

Abstract
The formation of memories relies on plastic changes at synapses between neurons. Although the mechanisms of synaptic plasticity have been studied extensively over several decades, many aspects of this process remain controversial. The cellular locus of expression of long-term potentiation (LTP), a major form of synaptic plasticity, is one of the most important unresolved phenomena. In this article, we summarize some recent advances in this area made possible by the development of new imaging tools. These studies have demonstrated that LTP is compound in nature and consists of both presynaptic and postsynaptic components. We also review some features of presynaptic and postsynaptic changes during compound LTP.
Introduction
The survival of individual organisms often depends on behavioral adaptation. Encountering a novel stimulus or circumstance may elicit a particular behavior, and subsequent responses to the same stimulus may then be altered for minutes, days, or even years. Change in behavior resulting from experience is an operational definition of learning, and the importance of learning to an organism's survival is obvious when we observe animal behaviors as diverse as aggressive displays, mating rituals, and feeding strategies. Neuroscientists believe that behavioral adaptation is linked to changes in patterns of communication between neurons resulting from previous synaptic activity and that learned behaviors are therefore the sum of changes at the synapse level, many of which may last a lifetime.
As early as the 1940s, clinical observations enabled investigators to link human memory dysfunction to the hippocampus (Scoville and Milner 1957; Olds 1972) and develop hypotheses about learning and memory at the neuronal level (Hebb 1949). These two factors stimulated research in the field of synaptic plasticity in the mammalian brain. Bliss and Lomo, while recording postsynaptic responses in the hippocampus of an anesthetized rabbit, demonstrated a pattern of enhanced neuronal responses that persisted for several hours after a single high-frequency tetanization of cortical afferents (Lomo 1966; Bliss and Lomo 1973). They recognized that such signaling enhancement, now known as long-term potentiation (LTP), could serve as a cellular substrate for information storage (Bliss and Lomo 1973).
Since Bliss and Lomo conducted those first studies, more than 8,000 papers on LTP mechanisms have appeared in the literature, as other groups have continued to explore this area. Demonstrations of many other forms of synaptic plasticity have also provided compelling evidence that neurons can alter their synaptic efficacy along a wide continuum of stimulus parameters and retention times. Thus, synaptic plasticity may be the rule throughout the brain, rather than the exception. Research has now shown that LTP (or LTP-like plasticities) occurs in all major cortices, the cerebellum, midbrain, brainstem, and peripheral ganglia. Synaptic plasticity is not limited to only the excitatory neurons of the brain, as evidence by the fact that inhibitory interneurons also express LTP (Kullmann and Lamsa 2007).
In the beginning of the LTP era, the explanation for this phenomenon was intuitively simple. The increased synaptic strength that occurs during LTP should arise from increased neurotransmitter release, originate from a postsynaptic cell that becomes more responsive to the same amount of neurotransmitter, or both. Surprisingly, this issue that was once thought to be “simple” still has not been resolved. Thus, unfortunately this topic has remained a great source of controversy for the LTP field. Although we still cannot claim complete clarity on this issue as we write this review, our understanding of LTP (and how to study LTP) is vastly better than it was previously, when the condition of the field was pronounced by some investigators as “frustrating or embarrassing”. As seen in other scientific areas, we owe this progress to the availability of novel technologies that have allowed us to examine LTP at unprecedented levels of detail.
As each new tool to investigate LTP has emerged, opinions in the field have been swayed, favoring either a presynaptic or postsynaptic origin of LTP. First, advances in whole-cell recording and quantal analysis indicated that presynaptic mechanisms predominate LTP. The subsequent availability of high-resolution imaging and the capability of manipulating genes of interests boosted the theory that LTP originates at postsynaptic neuronal compartments, which are easier to image and manipulate than presynaptic counterparts. Indeed, the combination of whole-cell recording from postsynaptic neurons and two-photon imaging of dendritic spines, the small protuberants that are the sites of excitatory input, has provided much clarity about what happens in the postsynaptic compartment during LTP. In contrast, the presynaptic sites are located considerably more remotely from the cell body and are much harder to manipulate; therefore, data on the presynaptic contribution to LTP are substantially scarcer.
It is not surprising that more abundant postsynaptic data predispose current thinking toward that site's contribution, as evidenced by synaptic plasticity review papers published during the last several years (Baudry and Lynch 2001; Malinow and Malenka 2002; Bredt and Nicoll 2003; Malenka and Bear 2004; Cavazzini and others, 2005; Segal 2005; Raymond 2007). However, although we understand quite a bit about postsynaptic mechanisms of LTP, the complete picture is far from clear. Do presynaptic mechanisms of LTP not exist, or are we simply unable to study them? With better imaging tools and better probes to study presynaptic function on the horizon, we are optimistic that we will answer this question, and the two pieces of the LTP puzzle will be joined.
As we pointed out above, the advancement of techniques will facilitate our eventual understanding of LTP, and many reviews have been written on postsynaptic function during this phenomenon. Therefore, because reviews of the presynaptic mechanisms of LTP are rare in the literature, we will focus on the tools that are (or soon will be) available to study changes in presynaptic function during LTP.
LTP refers to different phenomena
As one surveys the vast literature on LTP, it becomes clear that the term is now applied to various phenomena with distinct cellular and molecular mechanisms. Since the first characterization of LTP by Lomo and Bliss, use-dependent potentiations have been described in other parts of the central nervous system. It quickly became apparent that these LTPs may differ. The brain region, the neuron type in that region, and the types of inputs that synapse on a particular neuron type are all major determinants of a type of LTP. Furthermore, the technique used to prepare or isolate the neurons for study (dissociated neurons in culture vs. brain slice preparation) and the protocols used to induce LTP can influence findings and sometimes provide opposite results at the same synapse.
Even within the hippocampus, LTP occurs at multiple locations and uses different mechanisms. The excitatory synapses between CA3 and CA1 pyramidal neurons (CA3-CA1 synapses) in the hippocampus have received the majority of attention from LTP researchers, because of the availability of a brain slice preparation in which LTP can be studied with relative ease via extracellular field recordings. LTP at these synapses closely parallels the model for cellular learning and is analogous to the form of learning seen in classical conditioning (Hebb 1949). Therefore, to limit the scope of this review, we will restrict most of our discussion to the LTP mechanisms of CA3-CA1 synapses.
