

Abstract
1,1′-Bipyrrole is synthesized in four steps from hydrazine. A colorless solid, mp 52°C, it sublimes readily at room temperature and forms X-ray quality crystals in which the rings are not coplanar but are nearly orthogonal.
Bipyrroles1 are N-heterocyclic analogs of biphenyl, although far less well studied. Bipyrrole articles entered the literature only about a dozen times during the first half of the last century; yet, in the past 25 years there has been a relative explosion of interest in bipyrroles and bipyrrole-based more complex structures with more than 400 citations. Bipyrroles are found in nature as components of clinically interesting and important natural products for the treatment of cancer, viral and bacterial infection2; (in reduced form) in vitamin-B123; in marine natural products4; in macrocyclic oligopyrroles for use as ionophores5 and in medicine6; and in synthetic linear oligopyrrole conductive polymers.7
There are six different bond connections that can be drawn between two pyrrole molecules, leading to six constitutionally isomeric bipyrroles: 3 symmetric (1,1′; 2,2′; and 3,3′) and 3 nonsymmetric (1,2′; 1,3′; and 2,3′). All but one, 1,1′-bipyrrole, have C-C or N-C bonds linking the two pyrrole rings. Four of the six parent, unsubstituted bipyrroles (1,1′, 2,2′, 3,3′, and 2,3′) have been synthesized in 19768 and 1977,9 and subsequent spectroscopic10 and theoretical analyses11,12 were reported in only a few publications. Unlike biphenyls and other biaryls, rotational stereochemistry (atropisomerism) about the interconnecting bond of the six possible bipyrrole isomers is not well understood.1,12
In contrast to the well-studied atropisomeric stereochemistry of biaryls,13 there are only two known optically active bipyrroles: a 1,1′-bipyrrole (2,2′,5,5′-tetramethyl-1,1′-bipyrrole-3,3′-dicarboxylic acid)14a and a 2,2′-bipyrrole (1,1′,2,2′,5,5′-hexamethyl-2,2′-bipyrrole-3,3′-dicarboxylic acid).14b Molecular orbital calculations and photoelectron spectroscopy have indicated a preference for orthogonal rings in 1,1′-bipyrrole.10 Ab initio calculations on 2,2′-bipyrrole show it adopting preferentially an anti-clinal (ac) conformation at the global minimum with an N-2-2′-N’ torsion angle ∼148° and a 3-4 kcal/mole greater stability than the sc local minimum conformation, where the N-2-2′-N’ torsion angle is ∼46°.11,12b Theory also predicts the ac conformations of 3,3′- and 2,3′-bipyrrole to be the most stable.11a,c
Such theoretical predictions do not necessarily relate to the solid phase: an X-ray structure of 2,2′-bipyrrole shows it to adopt an ap planar conformation in the crystal.12 There are no crystal structures available of any of the other parent constitutional isomers of bipyrrole. And although crystallographic structures of octa-substituted 1,1′-bipyrroles may not reflect that of the parent, octamethyl- and octa(trifluoromethylthio)-1,1′-bipyrrole were found to have orthogonal rings (92.4° and 92.8°, respectively).15 Given the differences between the predicted conformation of 2,2′-bipyrrole and that found in its crystal, and the general prediction that bipyrroles are twisted, we were attracted to determine the crystal structure conformation of 1,1′-bipyrrole, which is a solid with a reported mp 57°C.9
1,1′-Bipyrroles were apparently first synthesized in 1904,16 the first bipyrroles (2,2′,5,5′-dimethyl-1,1′-bipyrrole-3,3′-dicarboxylic acid) to have been synthesized, by condensing 1,4-dione (ethyl 3-acetyl-5-oxohexanoate) with hydrazine in a reaction that proceeded stepwise through 1-aminopyrrole. Our syntheses of the parent 1,1′-bipyrrole (1) follows a similar path, as outlined in Scheme 1. It differs from the most recently published synthesis of 19 in two ways: (1) improved yields at each step shown, and (2) a shorter synthesis. The published synthesis converts 1-aminopyrrole (2)17 to its succinimide derivative by reaction with succinic anhydride (50% yield), followed by treatment with pyrocatecholphosphorous trichloride to give 2,5-dichloro-1,1′-bipyrrole in 59% yield and, finally, dechlorination by n-Bu3SnH in 30% yield. The overall yield was 1.6-2.3% in the longer route vs 22% in a new shorter route shown below. The authors9 indicate that a direct reaction of 1-aminopyrrole with 2,5-diethoxytetrahydrofuran to produce 1,1′-bipyrrole was not possible, that TLC of the reaction mixture showed two spots that gave the expected color reaction with Ehrlich’s reagent. In our route (Scheme 1), we too converted phthalimide in 3 steps to 1-aminopyrrole, via 3 and 4, in an overall yield of 49%. We found that the key final step, direct reaction of 2 with 2,5-dimethoxytetrahydrofuran gave an acceptable yield of 1. However, it should be noted that this low melting solid sublimes easily at room temperature and low yields can result from such losses during isolation.
