

Summary
The BimEL tumor suppressor is a potent pro-apoptotic BH3-only protein. We found that in response to survival signals BimEL was rapidly phosphorylated on three serine residues in a conserved degron, facilitating binding and degradation via the F-box protein βTrCP. Phosphorylation of the BimEL degron was executed by Rsk1/2 and promoted by the Erk1/2-mediated phosphorylation of BimEL on Ser69. Compared to wild type BimEL, a BimEL phosphorylation mutant unable to bind βTrCP was stabilized and consequently potent at inducing apoptosis by the intrinsic mitochondrial pathway. Moreover, although non-small cell lung cancer (NSCLC) cells often become resistant to gefitinib (a clinically relevant tyrosine kinase inhibitor that induces apoptosis through BimEL), silencing of either βTrCP or Rsk1/2 resulted in BimEL-mediated apoptosis of both gefitinib-sensitive and gefitinib-insensitive NSCLC cells. Our findings reveal that βTrCP promotes cell survival in cooperation with the ERK-RSK pathway, by targeting BimEL for degradation.
Introduction
Bim (Bcl-2 Interacting Mediator of cell death) is a powerful, proapoptotic member of the Bcl-2 protein family expressed mainly in hematopoietic, epithelial, neuronal, and germ cells (O’Reilly et al., 2000). Alternative mRNA splicing generates three major isoforms: short (BimS), long (BimL), and extra long (BimEL), with BimEL the predominant isoform in most tissues (O’Connor et al., 1998; O’Reilly et al., 2000). Bim plays a key role in linking stress-induced signals to the intrinsic (mitochondrial) apoptotic pathway. Upon exposure to stress, such as growth factor deprivation, Bim activates proapoptotic Bak and Bax that, in turn, permeabilize the mitochondrial membrane, causing the release of cytochrome C and the consequent activation of caspases to cause programmed cell death. Mechanistically, Bim is thought to activate Bax and Bak by direct binding and/or by binding and inhibiting anti-apoptotic members of the Bcl2 family (Mcl1 and Bcl-XL), which restrain Bak and Bax (Fletcher and Huang, 2008).
Several studies suggest that Bim functions as a tumor suppressor. In mice, inactivation of one allele of Bim accelerates Myc-induced B cell leukemia (Egle et al., 2004). In human cancers, Bim is eliminated via various mechanisms to provide a growth advantage to the tumor cells. Homozygous deletions of the Bim locus have been reported in mantle cell lymphomas, and methylation of the Bim promoter is found in certain Burkitt’s lymphomas and diffuse large B-cell lymphomas (Mestre-Escorihuela et al., 2007; Tagawa et al., 2005). Moreover, in a manner similar to other tumor suppressor proteins, such as p27 and p53, Bim levels are decreased in transformed cells via enhanced protein degradation, particularly when the ERK pathway is constitutively activated. For example, in transformed epithelial cells (both in culture and in animals), paclitaxel-induced apoptosis is mediated by Bim (Tan et al., 2005). When the H-Ras/ERK pathway is activated in tumor cells, BimEL is eliminated by proteasomal degradation, and cells become refractory to paclitaxel. Treatment with bortezomib, a proteasome inhibitor, restores BimEL levels, thereby re-sensitizing cells to paclitaxel.
Bim levels are also low in non-small cell lung cancer (NSCLC) cells harboring activating EGFR mutations (Costa et al., 2007; Cragg et al., 2007; Deng et al., 2007a; Gong et al., 2007). Inhibition of EGFR tyrosine kinase activity using drugs such as gefitinib results in BimEL accumulation and, consequently, induction of apoptosis. Similarly, BimEL accumulation mediates imatinib-induced cell death of Bcr/Abl+ leukemic cells (Belloc et al., 2007; Kuroda et al., 2006).
The proteasomal degradation of BimEL is dependent on phosphorylation by Erk1/2 on a specific serine (Ser69 in human) (Hubner et al., 2008; Ley et al., 2003; Ley et al., 2004; Ley et al., 2005; Luciano et al., 2003). However, despite the importance of BimEL in determining cell fate and the fact that its degradation enables tumor cells to escape chemotherapy-induced apoptosis, the cellular machinery responsible for BimEL degradation has not yet been identified. The study described herein identifies the ubiquitin ligase and kinases that target BimEL for proteasomal degradation, elucidating a critical control mechanism for the apoptotic response.