Stimulation protocols: timing is everything
From the very beginning of the LTP era, researchers have tried to find stimulation (i.e., induction) protocols that closely resemble neuronal activity in the intact brain. The original stimulus protocol used by Bliss and Lomo in the anesthetized rabbit preparation ranged from 10 to 100 Hz (Bliss and Lomo 1973). Since that time, a variety of LTP induction protocols from different research groups have emerged in the literature. Most involve high-frequency trains of stimulation (tetanization) that are delivered to presynaptic axons. The tetanization typically lasts several seconds and is delivered at frequencies of 25 to 400 Hz. To mimic hippocampal activity recorded in awake or sleeping animals, Larson and others developed the theta-burst stimulation (TBS) protocol, which also effectively produces LTP (Larson and others, 1986). Similar to tetanization, TBS is delivered to axons but with a more complex pattern. It usually consists of a short burst of 5 pulses delivered at 100 Hz. Multiple bursts are applied at 5-Hz and may be repeated several times to create a sequence, which may be repeated 2 to 3 times over a 10- to 15-min time frame. The total number of stimuli in all of the above-mentioned protocols varies from as few as 100 to more than 1000.
The alternative to tetanization or TBS approaches is to induce LTP by time-locked depolarization of the postsynaptic cell and stimulation of the presynaptic afferent (Wigstrom and Gustafsson 1986). This approach allows precise temporal manipulation of both presynaptic and postsynaptic neurons, as opposed to tetanic stimulation or TBS, which induce LTP as a form of spike-timing–dependent plasticity (Dan and Poo 2004). To induce LTP using the time-locked depolarization protocol, low-frequency (typically 0.1-1 Hz) presynaptic stimulation is paired with the postsynaptic depolarization of CA1 neurons via intracellular current injection.
Until recently, LTP was treated as a simple, 1-compartment phenomenon. Consequently, all induction protocols that increased synaptic strength were considered equivalent tools to achieve LTP. Moreover, the LTP field adopted the dogma that regardless of the induction protocol, LTP at CA3-CA1 synapses depended on the activation of NMDA receptors (NMDARs). We now realize that this dogma should be amended.
In a comparison of stimulus frequencies, Grover and Teyler discovered that at CA3-CA1 synapses, LTP induced by the 200-Hz tetanization was only partially blocked by NMDAR antagonists (Grover and Teyler 1990). Surprisingly, at the same synapses, LTP elicited by 25-Hz tetanization was completely blocked by an NMDAR antagonist. Thus, 2 components of LTP were identified: NMDAR-dependent LTP (NMDAR-LTP) and NMDAR-independent LTP. Induction of both forms of LTP (induced by either 200- or 25-Hz tetanizations) was blocked by dialyzing the postsynaptic neuron with calcium chelators (Grover and Teyler 1990). This finding suggested that calcium influx into the postsynaptic neuron initiates the induction of both forms of LTP. However, the pathways of calcium influx were different, i.e., NMDAR-LTP uses the NMDAR pathway, and NMDAR-independent LTP uses the L-type voltage-gated calcium channels (L-VGCCs) (Grover and Teyler 1990), specifically Cav1.2 L-type Ca2+ channel (Moosmang and others, 2005). These studies showed that LTP is not a unitary phenomenon, even at a single synapse, but rather is a group of plasticities. It was therefore proposed that multiple forms of LTP induced at a single synapse be called “compound LTP”. Later studies showed that not only different tetanizations but also different TBS protocols can produce NMDAR-LTP and NMDAR-independent LTP at CA3-CA1 synapses. Specifically, short versions of TBS induce NMDAR-LTP, and a long version of TBS induces compound LTP (Morgan and Teyler 2001).
Does calcium entering the postsynaptic cell through 2 different paths affect the expression of LTP? Are NMDAR-LTP and compound LTP different degrees of the same process that depends on the concentration of calcium in the postsynaptic cell, or are they controlled by 2 different mechanisms? Recent data unequivocally indicate that NMDAR-LTP and NMDAR-independent LTP are 2 different processes that involve distinct molecular mechanisms. The initial pharmacological study showed that the expression of NMDAR-LTP depends on the activation of serine/threonine kinases, whereas NMDAR-independent LTP also requires the activation of tyrosine kinases (Cavus and Teyler 1996). Recent data have shown even more profound molecular differences between these forms of LTP. NMDAR-independent LTP requires brain-derived neurotrophic factor, mitogen-activated protein kinase, and protein kinase A for its expression, whereas NMDAR-LTP is substantially less sensitive to the disruption of signaling involving these molecules (Zakharenko and others, 2003; Moosmang and others, 2005; Bayazitov and others, 2007).
Moreover, now it is apparent that the 2 forms of LTP are expressed at different cellular loci of the CA3-CA1 synapse. Using advanced imaging technologies, we have shown that NMDAR-LTP is expressed strictly postsynaptically, whereas NMDAR-independent LTP is expressed presynaptically (Zakharenko and others, 2001; Bayazitov and others, 2007). Thus, compound LTP requires the engagement of both presynaptic and postsynaptic neurons. This finding has 2 important ramifications: First, not all induction protocols that produce LTP should be treated equally. Second, the LTP produced by all protocols (unless specifically addressed) is compound LTP, with varying degrees of “contamination” of NMDAR-independent LTP.
The presence of 2 forms of LTP in hippocampal slices is not an “artificial” phenomenon; it has important behavioral consequences. NMDAR-LTP is important for certain forms of learning and memory (Morris and others, 1986; Tsien and others, 1996). However, inhibition of NMDARs only partially impairs spatial learning in task-naive animals, and pretraining in a spatial task overcomes the inhibition induced by NMDAR antagonists (Bannerman and others, 1995). Several studies have shown that pharmacologic or genetic blockade of L-VGCCs, which mediate NMDAR-independent LTP, severely impairs hippocampus-dependent spatial memory (Borroni and others, 2000; Moosmang and others, 2005). Thus, both forms of compound LTP are important for learning and memory.
It is becoming also clear that the existing of 2 forms of LTP at the same synapse is not a mechanism of LTP redundancy. NMDAR-LTP and NMDAR-independent LTP differ not only in their mechanisms and synaptic locus but also in their time course of expression. Recent data indicate that NMDAR-LTP develops rapidly (within the first minutes) and is strongest during the first hours after induction, whereas NMDAR-independent LTP develops substantially slower and stronger during later times after induction (Bayazitov and others, 2007). Behavioral experiments have confirmed the significance of this finding: NMDAR-independent LTP is necessary for the retention of information over longer times, whereas NMDAR-LTP is needed for memory retention over shorter times (Borroni and others, 2000).
Advances in tools to analyze LTP
What tools do researchers have to directly and objectively gauge either presynaptic or postsynaptic functions and monitor them during the course of LTP? Initial attempts were made to dissect presynaptic and postsynaptic functions by using the classic electrophysiological approach. Unfortunately, that approach has inherent limitations that resulted in a variety of interpretations and, as a consequence, long-lasting debates. Two electrophysiological methods were (and still are) used to dissect the contributions of presynaptic neurons in LTP: quantal analysis and pair-pulse facilitation (PPF).