Scheme 1.

A crystal grown by sublimation proved suitable for an X-ray crystallographic determination that showed (Fig. 1A) 1 to be twisted in the crystal, with an ∼80° interplanar angle. This result stands in stark contrast with that from the crystal structure of the 2,2′-bipyrrole isomer, in which the two rings lie coplanar.
Figure 1.
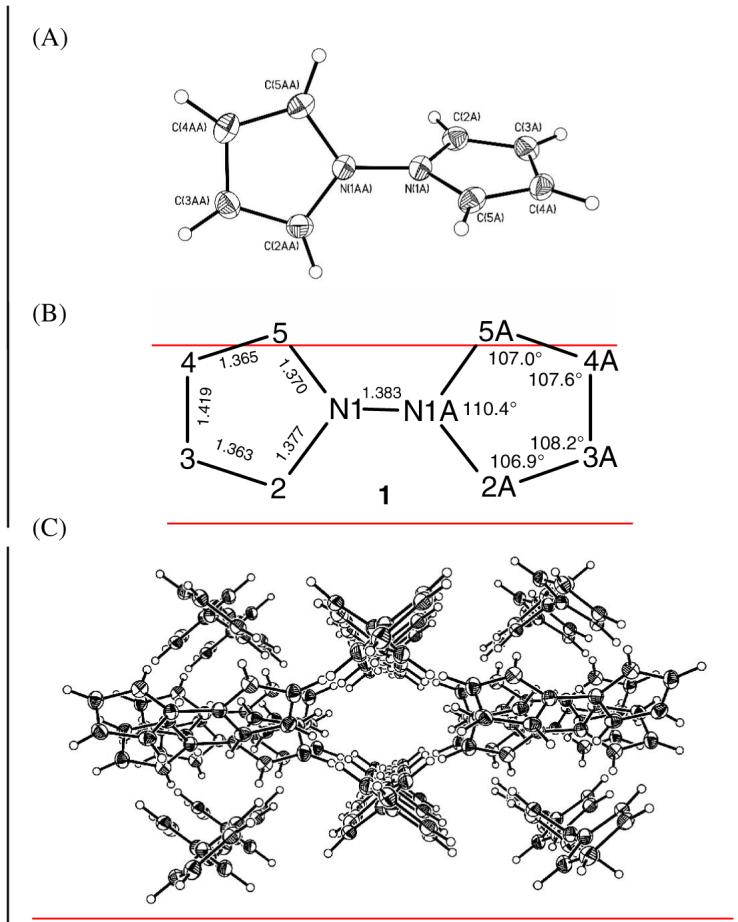
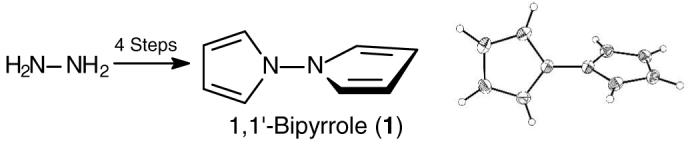
(A) Structural drawing of 1 in the crystal showing the atom numbering system. (B) Bond distances (Å) and angles (°)found in the X-ray crystallographic structure of 1,1′-bipyrrole (1) with the numbering system used. The atoms of the leftmost pyrrole ring lie in a plane, as do those of the rightmost. The C(2)-N(1)-N(1A)-C(5A) torsion angle was found to be ∼80°, and the C(5)-N(1)-N(1A)-C(5A) torsion angle is ∼100°. (C) Packing arrangement of 1 in the crystal.