Results
Degradation of BimEL is promoted by ERK-dependent phosphorylation on Ser69. As phosphorylation often targets proteins to SCF (Skp1-Cullin1-F-box protein) ubiquitin ligase complexes, we asked whether BimEL binds to Cul1 in HEK293 cells. We found that in the presence of PMA, an activator of the ERK pathway, Cul1, but not Cul2, was able to co-immunoprecipitate endogenous BimEL (Fig. S1). We then investigated which F-box protein specifically targets BimEL to the SCF. Screening of the FBXW (F-box, WD repeat) family proteins revealed that endogenous BimEL specifically interacts with βTrCP1 and βTrCP2 (Fig. 1A), two paralogous F-box proteins that (to date) share identical biochemical properties and substrates (Frescas and Pagano, 2008). (The term βTrCP will refer to both, unless specified.) The interaction of endogenous BimEL and βTrCP1 was also observed (Fig. S2). Addition of PMA to HEK293 cells promoted the binding of endogenous BimEL to βTrCP1 and binding was enhanced when the proteasome inhibitor MG132 blocked BimEL degradation (Fig. 1B). However, the binding of BimEL to βTrCP1 in the presence of PMA and MG132 was strongly reduced when PMA-induced ERK activation was inhibited with UO126, a MEK inhibitor.
Figure 1. βTrCP controls the degradation of BimEL.
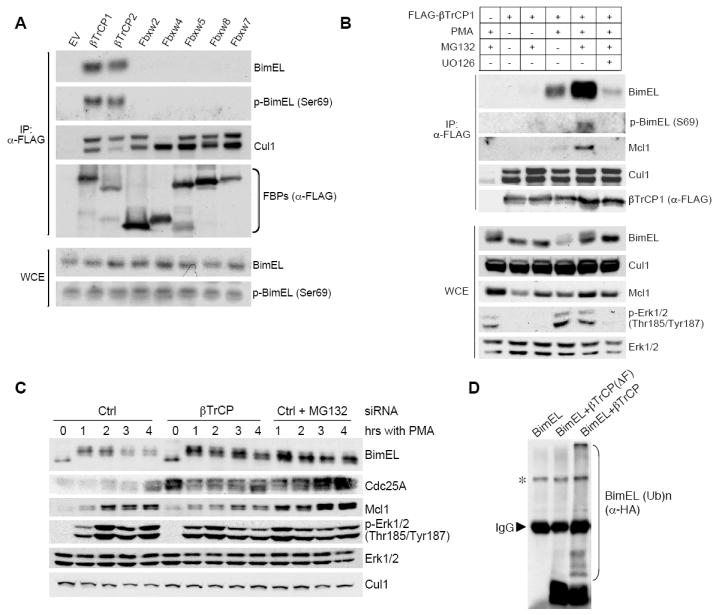
(A) BimEL specifically interacts with βTrCP. HEK293 cells were transfected with the indicated FLAG-tagged F-box proteins (FBPs) or an empty vector (EV). Twenty-four hours post-transfection, cells were treated for three hours with PMA and MG132 before harvesting. Whole cell extracts (WCE) were subjected to immunoprecipitation (IP) with anti-FLAG resin (α-FLAG) and immunobloting for the indicated proteins.
(B) The interaction of βTrCP1 with BimEL is promoted by PMA and MG132 and is inhibited by a MEK inhibitor. HEK293 cells were transfected with FLAG-tagged βTrCP or an empty vector. Twenty-four hours post-transfection, cells were treated with the indicated drugs for three hours before harvesting. Whole cell extracts (WCE) were subjected to immunoprecipitation (IP) with anti-FLAG resin (α-FLAG) and immunobloting for the indicated proteins.
(C) βTrCP silencing stabilizes BimEL. HEK293 cells were treated with a control siRNA (Ctrl) or an siRNA targeting βTrCP. Forty-eight hours post-transfection, cells were treated with either PMA alone or PMA and MG132 for the indicated times. Extracts were then immunoblotted for the indicated proteins. The accumulation of Cdc25A (a known βTrCP substrate) demonstrates efficient βTrCP knockdown [also confirmed by RT-PCR (Fig. S10)].