Quantal analyses was introduced to search for presynaptic mechanisms (Katz 1971). Initial studies using either minimal stimulation of or paired recordings from single CA3 or CA1 neurons observed that the number of failures of the AMPA receptor (AMPAR)-mediated postsynaptic response significantly decreased after LTP induction (Malinow and Tsien 1990; Bekkers and Stevens 1990; Bolshakov and Siegelbaum 1995). According to the assumptions of quantal analysis, this finding indicates an increase in the probability that a presynaptic action potential elicits the release of quanta of neurotransmitter. However, this interpretation was later called into question by the observation that the number of NMDAR-mediated synaptic failures was much lower than that of AMPAR responses, and induction of LTP caused little decrease in the number of NMDAR failures (Nicoll and Malenka 1999; Malinow and others, 2000). Furthermore, LTP stimulus protocols induced the rapid appearance of synaptic responses that could be blocked by AMPAR antagonists (Liao and others, 1995; Isaac and others, 1995).
The “silent synapse” hypothesis offered an alternative explanation to these observations by proposing that a significant number of sites of synaptic transmission lack functional AMPARs but contain functional NMDARs. At the normal resting potential, NMDARs are blocked by magnesium; thus, synapses lacking AMPARs are functionally silent. Induction of LTP leads to the insertion of new AMPARs in the membrane, which greatly increases the number of synapses that contain AMPARs and decreases the number of silent synapses (Liao and others, 1995; Isaac and others, 1995). Therefore, according to the silent synapse hypothesis, LTP is purely a postsynaptic phenomenon.
The PPF method is based on the observation that 2 stimuli delivered to the presynaptic axon in rapid succession (within 1 s) produce postsynaptic responses of different amplitudes, where the second response is larger. Although measured postsynaptically, it was agreed that the mechanisms of PPF are exclusively presynaptic (Foster and McNaughton 1991; Schulz and others, 1994). However, this assumption has also been challenged. Experiments with photolysis of caged glutamate have shown that PPF at CA3-CA1 synapses has a strong postsynaptic component (Bagal and others, 2005) that presumably originates from relieving the polyamine block of the AMPARs lacking the glutamate receptor GluR2 (Rozov and Burnashev 1999). Thus, the electrophysiological approach has not provided unambiguous monitoring of presynaptic function.
Efforts to discover more direct approaches to test the function of individual synapses and dissect presynaptic and postsynaptic functions have proven more successful with the advancement of imaging methods. Fluorescent markers can be used to visualize exclusively either a presynaptic terminal or a postsynaptic dendritic spine and are, therefore, more specific and less biased toward a postsynaptic cell than electrophysiological approaches. However, imaging synapses in hippocampal slices by using single-photon imaging tools such as epifluorescence or confocal scanning microscopy has proven to be challenging. The healthiest synapses are located at depths greater than 50 μm in the slice, where single-photon imaging is ineffective. Thus, multiphoton laser scanning microscopy (MPLSM), which allows high-resolution imaging several hundreds of microns into brain tissue, has become the imaging method of choice (Denk and others, 1990).
The first direct test of presynaptic function in hippocampal slices was done when MPLSM was combined with a fluorescent marker of synaptic vesicle cycling, FM 1-43 (Zakharenko and others, 2001; Stanton and others, 2001). The existence of presynaptic LTP at CA3-CA1 synapses was confirmed using the less direct imaging method optical quantal analysis of calcium transients in single dendritic spines (Emptage and others, 2003).
The evolution of fluorescent probes of presynaptic activity
Interest in presynaptic mechanisms of LTP has been renewed with the advent of new probes designed to directly assay synaptic vesicle cycling. These probes include fluorescent lipophilic styryl dyes (such as FM 1-43), green fluorescent proteins (GFPs) with enhanced pH sensitivity (pHluorins), and photoluminescent semiconductor nanocrystals (also known as quantum dots or Qdots).
FM 1-43 was first developed for optical analysis of synaptic vesicle recycling in the frog neuromuscular junction (Betz and Bewick 1992). This dye is not fluorescent in aqueous solutions but becomes fluorescent in lipids. FM 1-43 is taken up into vesicles in presynaptic terminals during endocytosis triggered by presynaptic activity (Fig. 1A). The dye is then released from the presynaptic terminal during a second round of synaptic stimulation, as vesicles fuse with the plasma membrane and release their contents via exocytosis. FM dyes are typically loaded into synaptic vesicles by triggering endocytosis using electrical stimulation, sucrose shock, or a high concentration of potassium. Exocytosis is then assayed by the rate of destaining (loss of fluorescence), as synaptic vesicles release the dye to the extracellular space (Fig. 1B, C). This initial work in hippocampal neurons maintained in culture provided a framework for the further analysis of synaptic vesicle recycling by using FM 1-43 assays (Ryan and others, 1993). Additional probes have been developed with unique properties that expand the usefulness of FM dyes. For example, reducing the length of the bifurcated hydrocarbon tail from 4 carbons (as in FM 1-43) to 2 (FM 2-10) increases its membrane dissociation rate; however, increasing the hydrocarbon tail to 5 carbons (FM 1-84) decreases the dissociation rate (Ryan and others, 1996). FM 4-64 is a red-shifted fluorescent variant of FM 1-43 in which the ring structure has been modified (Ryan 2001).
Figure 1. The FM 1-43 assay is a direct and reliable indicator of presynaptic activity in hippocampal slices.
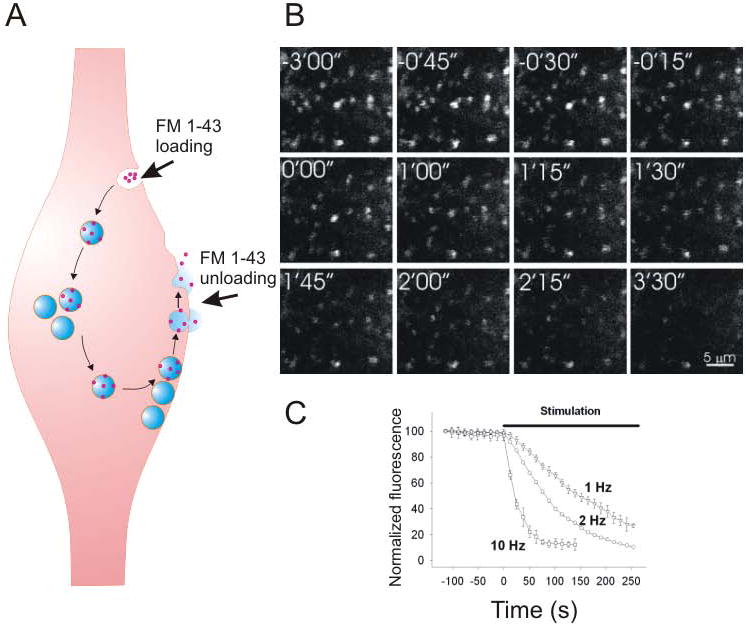
A. The presynaptic terminal is loaded with the FM 1-43 dye (red dots) through endocytosis. The dye is then unloaded with neurotransmitter (blue) from synaptic vesicles via exocytosis. B. Fluorescent images of presynaptic boutons loaded with FM 1-43 in hippocampal slices. FM 1-43 was unloaded during synaptic stimulation starting at 0'00”. C. The rate of FM 1-43 unloading is positively associated with”] the frequency of synaptic stimulation.
