt WR, Wyllie GRA, Ellison MK, Sage JT, Rai BK, Durbin SM, Sturhahn W, Alp EE. J Inorg Biochem. 2003;96:51–51. [Google Scholar]
- 156.Yang J, Liu XS, Bhalla K, Kim CN, Ibrado AM, Cai JY, Peng TI, Jones DP, Wang XD. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 157.Purring C, Zou H, Wang XD, McLendon G. J Am Chem Soc. 1999;121:7435–7436. [Google Scholar]
- 158.Martin AG, Fearnhead HO. J Biol Chem. 2002;277:50834–50841. doi: 10.1074/jbc.M209369200. [DOI] [PubMed] [Google Scholar]
- 159.Neupert W, Herrmann JM. Ann Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 160.Tetreau C, Lavalette D, Momenteau M, Fischer J, Weiss R. J Am Chem Soc. 1994;116:11840–11848. [Google Scholar]
- 161.Caignan GA, Deshmukh R, Zeng YH, Wilks A, Bunce RA, Rivera M. J Am Chem Soc. 2003;125:11842–11852. doi: 10.1021/ja036147i. [DOI] [PubMed] [Google Scholar]
- 162.Gruia F, Kubo M, Ye X, Ionascu D, Lu C, Poole RK, Yeh SR, Champion PM. J Am Chem Soc. 2008;130:5231–5244. doi: 10.1021/ja7104027. [DOI] [PMC free article] [PubMed] [Google Scholar]