(D) HEK293T cells were transfected with HA-tagged BimEL, Skp1, Cul1, and Roc1 in the presence of an empty vector (EV), FLAG-tagged βTrCP or FLAG-tagged βTrCP(ΔF-box). After immunopurification with anti-FLAG resin, in vitro ubiquitylation of BimEL was performed. Samples were analyzed by immunoblotting with an anti-HA antibody. The asterisk denotes a non-specific band.
These results indicated that the binding of BimEL to βTrCP is stimulated by the activation of Erk1/2, suggesting (consistent with previous evidence that Erk1/2 induce BimEL degradation) that βTrCP controls the phosphorylation-dependent degradation of BimEL. We therefore investigated this hypothesis by reducing the expression of both βTrCP1 and βTrCP2 in HEK293 cells using a previously validated siRNA. Fig. 1C shows that βTrCP silencing counteracted the effect of PMA on BimEL degradation, stabilizing BimEL. Finally, immunopurified βTrCP1, but not an inactive βTrCP1(ΔF-box) mutant, induced the in vitro ubiquitylation of BimEL (Fig. 1D), supporting the hypothesis that the effect of βTrCP on BimEL is direct.
βTrCP binds its substrates via phosphorylated residues in a conserved degron, typically the consensus sequence DpSGXXpS. In searching for a βTrCP degron in human BimEL, we found the conserved motif 91RSSSGYFSFD100 (Fig. S3A), in which the charge on the aspartic acid is potentially substituted by phosphorylated Ser93. This sequence fits into the three-dimensional structural space of the βTrCP1 substrate-binding surface, similar to the phospho-degron of βcatenin, a known substrate of βTrCP (Figs. S3C-E). To test whether BimEL binds βTrCP via this motif, we generated a number of serine to alanine mutants (Fig. S3B) and tested their binding to endogenous βTrCP1. Single mutations of Ser93, Ser94, and Ser98 to Ala or a double Ser94/98Ala mutation (S94/98A) abrogated the interaction between BimEL and endogenous βTrCP1, although they did not abolish BimEL binding to endogenous Mcl1 (Fig. 2A). Mutation of Ser92 did not inhibit BimEL binding to either βTrCP1 or Mcl1.
Figure 2. Identification of the BimEL degron.

(A) Ser93, Ser94, and Ser98 are required for the interaction of BimEL with βTrCP1. HEK293 cells were transfected with an empty vector (EV), HA-tagged βcatenin (positive control), HA-tagged wild type BimEL, or the indicated HA-tagged BimEL mutants. Twenty-four hours post-transfection, cells were treated for three hours with PMA and MG132. Whole cell extracts (WCE) were subjected to immunoprecipitation (IP) with anti-HA resin (α-HA) and immunobloting for the indicated proteins.
(B) The BimEL degron requires phosphorylation to bind βTrCP1. 35S-labeled, in vitro-translated βTrCP1, Fbxw2, and Fbxw4 were used in binding reactions with beads coupled to the BimEL peptide 88CLSRSSSGYFSFD100 (lane 2) or the phosphopeptide 88CLSRSpSpSGYFpSFD100 (lane 3). Beads were washed with lysis buffer, and bound proteins were eluted and subjected to SDS-PAGE and autoradiography. The first lane shows 10% of the in vitro-translated protein inputs.
(C) In vivo phoshorylation of BimEL on Ser93/94/98 is induced by mitogens. HEK293 cells were serum deprived (SD) for 24 hours (lane 1), and then either serum (S) (lanes 2-3) or PMA (lane 4-5) was added for 20 minutes in the absence or presence of UO126, as indicated. Whole cell extracts (WCE) were subjected to immunoprecipitation (IP) with an anti-Bim antibody and immunobloting for the indicated proteins.
(D) Ser69 promotes phoshorylation of BimEL on Ser93/94/98. HEK293 cells were transfected with HA-tagged wild type (WT) BimEL (lane 1-3), HA-tagged BimEL(S69A) (lane 4), or HA-tagged BimEL(S94/98A) (lane 5). Cells were serum deprived (SD) for 24 hours (lane 1), and then either serum (S) (lane 2) or PMA (lane 3-5) was added for 20 minutes. Whole cell extracts (WCE) were subjected to immunoprecipitation (IP) with anti-HA resin (α-HA) and immunobloting for the indicated proteins.