Studying presynaptic LTP in hippocampal slices by using FM dyes was challenging, because FM dyes nonspecifically bind to numerous membranous structures, which increases background fluorescence and reduces the visualization of targeted presynaptic boutons. This was alleviated somewhat by 2 methodological improvements: First, ADVASEP-7, a sulfobutylated derivative of β-cyclodextrin whose affinity for FM dyes is higher than that of plasma membranes, can be used to scavenge the dye from nonspecific extracellular binding sites (Kay and others, 1999). Second, sulforhodamine, a red-shifted fluorophore that when placed in close contact with FM 1-43 quenches FM 1-43 fluorescence, can be used to quench fluorescence of extracellular but not intracellular FM 1-43 levels (Pyle and others, 1999). Using these approaches, it became possible to see for the first time presynaptic changes that occurred during LTP in hippocampal slices (Zakharenko and others, 2001; Stanton and others, 2001; Zakharenko and others, 2003; Stanton and others, 2005).
In the first of such studies, we directly showed that after induction of LTP, presynaptic terminals more effectively release neurotransmitter (Zakharenko and others, 2001). FM 1-43 destaining from individual presynaptic terminals was 40 min faster after induction of LTP than before it. Interestingly, this accelerated FM 1-43 destaining happened only when LTP was induced with either 200-Hz or TBS protocols (for induction of compound LTP) but not with the 50-Hz protocol (for induction of NMDAR-LTP) (Zakharenko and others, 2001; Zakharenko and others, 2003) (Fig. 2). Antagonists of L-VGCC, which only partially inhibited compound LTP and did not inhibit NMDAR-LTP, completely eliminated the acceleration of FM 1-43 destaining. These were the first results demonstrating that compound LTP consists of both presynaptic and postsynaptic components and that the presynaptic component depends on L-VGCC, while the postsynaptic component depends on NMDAR-LTP.
Figure 2. Dissecting postsynaptic LTP from compound LTP.
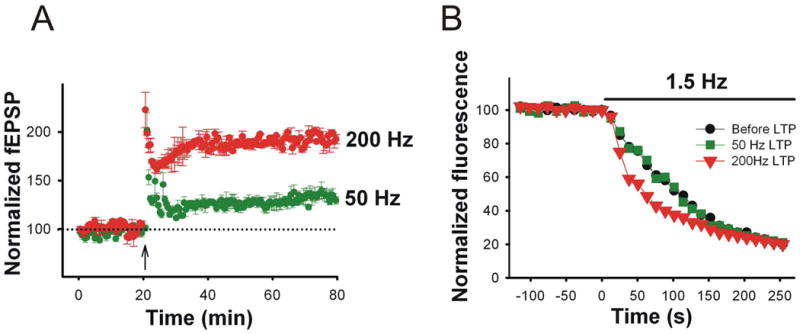
A. The FM 1-43 assay was used to dissect LTP induced at CA3-CA1 synapses with the 200-Hz (red) or 50-Hz (green) stimulation protocols. The arrow depicts the onset of LTP induction. B. Presynaptic function assayed as rate of FM 1-43 unloading in response to 1.5 Hz synaptic stimulation (bar) was enhanced during the 200-Hz LTP (red triangles) but not during the 50-Hz LTP (green squares) compared to that before LTP was induced (black circles).
The FM assay in acute hippocampal slices was a big step forward in dissecting the presynaptic component of compound LTP at CA3-CA1 synapses. However, FM dyes had a serious limitation that precluded using them for more detailed experiments, i.e., they cannot be used for continuous monitoring of presynaptic function, because the dye is lost to the extracellular medium upon exocytosis. Consequently, if multiple measurements are desired, the loading procedure must be repeated. With the recent development of pHluorins, pH-sensitive GFP molecules linked to synaptic vesicle proteins, it is now possible to quantify the rates of presynaptic exocytosis and endocytosis with repeated measures (Sankaranarayanan and others, 2000; Ryan 2001). As constructed by Miesenbock et al., ecliptic pHluorin molecules lose fluorescence as the pH decreases (Miesenbock and others, 1998). By linking pHluorin to VAMP-2, a luminal synaptic vesicle protein, Miesenbock et al. created the probe synaptopHluorin (spH), which changes its fluorescence from low when it is inside the acidic lumen of synaptic vesicles to high when it become exposed to the extracellular medium during exocytosis. The probe then becomes reinternalized and reincorporated into synaptic vesicle membranes during the process of endocytosis, and fluorescence is again quenched with synaptic vesicle reacidification (Fig. 3A).
Figure 3. The synaptopHluorin assay directly and continuously monitors presynaptic activity in hippocampal brain slices.
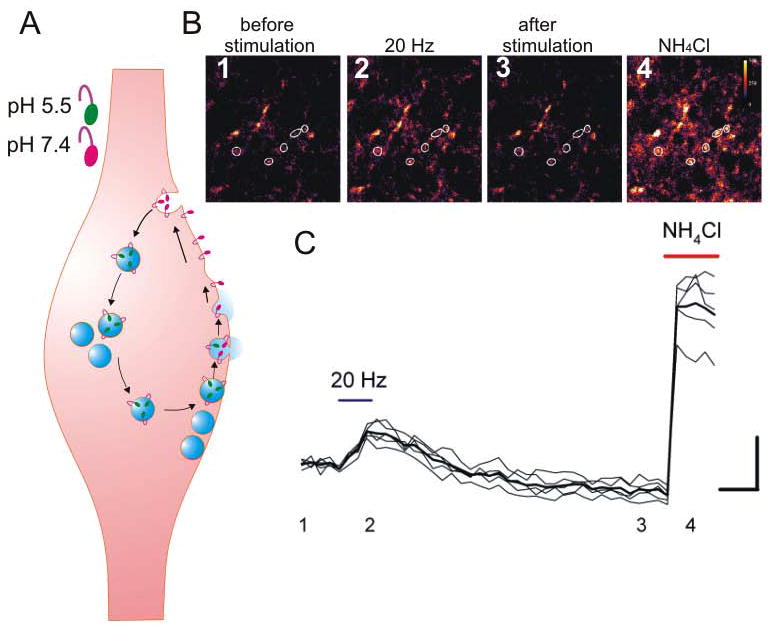
A. The presynaptic terminal expresses synaptopHluorin (spH) in synaptic vesicles. The spH fluorescence is quenched (green) inside of the acidic synaptic vesicles and increases (pink) when the synaptic vesicle lumen is exposed to the extracellular space. The spH remains within a presynaptic terminal during the synaptic vesicle cycle. B, C. Changes in spH fluorescence in CA3 presynaptic boutons (white circles) before (1), during (2), after (3) synaptic stimulation and after application of NH4Cl (4) in the hippocampal slices. Scale bars: vertical,10%; horizontal, 5 sec.
