

Introduction
Six viral proteins play major roles in the regulation of EHV-1 gene expression: the sole immediate-early protein (IEP), four early proteins (IR2P, EICP0P, UL5P [EICP27], and IR4 [EICP22]), and the late tegument protein, α-trans-inducing factor (ETIF) (O’Callaghan and Osterrieder, 2008). The 1,487aa IEP is the major regulatory protein of EHV-1 and serves a bifunctional regulatory role by: i) independently trans-activating EHV-1 early promoters (Caughman et al., 1985; Harty et al., 1990; Smith et al., 1992, 1994); and ii) trans-repressing its own promoter (Harty and O’Callaghan, 1991; Smith et al., 1994) and some late promoters (Kim et al., 1999). The IE gene is further regulated by the IR2 protein, an amino-terminal truncated (323-1487aa) version of the IEP encoded within the IE gene (Harty and O’Callaghan, 1991; Caughman et al., 1995; Kim et al., 2006) and by the IR3 RNA that is antisense to the IE transcript (Holden et al., 1992a; Ahn et al., 2007) The IEP also functions in concert with EHV-1 early regulatory proteins IR4 (Kim et al., 1997; Derbigny et al., 2000, 2002) and UL5P (Smith et al., 1993; Zhao et al., 1995; Albrecht et al., 2004, 2005) as well as with cellular transcription factors TATA box-binding protein (TBP) and TFIIB (Jang et al., 2001; Albrecht et al., 2003; Kim et al., 2003; Kim and O’Callaghan, unpublished results) and the nucleolar shuttle protein, EAP (Kim et al., 2001). Several IEP functional domains have been characterized, including the trans-activation domain (Smith et al., 1994; Buczynski et al., 1999), a site-specific DNA binding domain (Kim et al., 1995), a nuclear localization signal (NLS; Smith et al., 1995), a serine-rich tract (SRT; Kim et al., 2001), and a TFIIB-binding domain (Jang et al., 2001). Characterization of 17 EHV-1 IE mutants revealed that these functional domains are essential for virus replication (Buczynski et al., 2005).
IR4 is an early 293aa protein that in concert with the IE or the EICP0 protein significantly enhances the trans-activation of early viral promoters (Holden et al., 1992b, 1994, 1995; Kim et al., 1997). While not a DNA binding protein, IR4 greatly enhances the DNA-binding activity of the IEP, and is able to restore the DNA-binding ability of some IEP mutants (Kim et al., 1995). Additionally, IR4 interacts with itself to form dimers and higher-ordered complexes (Derbigny et al., 2000). The exact mechanisms by which IR4 regulates viral gene expression, however, remain poorly defined.
EHV-1 IR4 is unique in that it functions as a trans-activator during lytic infection yet may trans-repress EHV-1 genes during persistent infection mediated by defective interfering particles (DIP) (Ebner and O’Callaghan, 2006, 2008). The recombination events that generate DIP genomes produce one of two unique hybrid genes consisting of 5’ sequences of IR4 fused to 3’ sequences of UL5 (Baumann et al., 1987; Yalamanchili et al., 1990; Chen et al., 1996, 1999; Ebner and O’Callaghan, 2006, 2008). Both hybrid proteins (HYB1.0 and HYB2.0) are produced in large quantities during EHV-1 persistent infection (Chen et al., 1996, Ebner and O’Callaghan, 2006), and previous studies demonstrated that the HYB1.0 protein downregulates EHV-1 gene expression (Chen et al., 1999). Recent work revealed that IR4 residues within the HYB proteins are necessary for interference with standard virus replication, a hallmark of persistent infection (Ebner and O’Callaghan, 2008).
To examine the role of the IR4 protein during lytic and persistent infections, bacterial artificial chromosome (BAC) technology was used to delete both copies of the IR4 gene from the highly pathogenic RacL11 strain of EHV-1. The resulting IR4-null virus (RacL11ΔIR4) was characterized with regard to replication in diverse cell cultures, pathogenesis in the murine model, and alterations in the replication cycle of EHV-1 at the levels of protein production and DNA replication. Overall, the results reveal that IR4 is essential for EHV-1 replication in the mouse as well as most cell types and for efficient replication in equine NBL-6 cells that allow limited growth of the IR4-null virus.