(E) Cytokines induce phosphorylation of BimEL on Ser93/94/98. Activated primary mouse T cells were deprived of IL2 for 5 hours and Ba/F3 and FL5.12 cells were deprived of IL3 for 5 hours. Then cells were stimulated for 15 minutes with IL2 or IL3, in the absence or presence of UO126, as indicated. Whole cell extracts (WCE) were subjected to immunoprecipitation with an anti-Bim antibody and immunobloting for the indicated proteins.
To investigate whether phosphorylation plays a role in the interaction with βTrCP, we used immobilized, synthetic peptides spanning the candidate phospho-degron (aa 88-100). While a peptide containing phosphorylated Ser93, Ser94, and Ser98 efficiently bound βTrCP1 (but not FBXW2 and FBXW4), a corresponding, non-phosphorylated peptide was unable to bind βTrCP1 (Fig. 2B), in agreement with the idea that phosphorylation of Ser93, Ser94, and Ser98 directly mediates the interaction with βTrCP. Furthermore, only in vitro phosphorylated BimEL binds βTrCP2 (see later, Fig. 3F).
Figure 3. Rsk1/2 control BimEL stability.

(A) Inhibition of Rsk1/2 reduces phosphorylation of BimEL on Ser93/94/98. HEK293 were serum deprived (SD) and then treated for 15 minutes with PMA in the absence or presence of FMK, as indicated. Whole cell extracts (WCE) were subjected to immunoprecipitation with an anti-Bim antibody and immunobloting for the indicated proteins.
(B) Inhibition of Rsk1/2 reduces BimEL binding to βTrCP1. HEK293 cells were transfected with FLAG-tagged βTrCP1. Twenty-four hours post-transfection, cells were treated with PMA and, where indicated, either FMK or LY294002 (LY) was added. Whole cell extracts (WCE) were subjected to immunoprecipitation (IP) using FLAG resin (α-FLAG) and immunobloting for the indicated proteins.
(C) HEK293 cells were treated with a control siRNA (Ctrl) or an siRNA targeting both Rsk1 and Rsk2 mRNAs. Cells were deprived of serum for 24 hours (-) and then activated with PMA (+) for 20 minutes. Cell extracts were subjected to immunoprecipitation with an anti-Bim antibody and immunobloting for the indicated proteins.
(D) Silencing Rsk1/2 stabilizes BimEL. HEK293 cells were treated with a control siRNA (Ctrl) or an siRNA targeting both Rsk1 and Rsk2. Forty-eight hours after siRNA treatment, cells were treated with PMA for the indicated times. Extracts were immunoblotted for the indicated proteins.
(E) Rsk1 phosphorylates BimEL in vitro. Recombinant, purified BimEL was incubated for 30 minutes with ATP and the indicated purified kinases. Reaction products were subjected to immunoblotting for the indicated proteins.
(F) In vitro binding of BimEL to βTrCP2 is dependent on Rsk1. Recombinant, purified wild type BimEL and BimEL(S94/98A) were phosphorylated as in (E) and incubated with in vitro translated, FLAG-tagged βTrCP2 (except sample shown in the last lane). Proteins were immunoprecipitated (IP) using FLAG resin (α-FLAG), and immunoblotting for the indicated proteins was performed.
To further investigate the role of BimEL phosphorylation, we generated a phospho-specific antibody against the 88CLSRSpSpSGYFpSFD100 peptide with phospho-serines at positions 93, 94, and 98. This antibody recognized wild type BimEL but not a BimEL(S93/94/98A) mutant, while single and double amino acid BimEL mutants displayed decreasing levels of detection (Fig. S4A), indicating that all three serine residues are phosphorylated and contribute to recognition by this antibody. In addition, λ-phosphatase treatment of immunopurified BimEL abolished BimEL recognition by the phospho-specific antibody (Fig. S4B). Using this reagent, we tested BimEL phosphorylation under different conditions in vivo. We found that both endogenous and exogenous BimEL were rapidly phosphorylated in HEK293 cells in response to mitogenic stimulation (serum or PMA), which correlated with ERK activation (Figs. 2C,D). In contrast, BimEL was not phosphorylated in serum-starved cells or UO126-treated cells. Similarly, cytokines also promoted phosphorylation of BimEL on its degron, as shown with IL2 in primary mouse T cells or IL3 in FL5.12 and BaF/3 cell lines (Fig. 2E).