The bond lengths and bond angles of the pyrrole rings of 1 (Fig. 1B) find good correlation with those of pyrrole itself. Microwave spectroscopic analysis gave the following for pyrrole: N-C2, C2-C3 and C3-C4 bond lengths of 1.370, 1.382, and 1.417 Å, respectively; and C2-N-C5, N-C2-C3 and C2-C3-C4 bond angles of 109.8°, 107.7° and 107.4°, respectively.18 Molecular mechanics calculations19 agree reasonably well: 1.300, 1.335 and 1.469 Å; and 113.9°, 107.6° and 105.4°, respectively, and predict a 1.393 Å N-N bond length. The N-N bond length of crystalline 1 is shorter than that of hydrazine (1.449 Å), determined by electron diffraction,20 where the nitrogen geometry is pyramidal. A plot of the energy barrier19 to rotation about the N-N bond of 1 is shown in Fig. 2. Here, the minimum energy conformation also lies near a 90° interplanar angle. The higher barrier (∼7 kcal/mole above the minimum) corresponds to coplanar rings with the nitrogen lone pairs syn; the lower barrier (∼2 kcal/mole above the minimum) corresponds to coplanar rings with the nitrogen lone pairs anti. They are similar to the calculated (HF/6-31G*) barriers to rotation about the N-N bond of hydrazine.21 From hydrazine’s energy-minimum conformation, where the lp-N-N-lp torsion angle is 90°, the two barriers to rotation are: 0° (∼10.5 kcal/mole), where the lone pairs are syn and 180° (∼3.3 kcal/mole), where the lone pairs are anti.21
Figure 2.
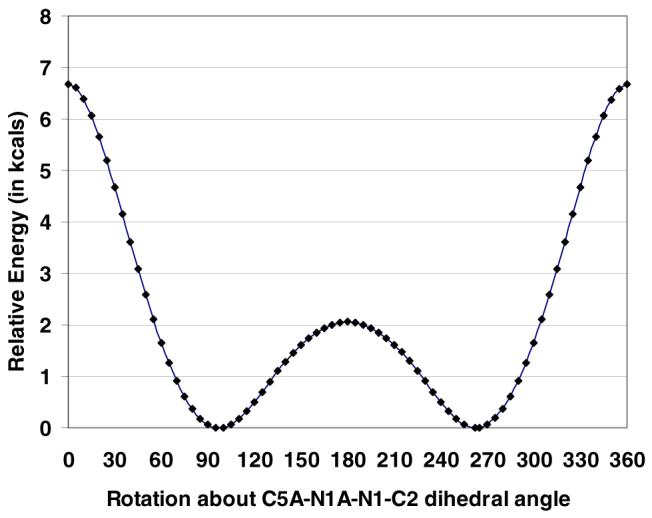
Plot of total energy (kcal/mole) vs angle of rotation about the N-N bond of 1. The minima lie at 95° and 265°. The maxima corresond to the planar molecule, with syn (0° rotation angle) and anti (180° rotation angle) nitrogen lone pairs.
The synthetic methodology of the current work suggests the possibility of making 2,2′-di-t-butyl-1,1′-bipyrrole for studies of atropisomerism and possible chiral resolution.
Experimental
For general procedures of the synthesis, see ref. 12.
N-Aminophthalimide (3)22
This variation of the reported procedure17 avoids heating, which causes 3 to rearrange to the phthalhydrazide. To an ice-cold suspension of 14.7 g (0.1 mol) of phthalimide in 100 mL of 95% ethanol at 5°C, with stirring 3.6 mL (0.11 mol) of 99% hydrazine was added dropwise. A slightly exothermic reaction was observed, and the mixture was allowed to stir at 5°C for 2 h. The mixture was then diluted with 200 mL of ice-water, stirred, filtered, washed with water and dried in air to give 75% of pure product 3, mp 199-200°C (lit.22 199-202°C). It had 1H NMR (500 MHz, CDCl3) δ 3.57 (brs, 2H), 7.73 (dd, 2H, J=5.0, 3.0 Hz), 7.85 (dd, 2H, J=5.0, 3.0 Hz) ppm and 13C NMR (125 MHz, CDCl3) δ 167.0(s), 134.5 (s), 130.5 (d), 123.4 (d) ppm.