The pHluorin-based indicators are genetically encoded, and animal models expressing these indicators are already available (Araki and others, 2005; Li and others, 2005; Tabares and others, 2007). Using mouse models that express spH in a subset of pyramidal neurons, investigators have monitored changes in presynaptic function over several hours, before and after the induction of LTP (Bayazitov and others, 2007) (Fig. 3B, C). Simultaneously, changes were measured in postsynaptic potentials as a measure of total changes in presynaptic and postsynaptic responses. Changes in presynaptic function were continuously assessed as changes in peak spH fluorescence in individual boutons in response to test stimuli, and changes in postsynaptic function could be deduced as a difference between postsynaptic potentials and spH fluorescence. This approach showed that after induction of compound LTP, peak spH fluorescence increased gradually (half-time of increase was approximately 35 min) and then plateaued approximately 90 min after LTP induction (Fig. 4A). In contrast, changes in postsynaptic function were rapid (half-time of increase was approximately 1 min). The increase peaked within several minutes after induction and decreased thereafter.
Figure 4. Simultaneous monitoring of synaptic strength and presynaptic function during LTP.
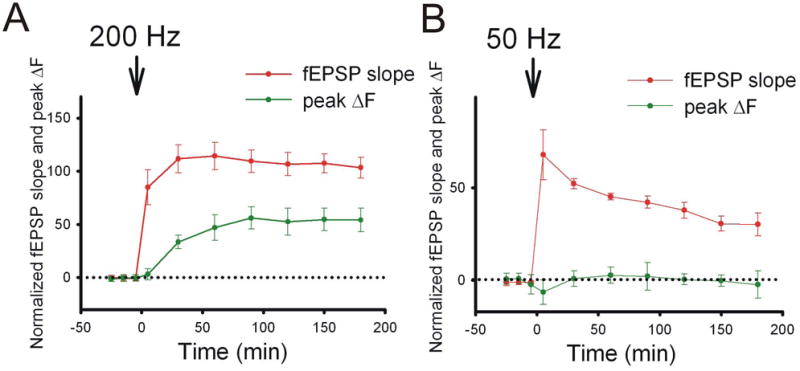
A. Presynaptic function, as indicated by peak ΔF, was slowly enhanced during compound LTP induced by 200-Hz tetanization. B. NMDAR-LTP induced by 50-Hz tetanization resulted in a lack of presynaptic enhancement.
These experiments demonstrated that not only do 2 neurons participate in compound LTP, but also events in presynaptic and postsynaptic neurons are temporally separated. This double dissociation may help to explain many discrepancies in the LTP field: testing roles of various biochemical cascades in LTP at different times (for instance, 30 min vs. 180 min, as has been done in many such works) after induction may provide diametrically different results. Thus, if a molecule of interest is involved in presynaptic mechanisms, its role will be more evident if tested at later times rather than during the first 30 min. Conversely, the contribution of postsynaptic mechanisms should be tested at earlier times. Therefore, the lack of an effect of a specific inhibitor or mutation on LTP at one time point should not be considered a final verdict, but rather it should be retested in a more systematic way.
Although the spH assay has proven useful in studying presynaptic changes during LTP in slices, a major drawback prevents using this indicator to measure the release of individual quanta. Specifically, spH has a high surface membrane expression (15%-24%) that reduces the signal-to-noise ratio and thus interferes with the detection of membrane fusion events (Granseth and others, 2006; Balaji and Ryan 2007). Progress in the development of pHluorin-based fluorescent probes has increased the signal-to-noise ratio, now making it possible to assay neurotransmitter release in a presynaptic bouton evoked by single action potentials (Balaji and Ryan 2007). Granseth et al. linked an enhanced super-ecliptic GFP to the second intraluminal loop of the synaptic vesicle protein synaptopHhysin (sypHy) (Granseth and others, 2006), which decreased surface expression of SypHy to 8% to 9% (Granseth and others, 2006; Balaji and Ryan 2007). Most recently, pHluorin has been fused to an intraluminal loop of the vesicular glutamate transporter protein VGLUT1 (Voglmaier and others, 2006). The level of VGLUT1-pHluorin (VGpH) expression on the surface is only 2%, which allows the visualization of strong fluorescent peaks attributable to exocytosis of a single synaptic vesicle (Balaji and Ryan 2007).
Although these pHluorin probes allow for continuous monitoring of presynaptic exocytosis and endocytosis with surprising spatial and temporal resolution, their usage is not entirely limitless. All conventional fluorescent dyes are subject to photobleaching; therefore, repeated illumination of the dyes can result in eventual loss of fluorescence. Furthermore, converting fluorescent signals to high-resolution images suitable for electron microscopy is very challenging, especially in brain slices. A new class of biocompatible fluorescent probes, semiconductor nanocrystals or quantum dots (Qdots), provides a high degree of fluorescence with broad absorption spectra and narrow emission spectra. Qdots have recently been used to fluorescently tag synaptic vesicles, and their fluorescent emissions are highly resistant to photobleaching (Zhang and others, 2007b). Moreover, these probes do not require any additional processing to be visible by electron microscopy. Qdots have been recently loaded into synaptic vesicles of hippocampal neurons maintained in culture, thereby providing a proof of principle that Qdots are a new generation of probes that can be used for directly testing presynaptic function (Zhang and others, 2007a). Indeed, Qdots fluorescence showed a high degree (96.1%) of coincident labeling with FM dyes. Electron micrographs of presynaptic boutons demonstrated that 1 Qdot can label a single synaptic vesicle. Thus, if successful in slices, the Qdot method will provide 3 advantages over conventional fluorophores for testing presynaptic function: First, Qdots do not lose their fluorescence during prolonged illumination; second, they are easily visible with electron microscopy; and third, they indicate the exact number of stained synaptic vesicles. However, similar to FM dyes, Qdots should be repeatedly loaded into synaptic vesicles for continuous monitoring presynaptic function.
In summary, the development of lipophilic styryl dyes, synaptic proteins tagged with pH-sensitive GFPs, and fluorescent Qdots has created a set of tools that can be used to directly gauge presynaptic function during LTP. These probes clearly identify properties and behaviors that are not the result of changes in postsynaptic function; therefore, they can be used to dissect presynaptic LTP from compound LTP. These probes are also being used to study the roles of specific molecules in presynaptic mechanisms of synaptic plasticity.