Interestingly, mutation of Ser69 to Ala strongly inhibited phosphorylation of the BimEL degron despite PMA treatment (Fig. 2D). Accordingly, the BimEL(S69A) mutant bound less efficiently to endogenous βTrCP1 (Fig. 2A). These results indicate that phosphorylation on Ser69 promotes the phosphorylation of Ser93, Ser94, and Ser98 (see also later, Figs. 3E and S7A). Such bimodal activation has been previously demonstrated for other substrates of βTrCP that require combinatorial phosphorylation by two cooperative kinases (Hunter, 2007).
Erk1/2-mediated phosphorylation of Ser69 is well-established, so we pursued the identification of the kinase that phosphorylates the BimEL degron. GPS (Group Based Prediction System), a kinase prediction program (Xue et al., 2008), detected consensus sites for Rsk1/2 and S6k1/2 in the BimEL degron. To gain insight into the kinase involved in BimEL phosphorylation, we used pharmacological inhibitors and found that FMK [an RSK inhibitor (Cohen et al., 2005)], but not LY294002 (a PI-3K inhibitor, which, consequently, inhibits also S6k1/2), strongly reduced both the phosphorylation of BimEL on Ser93/Ser94/Ser98 and the binding of BimEL to βTrCP1 (Figs. 3A,B). Similarly, another RSK inhibitor, BI-D1870 (Sapkota et al., 2007) reduced BimEL-βTrCP1 interaction too (not shown). We also found that Rsk1, but not S6k1, was co-immunoprecipitated with BimEL in vivo (Fig. S5). All these results suggest that Rsk1/2 phosphorylate the BimEL degron. Accordingly, knockdown of both Rsk1 and Rsk2 with two validated siRNAs inhibited the PMA-induced phosphorylation of Ser93/Ser94/Ser98 in both endogenous and exogenous BimEL (Figs. 3C and S6A). Importantly, downregulation of Rsk1/2 inhibited BimEL degradation (Fig. 3D) and BI-D1870 treatment induced BimEL accumulation (Fig. S6B).
To test whether Rsk1 can directly phosphorylate the BimEL degron, we performed an in vitro kinase assay using recombinant, bacterially-expressed, purified BimEL and kinases. Rsk1 phosphorylated the degron of BimEL, as shown by the appearance of a slow migrating band and recognition by our phospho-specific antibody, and this event was promoted by Erk1 (Fig. 3E). In contrast, neither Erk1 nor S6k1 alone was able to induce phosphorylation of BimEL on Ser93/Ser94/Ser98. Addition of Erk1 did not increase the activating phosphorylation of Rsk1 (bottom panel of Fig. 3E), and when Erk1 was first used to phosphorylate BimEL (and washed away prior to Rsk1 addition), the stimulation by ERK was observed for wild type BimEL but not for BimEL(S69A) (Fig. S7A). Thus, the enhancement of the Rsk1-dependent phosphorylation of BimEL is not due to the activation of Rsk1 by Erk1; instead it is promoted by the phosphorylation of BimEL on Ser69 by Erk1. We also used single serine mutants and found that Rsk1 was able to phosphorylate (in a ERK-dependent manner) each of the three serines in the BimEL degron (Figs. S7B-D), suggesting that, like other established substrates (Anjum and Blenis, 2008), Rsk1/2 target multiple residues in BimEL. Finally, in agreement with the phosphorylation results, the in vitro binding of βTrCP2 to phosphorylated BimEL was dependent on Rsk1 and stimulated by Erk1 (Fig. 3F).