Results
Construction of an EHV-1 IR4 deletion mutant
Our previous studies revealed that IR4 is an auxiliary regulatory protein that physically interacts with the IE protein and enhances its ability to trans-activate EHV-1 promoters (Kim et al., 1995, 1997). We therefore hypothesized that the IR4 protein plays an essential role for viral replication. To address this hypothesis, the IR4 genes contained in the terminal and internal repeats of the two isomeric EHV-1 genome were replaced with DNA cassettes harboring kanamycin (KAN) and zeocin (ZEO) antibiotic resistance genes using bacterial artificial chromosome techniques (Rudolph et al. 2002). Comparison of the BamHI restriction enzyme digestion pattern of the RacL11 BAC to those of the single IR4 knockout and the double IR4 knockout mutant BAC revealed the appearance of a high molecular weight band and the loss of a lower molecular weight band in the IR4 mutants, observations consistent with the predicted genomic changes resulting from IR4 deletion as noted in Figure 1A. Southern blot analysis of the parent BAC genome and the single and double IR4 knockout mutant BAC genomes indicated that a single copy of the IR4 gene was successfully replaced by the KAN gene (Fig. 1B and C) and that the second copy of the IR4 gene was successfully replaced by the ZEO gene. This was demonstrated by a failure of the IR4 probe to hybridize to pRacL11ΔIR4(2x) DNA (Fig. 1B) and the appearance of bands of the expected size in samples incubated with KAN (Fig. 1C) and ZEO (Fig. 1D) specific probes. The EUs4 gene, which encodes glycoprotein 2, was deleted in the construction of the BAC and was restored to RacL11ΔIR4ΔEUs4 and RacL11BDΔEUs4, creating RacL11ΔIR4 and RacL11BD, respectively. PCR analysis (Fig. 1F) and DNA sequencing of the EHV-1/antibiotic resistance cassette junctions (data not shown) confirmed the insertion sites of the antibiotic resistance genes and the restoration of EUs4 to both RacL11ΔIR4 and RacL11BD. Growth kinetics of RacL11BD and wt RacL11 were monitored over a 72h time course and found to be indistinguishable (data not shown). Virus genotypes are summarized in Table 1.
Fig. 1.
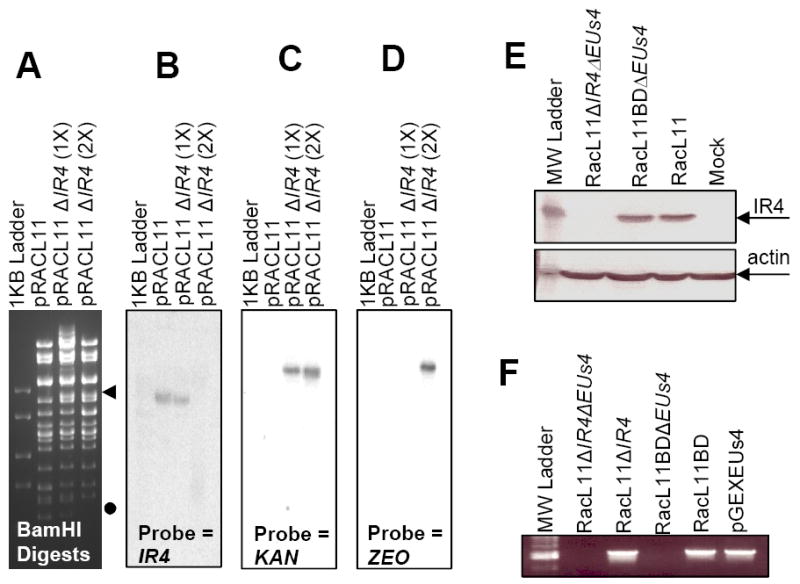
Confirmation of IR4 deletion mutant BAC. Panel A: BamHI restriction enzyme analysis of BAC DNA. ◀ = band added as result of IR4 deletion; ● = band removed as result of IR4 deletion. Panels B to D: Southern blot analyses of digested BAC DNAs. Panel E: western blot analysis of extracts of NBL-6 cells infected with EHV-1 wt (RacL11), EHV-1 RacL11 deleted of both the IR4 and EUs4 genes (RacL11ΔIR4ΔEUs4), or EHV-1 RacL11 deleted of EUs4 (RacL11ΔEUs4). Panel F: PCR amplification using primers specific for EUs4 of viral DNA lacking both IR4 and EUs4 genes (RacL11ΔIR4ΔEUs4), viral DNA lacking IR4 with the EUs4 gene restored (RacL11ΔIR4), viral DNA lacking EUs4 gene (RacL11ΔEUs4), viral DNA with the EUs4 gene restored (RacL11BD), and a control plasmid harboring the EUs4 ORF (pGEXEUs4). Methods for restriction enzyme digestion, Southern blot analysis, and PCR protocols are described in the Materials and Methods.