A summary of LTP mechanisms
LTP proceeds through several steps or phases that involve the activation of unique physiological processes. Tetanic stimulation leads to a calcium-dependent induction phase, which is then followed by expression and subsequent maintenance phases with 2 (early- and late-phase LTP) (Malenka and Bear 2004) or 3 time constants of decay (LTP1, LTP2, LTP3) (Raymond 2007). Early-phase LTP (or LTP1) may last only 30 to 60 min and occurs independent of protein synthesis. Late-phase LTP (LTP2 and LTP3) involves protein synthesis. Raymond (2007), who equated late-phase LTP with LTP2 and LTP3, distinguished LTP2 from LTP3 as follows: LTP2 requires protein synthesis but no change in gene transcription, and LTP3 requires both.
The induction of LTP requires specific activation of postsynaptic receptors that allow calcium into dendritic spines. Initially, NMDA receptors were identified as a major source of calcium influx during LTP induction (Collingridge 2003). Postsynaptic calcium was determined to be a requirement for LTP, because intracellular injection of the calcium chelators into CA1 pyramidal neurons was sufficient to block LTP induction (Lynch and others, 1983b). Activation of AMPARs during LTP induction provides sufficient depolarization to relieve the magnesium blockade of NMDARs and let calcium in. Therefore, in the older, “classical” version, calcium influx through NMDARs initiates the expression of LTP.
As we know now, LTP is not a unitary phenomenon, even during induction. In addition to NMDARs, dendritic spines have additional channels that allow calcium entry. Specifically, L-VGCCs allow calcium influx when dendritic spines are sufficiently depolarized. As discovered in 1990, “classical” NMDAR-LTP is only part of compound LTP, which depends on both NMDAR and L-VGCCs (Grover and Teyler 1990). NMDAR-LTP and VGCC-LTP are independent phenomena and constitute 2 parts of compound LTP. Therefore, a previous view should be amended as follows: calcium influx through NMDARs leads to the expression of NMDAR-LTP, and calcium influx through L-VGCCs leads to the expression of VGCC-LTP. Enhanced calcium influx through these 2 pathways into dendritic spines leads to the expression of 2 independent forms of LTP (Fig. 5). A great deal is known about the molecular mechanisms of NMDAR-dependent LTP, which has been discussed in numerous review articles. In summary, calcium-triggered activation of certain protein kinases leads to the redistribution of AMPARs (and/or enhancing their properties) in dendritic spines and eventually increases the sensitivity of dendritic spines to glutamate, boosting the response of functional synapses and converting “silent synapses” into functional synapses.
Figure 5. Models of NMDAR-LTP and compound LTP at CA3-CA1 synapses.
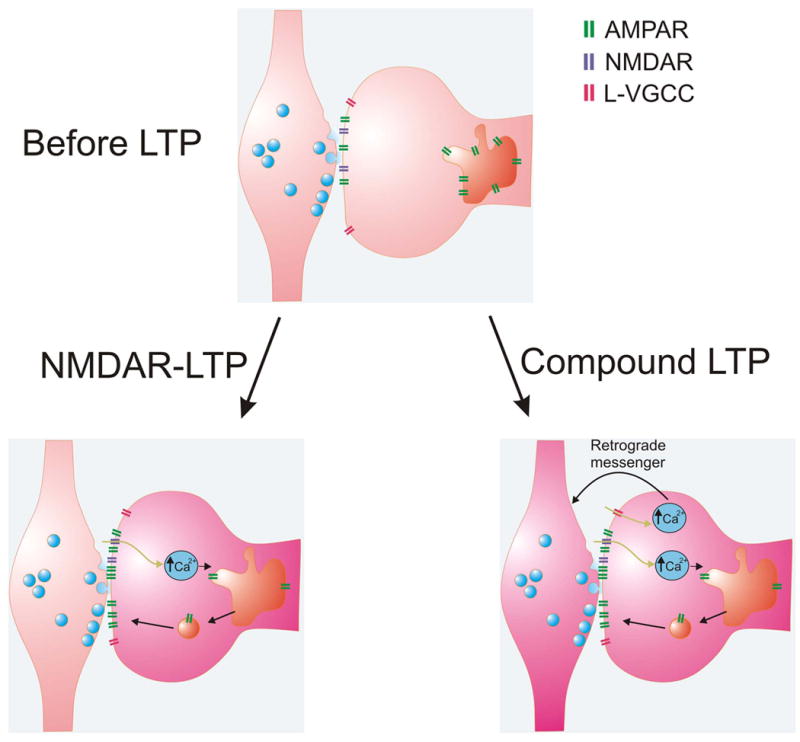
Top panel. A synapse before LTP occurs. Bottom left panel. Weak-induction protocols produce postsynaptic NMDAR-LTP. Calcium enters the postsynaptic dendritic spine via NMDARs (blue) and triggers the insertion of AMPARs (green) onto the postsynaptic plasma membrane. Bottom right panel. Strong-induction protocols produce compound LTP. During compound LTP, calcium enters the postsynaptic dendritic spine via NMDARs and L-VGCCs (pink). Calcium entering through L-VGCCs is used to generate a retrograde signal, which enhances presynaptic function.
Because NMDAR-LTP was considered the only LTP at CA3-CA1 synapses for several decades, many scientists viewed all LTP-related mechanisms through the prism of the silent synapse hypothesis. Therefore, temporally distinct forms of LTP were viewed as strictly postsynaptic LTPs with different requirements for protein synthesis (Raymond 2007). Most recent data have shown that LTP at CA3-CA1 synapses is compound in nature and consists of a rapidly developing postsynaptic component and slowly developing presynaptic component (Bayazitov and others, 2007). The degrees of expression of these 2 components depend on stimulation activity; therefore, they are inherently variable across studies because of the different induction protocols used. Low-frequency tetanizations (25-50 Hz) or a brief version of TBS induces postsynaptic NMDAR-LTP. Increased frequency of tetanization or duration of TBS “contaminates” this form of LTP with L-VGCC–dependent presynaptic LTP.
Given that most laboratories adopted 100-Hz tetanization as the primary induction protocol for their LTP studies, we should assume that those groups studied compound LTP. Therefore, temporally distinct phases of LTP associated with protein synthesis requirements in the postsynaptic neuron should be viewed as slow presynaptic phase and fast postsynaptic phase, with their own protein synthesis requirements. Given that postsynaptic LTP develops and declines quickly, and presynaptic LTP develops slowly and is sustained for a long period, we believe that presynaptic LTP is more sensitive to the production of new proteins. Because delivery of new proteins most likely depends on active transport from the soma along neurites, the focus of research in this area should be shifted toward axonal transport rather than dendritic transport.