The above data strongly support a model in which phosphorylation of BimEL on Ser93/Ser94/Ser98 mediates binding to βTrCP and degradation via SCFβTrCP. Therefore, failure to bind βTrCP should result in stabilization of BimEL. To test this hypothesis, we transfected wild type BimEL or BimEL(S94/98A) into HEK293 cells and subsequently treated with PMA and cycloheximide. As predicted, in contrast to wild type BimEL, BimEL(S94/98A), which does not bind βTrCP (Fig. 2A), was not degraded upon PMA treatment (Fig. 4A). Importantly, expression of BimEL(S94/98A) in immortalized Bim-/- mouse embryo fibroblasts (MEFs) triggered a much more robust apoptotic response than that obtained by expressing wild type BimEL or even BimEL(S69A) (Fig. 4B). Neither wild type BimEL or BimEL mutants induced apoptosis in immortalized Bak-/-;Bax-/- MEFs, confirming that BimEL(S94/98A)-dependent cell death occurs via the intrinsic mitochondrial pathway.
Figure 4. SCFβTrCP- and Rsk-mediated degradation of BimEL controls the apoptotic response.

(A) Mutation of Ser94/98 stabilizes BimEL despite ERK activation. Cells were transfected with either wild type BimEL or BimEL(S94/98A) mutant. Twenty-four hours post-transfection, cells were treated with PMA and cyclohexamide (CHX) for the indicated times before immunoblotting for the indicated proteins.
(B) Mutation of Ser94/98 augments the apoptotic activity of BimEL. Bim-/- and Bax-/-;Bak-/- MEFs were infected with a retrovirus expressing either wild type BimEL or different BimEL mutants. Apoptosis was measured 48 hours following infection using propidium iodide and Annexin-V staining, with flowcytometric analysis (n = 3, ± SD).
(C) Silencing Rsk1/2 or βTrCP promotes Bim-dependent apoptosis in primary human T cells. Human T cells were transfected twice with the indicated siRNAs and collected 24 hours thereafter. Apoptosis (left panel) was determined as in (B), and cell extracts were analyzed by immunoblotting for the indicated proteins (right panel).
(D) Silencing Rsk1/2 or βTrCP promotes apoptosis in NSCLC cells independent of their sensitivity to gefitinib. HCC827 and H1650 cells were transfected with the indicated siRNAs and collected at the indicated times. Apoptosis was determined as in (B) (n = 3, ± SD).
(E) Silencing Bim rescues apoptosis induced by downregualtion of Rsk1/2 or βTrCP. HCC827 and H1650 cells were treated with gefitinib for 24 hours or transfected with the indicated siRNAs and collected 48 hours thereafter. Apoptosis was determined as in (B) (n = 3, ± SD).
(F) HCC827 and H1650 cells, treated as in (E), were collected, and cell extracts were analyzed by immunoblotting for to the indicated proteins.
We also asked whether RSK and βTrCP mediate survival of primary human CD4+ T cells. Fig. 4C shows that the silencing of either Rsk1/2 or βTrCP in these cells resulted in BimEL accumulation and BimEL-mediated apoptosis (as demonstrated by the return of cell death to background levels when BimEL was downregulated together with Rsk1/2 or βTrCP). Accordingly, primary mouse T cells from wild type mice, but not from Bim-/- mice, died in response to pharmacologic inhibition of RSK (Fig. S8).
To further study the biological significance of the βTrCP- and Rsk1/2-mediated degradation of BimEL, we used non-small cell lung cancer (NSCLC) cells that harbor activating mutations in the Epidermal Growth Factor Receptor (EGFR). Initially, clinically-relevant inhibitors of EGFR tyrosine kinase activity, such as gefitinib, trigger a BimEL-dependent apoptotic response in NSCLCs with EGFR mutations (Costa et al., 2007; Cragg et al., 2007; Deng et al., 2007b; Gong et al., 2007). However, these tumors eventually become resistant to tyrosine kinase inhibitors and lose their ability to die via BimEL upregulation. We examined two EGFR mutant NSCLC cell lines, HCC827 (which are known to be sensitive to gefitinib) and H1650 (which are not) (see also Fig. 4E). Significantly, in the absence of gefitinib, apoptosis was promoted in both HCC827 and H1650 cells when either Rsk1/2 or βTrCP were downregulated (Figs. 4D-F). When BimEL was also silenced, cell death returned to background levels (Figs. 4E,F). Notably, up-regulation of BimEL correlated with the induction of apoptosis in H1650 and HCC827 cells. These experiments showed that restoration of BimEL levels in cells harboring activating mutations in EGFR promotes apoptosis in both gefitinib-sensitive and gefitinib-insensitive NSCLC cells.