Table 1.
Virus Genotypes
| Name | Genotype |
|---|---|
| RacL11 | wild-type virus |
| RacL11BDΔEUs4 | RacL11 derived from BAC DNA, EUs4 deleted in the construction of the BAC |
| RacL11BD | RacL11 derived from BAC DNA, EUs4 restored |
| RacL11ΔIR4ΔEUs4 | RacL11 derived from BAC DNA, ΔIR4, EUs4 deleted in the construction of the BAC |
| RacL11ΔIR4 | RacL11 derived from BAC DNA, ΔIR4, EUs4 restored |
Determination of the replication of RacL11ΔIR4 in equine cells
Transfection of equine fibroblast-like NBL-6 cells with the RacL11ΔIR4ΔEUs4 construct resulted in the production of viable virus. Western blot analysis performed using extracts of cells infected with RacL11ΔIR4ΔEUs4 demonstrated that the IR4 protein was not produced by the IR4-null virus (Fig. 1E). Monolayers of NBL-6 cells were infected with RacL11ΔIR4, RacL11BD, or RacL11 and incubated to allow for plaque formation. Plaque morphology (Fig. 2A) and plaque size (Fig. 2B) were not altered in the absence of IR4 in this equine cell type. Growth kinetics were analyzed by infecting NBL-6 cells with RacL11BD and RacL11ΔIR4, and virus production was measured at 3, 6, 12, 18, 24, 36, 48, and 72h post-infection. The replication of the IR4 deleted virus was delayed and its titers were consistently one log or more below those of RacL11BD in the NBL-6 cells (Fig. 3). Interestingly, studies with equine ETCC cells revealed that this tumor cell line failed to support the replication of RacL11ΔIR4, whereas the parent virus replicated to high titers (Table 2).
Fig. 2.

Plaques morphology and size of wt EHV-1 RacL11, BAC-derived EHV-1 deleted of the IR4 gene with EUs4 restored (RacL11ΔIR4), and BAC-derived EHV-1 RacL11 with the EUs4 restored (RacL11BD). Panel A shows representative plaque morphology. Panel B shows plaque size. Bars represent means of 50 plaques of each virus; error bars represent standard deviation.
Fig. 3.
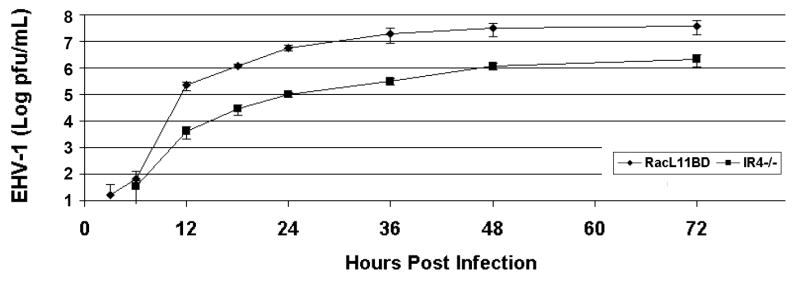
Growth kinetics of EHV-1 RacL11 derived from BAC DNA (RacL11BD) and RacL11 deleted of IR4 (RacL11ΔIR4) in equine NBL-6 cells. Infected cells were harvested at 3, 6, 12, 18, 24, 36, and 72h post-infection, and virus was quantitated by standard plaque assay on NBL-6 cells.