N-Phthalimidopyrrole (4) 17,23
A solution of N-aminophthalimide 3 (2.5 g, 2.54 mmol) and 2.5 mL (19.3 mmol) of 2,5-dimethoxytetrahydrofuran in dioxane (25 mL) was heated at reflux until a yellow solution was obtained. While maintaining heating, 2.5 mL of 5 N HCl was carefully added, and the yellow solution became darker. The mixture was cooled and the resultant precipitate was filtered, and washed with a 1:3 mixture of dioxane-water to yield 87% of the product 4, mp 208-209°C (lit.24 mp 218.5°C). It had 1H NMR (500 MHz, CDCl3) δ 6.37 (dd, 2H, J=2.0, 2.5 Hz), 6.75 (dd, 2H, J=2.0, 2.5 Hz), 7.86 (dd, 2H, J=5.5, 3.0 Hz) and 7.99 (dd, 2H, J=5.5, 3.0 Hz) ppm and 13C NMR (125 MHz, CDCl3) δ 164.6 (s), 135.3 (s), 129.8 (d), 124.6 (d), 121.7 (d), 109.3 (d) ppm.
1-Aminopyrrole (2) 23,24
N-phthalimidopyrrole 4 (11.5 g, 54.2 mmol) was dissolved in ∼150 mL of CH3OH, and to this solution was added ∼5 mL of 99% hydrazine monohydrate. The reaction was heated at reflux for 1 h and after cooling was treated with AcOH (3 mL). The mixture was further heated at reflux for 15 min, then it was filtered, and the resultant white precipitate was washed with CH3OH. The filtrate was evaporated in vacuo, and the solid residue was treated with an excess of 40% aq. NaOH until the solid residue dissolved. The aqueous layer was evaporated in vacuo to give 4.00 g of pure product (91%). It had 1H NMR (500 MHz, CDCl3) δ 4.86 (brs, 2H), 6.05 (dd, 2H, J=2.0, 2.5 Hz) and 6.70 (dd, 2H, J=2.0, 2.5 Hz) ppm; 13C NMR (125 MHz, CDCl3) δ 106.6 (α-C) and 122.2 (β-C) ppm.
1,1′-Bipyrrole (1)
1-Aminopyrrole 2 (0.5 g, 6.1 mmol) was dissolved in 10 mL of 1,4-dioxane and to this solution was added ∼0.9 g (6.9 mmol) of 2,5-dimethoxytetrahydrofuran. The solution was heated at reflux for 72 h; then, while keeping it warm, ∼2 mL of 5 N HCl was added, and the entire solution turned brown. The solution was then cooled and extracted (6 × 50 mL) with n-hexane. The organic layer was dried over anh. Na2SO4 and evaporated to a volume of ∼10 mL by a stream of air. Colorless 1,1′-bipyrrole crystallizes out but goes back to solution. The reduced volume of hexane-containing product was transferred to a sublimator. The cold finger was cooled to -30°C, and the sublimator was connected to a high vacuum pump and the bipyrrole crystals were collected shortly on the cold finger. The cold finger was removed carefully, and the solid was scraped off and collected to give 295 mg (37% yield) of product 1 [mp 52°C (sealed tube)] (lit.9 mp 57°C). (n.b.: product is very volatile; even keeping at RT reduces the weight of the product.) 1H NMR (500 MHz, CDCl3) δ 6.20 (dd, 4H, J=2.0, 2.5 Hz) and 6.91 (dd, 4H, J=2.0, 2.5 Hz) ppm; 13C NMR (125 MHz, CDCl3) δ 107.6 (α-C), 121.6 (β-H) ppm.
Supplementary Material
ACKNOWLEDGMENTS
We thank the U.S. National Institutes of Health (HD 17779) for generous support of this research. We also thank the National Science Foundation (CHE-0226402) for providing funding to purchase the X-ray diffractometer used in this work and Prof. Brian Frost for assistance with the crystallographic determination of this work. SKD is an R.C. Fuson Graduate Fellow.
REFERENCES
- (1).Falk H. The Chemistry of Linear Oligopyrroles and Bile Pigments. Springer Verlag; New York: 1989. , and references therein.