Discussion
Despite the importance of BimEL in controlling apoptotic responses, the ubiquitin ligase responsible for its degradation had remained elusive. Two ligases, c-Cbl and a Cul2 complex, have been proposed to target BimEL (Akiyama et al., 2003; Zhang et al., 2008) but these findings have not been confirmed by others (El Chami et al., 2005; Wiggins et al., 2007; Fig. S1). Indeed, we show here that in response to survival signals SCFβTrCP promotes the degradation of BimEL in cooperation with Erk1/2 and Rsk1/2.
Degradation of BimEL enables tumor cells to escape chemotherapy-induced apoptosis. We found that silencing of either βTrCP or Rsk1/2 induces Bim-dependent apoptosis in NSCLC cells harboring activating mutations in EGFR, irrespective of their sensitivity to gefitinib. In a clinical setting, such an increase in cell death could positively affect long-term outcomes, so our findings suggest that inhibition of RSK or βTrCP should be pursued as a rational and valid therapeutic strategy to induce apoptosis of tumor cells in NSCLC and, possibly, other malignancies. Furthermore, since BimEL degradation is dependent on Rsk1/2, but only stimulated by Erk1/2, it is expected that tumor cells that undergo BimEL-dependent death (e.g., in NSCLCs, Bcr/Abl+ leukemia’s, and certain breast cancers) may be more sensitive to RSK inhibitors than to ERK inhibitors.
In summary, we describe the biochemical and molecular details of the mechanisms controlling the degradation of BimEL in both normal and cancer cells. When cells are stimulated with mitogens, βTrCP directs the degradation of BimEL in cooperation with the ERK-RSK pathway, resulting in the inhibition of cell death. At the same time, in cooperation with the PI3K-S6K pathway, βTrCP targets Pdcd4 for degradation, allowing efficient protein synthesis and, consequently, cell growth (Dorrello et al., 2006). Thus, βTrCP coordinates cell survival and cell growth in response to mitogenic stimuli (Fig. S9).
Experimental Procedures
Biochemical methods
Extract preparation, immunoprecipitation, and immunoblotting were previously described (Dorrello et al., 2006). In vitro ubiquitylation was previously described (Busino et al., 2007).
Antibodies
Mouse monoclonal antibodies were from Invitrogen (Cul1, Erk1/2, Rsk1, Rsk2, βTrCP1), Sigma (anti-FLAG), Santa Cruz Biotechnology (Cdc25A), BD Biosciences (βcatenin) and Covance (anti-HA). Rabbit polyclonal antibodies were from Invitrogen (Cul1, Bim, Skp2), Biosource [phospho-Bim(Ser69), phospho-Erk1/2(Thr185/Tyr187)], Cell Signaling [phosho-RSK(Ser380), Bim, Caspase 3, cleaved Caspase 3], Bethyl (βTrCP1), and Santa Cruz (Mcl1). For IP of endogenous Bim we used a rat monoclonal antibody from Millipore. The phospho-specific antibody to BimEL was generated using the phosphopeptide CLSRSpSpSGYFpSFD.
Plasmids
BimEL mutants were generated using QuickChange Site-directed Mutagenesis (Stratagene). Both wild type BimEL and BimEL mutants were subcloned into the pBabe retroviral vector. All cDNAs were completely sequenced.
Transient transfections and retrovirus-mediated gene transfer
Transfections using the calcium phosphate and retrovirus-mediated gene transfer were previously described (Dorrello et al., 2006).
Apoptosis Assay
Apoptosis was assessed using AnnexinV-FITC and propidium iodide staining (BD Pharmigen).