Protein synthesis has also been implicated in morphological changes during LTP. Swelling of dendritic spines during LTP was detected via electron microscopy (Fifkova and Van 1977). Initially, dendritic spine swelling (or dendritic spine expansion) was proposed as a phenomenon associated with the late phase of LTP, which depends on protein synthesis (Fifkova and others, 1982). However, recent MPLSM data indicate that spine expansion occurs within 2 min after LTP induction (Lang and others, 2004). Upon rediscovery, spine expansion was immediately called a “structural basis of LTP” (Matsuzaki and others, 2001), again assuming that LTP at CA3-CA1 synapses is postsynaptic in nature. It was thought that the insertion of AMPARs delivers new membrane material to a dendritic spine, thereby increasing its size. However, the increased size of dendritic spines does not directly correlate with increased postsynaptic potentials during synaptic plasticity (Lang and others, 2004; Kopec and others, 2006; Wang and others, 2007). This finding suggests that spine expansion does not solely serve postsynaptic purposes. As seen by many researchers, dendritic spine expansion during LTP consists of 2 temporal phases, a transient phase and a gradual phase. The transient phase is the most spectacular part of dendritic spine expansion in which the spine may grow 4-fold in 2 min and then slowly decline (Lang and others, 2004). This slow decline, which lasts from 20 min to many hours, is the gradual phase (Lang and others, 2004; Matsuzaki and others, 2004; Tanaka and others, 2008).
Protein synthesis is not involved in mechanisms of spine expansion when LTP is induced by tetanization or glutamate uncaging at dendritic spines (Lang and others, 2004; Tanaka and others, 2008). The only evidence that protein synthesis is required for the gradual phase of dendritic spine enlargement came from a study in which LTP was induced by pairing glutamate uncaging with postsynaptic spikes (Tanaka and others, 2008). These data suggest that, in most cases, during LTP, protein synthesis is not required postsynaptically. Therefore, we assume that the role of protein synthesis is to sustain presynaptic LTP. Further experiments using direct presynaptic tools are required to either confirm or refute this hypothesis.
Spine expansion, especially during its robust transient phase, may mechanically affect the entire neuropil, including the synaptic cleft, which subsequently alters the concentrations of glutamate and extracellular modulators of synaptic function that influence LTP. But most importantly, the mechanical force exerted by dendritic spines may affect the cognate presynaptic terminal, thereby providing a link between postsynaptic and presynaptic LTP. The idea of a retrograde messenger mechanism was proposed at the very beginning of the LTP era. The postsynaptic induction of all LTPs at CA3-CA1 synapses (Lynch and others, 1983a; Grover and Teyler 1990) and partial presynaptic expression of LTP, by definition, requires a retrograde messenger. However, the identification of this mechanism was delayed, mostly because of the lack of adequate tools to directly gauge changes in presynaptic function during LTP. Initially, a chemical retrograde messenger was proposed. Several molecules (nitric oxide, BDNF, adhesion molecules, etc.) have been suggested (Fitzsimonds and Poo 1998); however, despite numerous efforts to prove their role in LTP, the presence of a retrograde messenger remains uncertain.
A perfect candidate for retrograde signaling during LTP must meet 3 requirements: (1) the retrograde signal must originate from the postsynaptic CA1 neuron; (2) it must transfer information from the activated dendritic spine to the presynaptic terminal; and (3) the signal must be spatially and temporally specific to enhance the function of the cognate presynaptic terminal. Dendritic spine expansion meets 2 requirements for a retrograde signal: Spine expansion originates from the postsynaptic neuron, and it is temporally locked to LTP induction and spatially restricted to a single synapse. However, several gaps in our knowledge must be filled before we claim that spine expansion is the retrograde messenger during LTP. Presynaptic terminals should be shown to possess mechanosensitive receptors that are activated by mechanical force during spine expansion, and this mechanotransduction mechanism should enhance neurotransmitter release. It is also conceivable that during this process mechanotransduction is accompanied by chemical mechanisms. We are confident that the rapid development of novel imaging tools will soon provide answers to these questions.
Future directions
The recent development of novel presynaptic probes and imaging technologies has enabled us to dissect the presynaptic and postsynaptic components of LTP. Therefore, direct studies of the molecular mechanisms of LTP are now possible, on both sides of a synapse. The availability of these new methods and tools also directs the field to revisit the roles of molecules implicated in LTP by electrophysiological studies. We now look forward to studies that will examine LTP as a convolution of well-coordinated presynaptic and postsynaptic processes that independently strengthen the synapse.
Acknowledgments
We thank Angela McArthur for editing the manuscript. This work was supported in part by the Whitehall Foundation, National Institute of Mental Health (R01MH079079), National Cancer Institute Cancer Center Support (P30 CA021765), and by the American Lebanese Syrian Associated Charities (S.S.Z.). S.S.Z. is a Searle Scholar.
References
- Araki R, Sakagami H, Yanagawa Y, Hikima T, Ishizuka T, Yawo H. Transgenic mouse lines expressing synaptopHluorin in hippocampus and cerebellar cortex. Genesis. 2005;42:53–60. doi: 10.1002/gene.20125. [DOI] [PubMed] [Google Scholar]
- Bagal AA, Kao JP, Tang CM, Thompson SM. Long-term potentiation of exogenous glutamate responses at single dendritic spines. Proc Natl Acad Sci U S A. 2005;102:14434–9. doi: 10.1073/pnas.0501956102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaji J, Ryan TA. Single-vesicle imaging reveals that synaptic vesicle exocytosis and endocytosis are coupled by a single stochastic mode. Proc Natl Acad Sci U S A. 2007;104:20576–81. doi: 10.1073/pnas.0707574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman DM, Good MA, Butcher SP, Ramsay M, Morris RG. Distinct components of spatial learning revealed by prior training and NMDA receptor blockade. Nature. 1995;378:182–6. doi: 10.1038/378182a0. [DOI] [PubMed] [Google Scholar]
- Baudry M, Lynch G. Remembrance of arguments past: how well is the glutamate receptor hypothesis of LTP holding up after 20 years? Neurobiol Learn Mem. 2001;76:284–97. doi: 10.1006/nlme.2001.4023. [DOI] [PubMed] [Google Scholar]