- (2)(a).Manville RA. Curr. Med. Chem.: Anti-Cancer Agents. 2001;1:195–218. doi: 10.2174/1568011013354688. [DOI] [PubMed] [Google Scholar]; Murthy MSR, Steenart NAE, Johnson RA, Gordon C. Pyrrole-type compounds, compositions, and methods for treating cancer or viral diseases. Patent No. WO2001055131, U.S. 2000-491712. Gemin X Biotechnol. Canada; Chem. Abstr. 2001;135:152662.; (c) Bhovi MG, Gadaginamath GS. Asian J. Chem. 2005;17:511–517. [Google Scholar]
- (3)(a).Smith KM. In: Comprehensive Heterocyclic Chemistry. Katritzky AR, Rees CW, editors. Vol. 4. Pergamon Press; Oxford: 1984. [Google Scholar]; (b) Kräutler B, Ostermann S. In: The Porphyrin Handbook, Vol. 11, Bioinorganic and Bioorganic Chemistry. Kadish KM, Smith KM, Guilard R, editors. Academic Press, Inc.; 2003. [Google Scholar]
- (4)(a).Vetter W, Gaul S, Olbrich D, Gaus C. Chemosphere. 2007;66:2011–2018. doi: 10.1016/j.chemosphere.2006.07.054. [DOI] [PubMed] [Google Scholar]; (b) Blank DH, Gribble GW, Schneekloth JS, Jr., Jasinski JP. J. Chem. Crystallogr. 2002;32:541–546. [Google Scholar]; (c) Vetter W. Rev. Environ. Contamin. Toxicol. 2006;188:1–57. doi: 10.1007/978-0-387-32964-2_1. [DOI] [PubMed] [Google Scholar]
- (5)(a).Bricker C, Devillers CH, Moutet J-C, Percaut J, Sessler JL. J. Chem. Soc. Chem. Comm. 2006:3981–3983. [Google Scholar]; (b) Sessler JL, An D, Cho W-S, Lynch V, Yoon D-W, Hong S-J, Lee C-H. J. Org Chem. 2005;61:321–347. doi: 10.1021/jo048480q. [DOI] [PubMed] [Google Scholar]; (c) Sessler JL, An D, Cho W-S, Lynch V. Angew. Chem. Intl Ed. 2003;42:2278–2281. doi: 10.1002/anie.200350941. [DOI] [PubMed] [Google Scholar]
- (6)(a).Naumovski L, Sirisawad M, Lecane P, Chen J, Ramos J, Wang Z, Cortez C, Magda D, Thiemann P, Boswell G, Miles D, Cho DG, Sessler JL, Miller R. Molec. Cancer Therapeut. 2006;5:2798–2805. doi: 10.1158/1535-7163.MCT-06-0246. [DOI] [PubMed] [Google Scholar]; (b) Tanada M, Shibata Y, Maeda M, Saski S. Heterocycles. 2004;63:29–39. [Google Scholar]
- (7)(a).Wenbo E, Ohkubo K, Sanchez-Garcia D, Zhang M, Sessler JL, Fukuzumi S, Kadish KM. J. Phys. Chem. 2007;590:49–54. doi: 10.1021/jp068717h. [DOI] [PubMed] [Google Scholar]; (b) Erben C, Will S, Kadish KM. In: The Porphyrin Handbook, Vol. 2, Heteroporphyrins, Expanded Porphyrins and Related Macrocyles. Kadish KM, Smith KM, Guilard R, editors. Academic Press, Inc.; 1999. [Google Scholar]; (c) Benicori T, Brenna E, Sannicolo F, Zotti G, Zechin S, Schiavon G, Gatti C, Frigerio G. Chem. Mater. 2000;12:1480–1489. [Google Scholar]; (d) Street GB, Lindsey SE, Nazzal AI, Wynne KJ. Mol. Cryst. Liq. Cryst. 1985;118:137–148. [Google Scholar]; (e) Ford WK, Duke CB, Salaneck WR. J. Chem. Phys. 1982;77:5030–5039. [Google Scholar]
- (8).Farnier M, Soth S, Fournari P. Can. J. Chem. 1976;54:1083–1086. [Google Scholar]
- (9).Flitsch W, Schulten W. Synthesis. 1977:414–415. [Google Scholar]