Supplementary Material
Acknowledgments
We thank M. McMahon, D. Ryoo, and J. Skaar for suggestions and critically reading the manuscript; E. McIntush and Bethyl Laboratories for providing βTrCP1 (BL726b) antibody, and I. Aifantis, J. Blenis, S. Buonamici, T. Cardozo, C. Lee, S. Fuchs, K. Kinnally, S. Korsmeyer L. Liebes, J. Maller, W. Pao, S. Valvo, and H.G. Wang for reagents and/or suggestions. M.P. is grateful to T. M. Thor for continuous support. This work was supported by a DOD fellowship to ED, an AACR fellowship to FB, a fellowship from the America Italian Cancer Foundation and from Provincia di Benevento to DG, grants from the National Institutes of Health to M.P. (R01-GM57587, R37-CA76584, and R21-CA125173), M.L.D. (R01-AI43542), and J.T. (GM071434), and an Australian NHMRC Fellowship, an Australian NHMRC Program Grant, and an Leukemia and Lymphoma Society SCOR grant to D.C.S.H. J.T. and M.P. are Investigators with the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama T, Bouillet P, Miyazaki T, Kadono Y, Chikuda H, Chung UI, Fukuda A, Hikita A, Seto H, Okada T, et al. Regulation of osteoclast apoptosis by ubiquitylation of proapoptotic BH3-only Bcl-2 family member Bim. Embo J. 2003;22:6653–6664. doi: 10.1093/emboj/cdg635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- Belloc F, Moreau-Gaudry F, Uhalde M, Cazalis L, Jeanneteau M, Lacombe F, Praloran V, Mahon FX. Imatinib and nilotinib induce apoptosis of chronic myeloid leukemia cells through a Bim-dependant pathway modulated by cytokines. Cancer Biol Ther. 2007;6:912–919. doi: 10.4161/cbt.6.6.4101. [DOI] [PubMed] [Google Scholar]
- Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, Draetta GF, Pagano M. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316:900–904. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:1669–1679. doi: 10.1371/journal.pmed.0040315. discussion 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007;4:1681–1689. doi: 10.1371/journal.pmed.0040316. discussion 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007a;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Deng J, Shimamura T, Perera S, Carlson NE, Cai D, Shapiro GI, Wong KK, Letai A. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007b;67:11867–11875. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Chami N, Ikhlef F, Kaszas K, Yakoub S, Tabone E, Siddeek B, Cunha S, Beaudoin C, Morel L, Benahmed M, Regnier DC. Androgen-dependent apoptosis in male germ cells is regulated through the protooncoprotein Cbl. J Cell Biol. 2005;171:651–661. doi: 10.1083/jcb.200507076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JI, Huang DC. Controlling the cell death mediators Bax and Bak: puzzles and conundrums. Cell Cycle. 2008;7:39–44. doi: 10.4161/cc.7.1.5178. [DOI] [PubMed] [Google Scholar]
- Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins Skp2 and βTrCP: Tipping the scales of cancer. Nature Reviews Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, Pao W. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell. 2008;30:415–425. doi: 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907–14912. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- Ley R, Ewings KE, Hadfield K, Howes E, Balmanno K, Cook SJ. Extracellular signal-regulated kinases 1/2 are serum-stimulated “Bim(EL) kinases” that bind to the BH3-only protein Bim(EL) causing its phosphorylation and turnover. J Biol Chem. 2004;279:8837–8847. doi: 10.1074/jbc.M311578200. [DOI] [PubMed] [Google Scholar]
- Ley R, Hadfield K, Howes E, Cook SJ. Identification of a DEF-type docking domain for extracellular signal-regulated kinases 1/2 that directs phosphorylation and turnover of the BH3-only protein BimEL. J Biol Chem. 2005;280:17657–17663. doi: 10.1074/jbc.M412342200. [DOI] [PubMed] [Google Scholar]
- Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, Beltran E, Agirre X, Marugan I, Marin M, et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109:271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. Embo J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly LA, Cullen L, Visvader J, Lindeman GJ, Print C, Bath ML, Huang DC, Strasser A. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol. 2000;157:449–461. doi: 10.1016/S0002-9440(10)64557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, et al. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J. 2007;401:29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, Morishima Y, Nakamura S, Seto M. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene. 2005;24:1348–1358. doi: 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7:227–238. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Wiggins CM, Band H, Cook SJ. c-Cbl is not required for ERK1/2-dependent degradation of BimEL. Cell Signal. 2007;19:2605–2611. doi: 10.1016/j.cellsig.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0: Prediction of kinase-specific phosphorylation sites in hierarchy. Mol Cell Proteomics. 2008 doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Cheng GZ, Gong J, Hermanto U, Zong CS, Chan J, Cheng JQ, Wang LH. RACK1 and CIS mediate the degradation of BimEL in cancer cells. J Biol Chem. 2008;283:16416–16426. doi: 10.1074/jbc.M802360200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.