- Bayazitov IT, Richardson R, Fricke RG, Zakharenko SS. Slow presynaptic and fast postsynaptic components of compound long-term potentiation. J Neurosci. 2007 doi: 10.1523/JNEUROSCI.3077-07.2007. in print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–9. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–3. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–56. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–4. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- Borroni AM, Fichtenholtz H, Woodside BL, Teyler TJ. Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J Neurosci. 2000;20:9272–6. doi: 10.1523/JNEUROSCI.20-24-09272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–79. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Cavazzini M, Bliss T, Emptage N. Ca2+ and synaptic plasticity. Cell Calcium. 2005;38:355–67. doi: 10.1016/j.ceca.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol. 1996;76:3038–47. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- Collingridge GL. The induction of N-methyl-D-aspartate receptor-dependent long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003;358:635–41. doi: 10.1098/rstb.2002.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–6. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A, Bliss TV. Optical quantal analysis reveals a presynaptic component of LTP at hippocampal Schaffer-associational synapses. Neuron. 2003;38:797–804. doi: 10.1016/s0896-6273(03)00325-8. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Anderson CL, Young SJ, Van HA. Effect of anisomycin on stimulation-induced changes in dendritic spines of the dentate granule cells. J Neurocytol. 1982;11:183–210. doi: 10.1007/BF01258243. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Van HA. Long-lasting morphological changes in dendritic spines of dentate granular cells following stimulation of the entorhinal area. J Neurocytol. 1977;6:211–30. doi: 10.1007/BF01261506. [DOI] [PubMed] [Google Scholar]
- Fitzsimonds RM, Poo MM. Retrograde signaling in the development and modification of synapses. Physiol Rev. 1998;78:143–70. doi: 10.1152/physrev.1998.78.1.143. [DOI] [PubMed] [Google Scholar]
- Foster TC, McNaughton BL. Long-term enhancement of CA1 synaptic transmission is due to increased quantal size, not quantal content. Hippocampus. 1991;1:79–91. doi: 10.1002/hipo.450010108. [DOI] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–86. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–9. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Hebb DO. The Organization of Behaviour. New York: Wiley; 1949. [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–34. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Katz B. Quantal mechanism of neural transmitter release. Science. 1971;173:123–6. doi: 10.1126/science.173.3992.123. [DOI] [PubMed] [Google Scholar]
- Kay AR, Alfonso A, Alford S, Cline HT, Holgado AM, Sakmann B, et al. Imaging synaptic activity in intact brain and slices with FM1-43 in C. elegans, lamprey, and rat. Neuron. 1999;24:809–17. doi: 10.1016/s0896-6273(00)81029-6. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–9. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Lamsa KP. Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci. 2007;8:687–99. doi: 10.1038/nrn2207. [DOI] [PubMed] [Google Scholar]
- Lang C, Barco A, Zablow L, Kandel ER, Siegelbaum SA, Zakharenko SS. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc Natl Acad Sci U S A. 2004;101:16665–70. doi: 10.1073/pnas.0407581101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Wong D, Lynch G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986;368:347–50. doi: 10.1016/0006-8993(86)90579-2. [DOI] [PubMed] [Google Scholar]
- Li Z, Burrone J, Tyler WJ, Hartman KN, Albeanu DF, Murthy VN. Synaptic vesicle recycling studied in transgenic mice expressing synaptopHluorin. Proc Natl Acad Sci U S A. 2005;102:6131–6. doi: 10.1073/pnas.0501145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–4. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lomo T. Frequency potentiation of excitatory synaptic activity in the dentate area of the hippocampal formation. Acta Physiol Scand. 1966;68:128. [Google Scholar]
- Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983b;305:719–21. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983a;305:719–21. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Mainen ZF, Hayashi Y. LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol. 2000;10:352–7. doi: 10.1016/s0959-4388(00)00099-4. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–80. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–92. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–6. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–5. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–92. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SL, Teyler TJ. Electrical stimuli patterned after the theta-rhythm induce multiple forms of LTP. J Neurophysiol. 2001;86:1289–96. doi: 10.1152/jn.2001.86.3.1289. [DOI] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–6. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann N Y Acad Sci. 1999;868:515–25. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- Olds J. Learning and the hippocampus. Rev Can Biol. 1972;31:Suppl–38. [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Choi S, Tsien RW. Visualization of synaptic activity in hippocampal slices with FM1-43 enabled by fluorescence quenching. Neuron. 1999;24:803–8. doi: 10.1016/s0896-6273(00)81028-4. [DOI] [PubMed] [Google Scholar]
- Raymond CR. LTP forms 1, 2 and 3: different mechanisms for the “long” in long-term potentiation. Trends Neurosci. 2007;30:167–75. doi: 10.1016/j.tins.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Rozov A, Burnashev N. Polyamine-dependent facilitation of postsynaptic AMPA receptors counteracts paired-pulse depression. Nature. 1999;401:594–8. doi: 10.1038/44151. [DOI] [PubMed] [Google Scholar]
- Ryan TA. Presynaptic imaging techniques. Curr Opin Neurobiol. 2001;11:544–9. doi: 10.1016/s0959-4388(00)00247-6. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–24. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Smith SJ, Reuter H. The timing of synaptic vesicle endocytosis. Proc Natl Acad Sci U S A. 1996;93:5567–71. doi: 10.1073/pnas.93.11.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Can molecules explain long-term potentiation? Nat Neurosci. 1999;2:597–604. doi: 10.1038/10154. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J. 2000;79:2199–208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci. 1994;14:5325–37. doi: 10.1523/JNEUROSCI.14-09-05325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–84. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Heinemann U, Muller W. FM1-43 imaging reveals cGMP-dependent long-term depression of presynaptic transmitter release. J Neurosci. 2001;21:RC167. doi: 10.1523/JNEUROSCI.21-19-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Zhang XL, Muller W. Imaging LTP of presynaptic release of FM1-43 from the rapidly recycling vesicle pool of Schaffer collateral-CA1 synapses in rat hippocampal slices. Eur J Neurosci. 2005;22:2451–61. doi: 10.1111/j.1460-9568.2005.04437.x. [DOI] [PubMed] [Google Scholar]
- Tabares L, Ruiz R, Linares-Clemente P, Gaffield MA, varez de TG, Fernandez-Chacon R, et al. Monitoring synaptic function at the neuromuscular junction of a mouse expressing synaptopHluorin. J Neurosci. 2007;27:5422–30. doi: 10.1523/JNEUROSCI.0670-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–7. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–38. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, et al. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Wang XB, Yang Y, Zhou Q. Independent expression of synaptic and morphological plasticity associated with long-term depression. J Neurosci. 2007;27:12419–29. doi: 10.1523/JNEUROSCI.2015-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigstrom H, Gustafsson B. Postsynaptic control of hippocampal long-term potentiation. J Physiol (Paris) 1986;81:228–36. [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, et al. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron. 2003;39:975–90. doi: 10.1016/s0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–7. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Cao YQ, Tsien RW. Quantum dots provide an optical signal specific to full collapse fusion of synaptic vesicles. Proc Natl Acad Sci U S A. 2007a;104:17843–8. doi: 10.1073/pnas.0706906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kaji N, Tokeshi M, Baba Y. Nanobiotechnology: quantum dots in bioimaging. Expert Rev Proteomics. 2007b;4:565–72. doi: 10.1586/14789450.4.4.565. [DOI] [PubMed] [Google Scholar]