- (10)(a).Flitsch W, Peeters H, Schulten W, Radmacher P. Tetrahedron. 1978;34:2301–2304. [Google Scholar]; (b) André JM, Vercauteren DP, Street GB, Brédas JL. J. Chem. Phys. 1984;80:5643–5648. [Google Scholar]; (c) Nazzal IA, Street GB, Wynne KJ. Mol. Cryst. Liq. Cryst. 1985;125:303–307. [Google Scholar]
- (11)(a).Sancho-Garcia JC, Karpfen A. Chem. Phys. Lett. 2005;411:3421–326. [Google Scholar]; (b) Orti E, Sánchez-Marin J, Viruela-Martin PM, Tomás F. Chem. Phys. Lett. 1986;130:285–290. [Google Scholar]; (c) Rabias I, Howlin BJ, Provata A, Theodore D. Mol. Simul. 2000;24:95–109. [Google Scholar]; (c) Orti E, Sánchez-Marin J, Tomás F. Theor. Chim. Acta. 1986;69:41–49. [Google Scholar]; (d) Falk H, Stressler G, Müller N. Monatsh. Chem. 1988;119:505–508. [Google Scholar]; (d) Millefiori S, Alparone A. J. Chem. Soc. Faraday Trans. 1988;94:25–32. [Google Scholar]
- (12).Skowronek P, Lightner DA. Monatsh. Chem. 2003;134:889–899. [Google Scholar]
- (13).Eliel EL, Wilen SH. Stereochemistry of Organic Compounds. John Wiley & Sons, Inc.; New York: 1994. pp. 1142–1150. [Google Scholar]; Eliel EL. Stereochemistry of Carbon Compounds. McGraw-Hill, Inc.; 1962. pp. 156–179. [Google Scholar]; Shriner RL, Adams R, Marvel CS. In: Organic Chemistry. An Advanced Treatise. Gilman H, editor. I. John Wiley & Sons; New York: 1943. pp. 343–377. [Google Scholar]
- (14)(a).Chang C, Adams R. J. Am. Chem. Soc. 1931;53:2353–2357. [Google Scholar]; (b) Webb JLA. J. Org. Chem. 1953;18:1423–1427. [Google Scholar]
- (15)(a).Kuhn N, Kotowski H, Steimann M, Speiser B, Worde M, Henkel G. J. Chem. Soc. Perkin. 2000;2:353–363. [Google Scholar]; (b) Gerstenberger MRG, Haas A, Kirste B, Kruger C, Kurreck H. Chem. Ber. 1982;115:2540–2547. [Google Scholar]
- (16).Korschun C. Ber. Dtsch Chem. Ges. 1904;37:2183–2192. [Google Scholar]
- (17).Flitsch W, Krämer U, Zimmerman H. Chem. Ber. 1969;102:3268–3276. [Google Scholar]
- (18).Nygaard L, Nielsen JT, Kircheimer J, Maltesen G, Rastrup-Andersen J, Soerensen GO. J. Mol. Struct. 1969;3:491–506. [Google Scholar]
- (19).Molecular mechanics calculations were carried out on an SGI Work Station using version 7.1 of SYBYL (Tripos Assoc., St. Louis, MO, USA), using the MM3 forcefield.
- (20).Kohata K, Fukuyama T, Kuchitsu K. J. Phys. Chem. 1982;86:602–606. [Google Scholar]
- (21).Schlegel HB, Skancke A. J. Am. Chem. Soc. 1993;115:7465–7471. [Google Scholar]
- (22) (a).Kirkpatrick JL, Patel NR, Rulter JL. Isothioureidoisoindolediones and their use as plant growth regulators. U.S. Patent No. 4292071. Chem. Abstr. 1982;96:64200.Patel NR, Rulter JL. N-(Arylthiocarbamoyl)-2-amino-1H-isoindole-1,3(2H)-diones and their use as plant growth regulators. U.S. Patent No. 4264502. Chem. Abstr. 1981;95:80725.
- (23).Flitsch H, Lült F-J. Liebig’s Ann. Chem. 1987:893–894. [Google Scholar]
- (24).Drew HD, Halt HH. J. Chem. Soc. 1937:16–26. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.