Table 2.
Replication of RacL11ΔIR4 and RacL11BD in Diverse Cell Types
| Cell Type | Virus Strain Tested | |
|---|---|---|
| RacL11BD | RacL11ΔIR4 | |
| L-M | 3.83×106 ± 4.71×105 | < 10 |
| RK13 | 6.33×106 ± 6.24×105 | < 10 |
| RK13+pSVIR4 | ND | 7.7×104 ± 1.04×104 |
| ETCC | 2.33×106 ± 8.50×105 | < 100 |
| NBL-6 | 2.43×106 ± 2.50×105 | 1.05×105 ± 1.32×104 |
| Vero | 1.58×106 ± 3.12×105 | < 100 |
| HeLa | 1.07×105 ± 1.43×104 | < 200 |
Cells were infected at a MOI of 1, and virus titers were determined at various times post infection by plaque assay on equine NBL-6 cell monolayers. Table shows results at 48 hours p.i.
EHV-1 RacL11ΔIR4 is non-pathogenic in the CBA mouse
The effects of the deletion of IR4 on EHV-1 pathogenesis were assessed by infecting groups of ten CBA mice with sterile medium, RacL11ΔIR4, or RacL11BD, and the percentage of body weight lost or gained in comparison to weight at the time of infection was determined at 24, 48, 72, and 96 hours post-infection. No statistical difference was observed between the mock group and the mice infected with RacL11ΔIR4, both of which gained close to 10% body weight by the termination of the experiment. Further, mice infected with the IR4-null virus failed to exhibit any clinical signs of infection. Consistent with our previous studies (Smith et al., 2005), mice infected with RacL11 derived from BAC DNA (RacL11BD) lost approximately one-fifth of their body weight by the termination of the experiment (Fig. 4). The in vivo replication capacity of the IR4 deletion mutant was measured by quantifying the concentration of virus in lungs of infected mice. Two groups of 20 mice per group were infected intranasally with 5.0 × 105 pfu of RacL11ΔIR4 or RacL11BD, and five mice from each group were sacrificed at 4, 48, 96, and 144h post-infection. Lungs were harvested from each mouse, and virus was quantitated by standard plaque assay on equine NBL-6 cells. RacL11ΔIR4 was not recovered at any time point in any of the mice. In contrast, mice infected with RacL11BD produced an average of 4 × 106 pfu per lung at 48h post-infection and exhibited clinical signs of severe disease with mortality rates of 20%-40% (Table 3). Thus, the deletion of the IR4 gene rendered EHV-1 non-pathogenic in the CBA mouse model of EHV-1 pathogenesis.
Fig. 4.
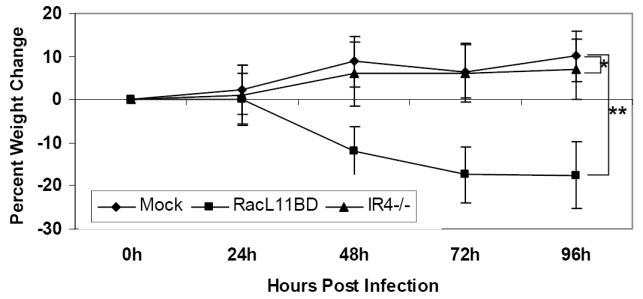
Weight loss or gain of CBA mice mock-infected or infected with EHV-1 RacL11BD or RacL11ΔIR4 (IR4-/-). Mice were inoculated intranasally and weighed every 24h for 96h as described in the Materials and Methods. Data points represent means of ten mice; weight loss or gain was calculated in relation to the initial weight at the time of infection. Error bars represent standard deviation. (*) P = .44; (**) P < .001.
Table 3.
RacL11ΔIR4 Fails To Replicate in the Lungs of CBA Mice.
| PFU Titer Determined in Equine NBL6 Cells |
||||
|---|---|---|---|---|
| Virus | inoculum† | 2 days pi‡ | 4 days pi‡ | 6 days pi‡ |
| RacL11ΔIR4 | 5 × 105 | <50 | <50 | <50 |
| RacL11BD | 5 × 105 | 4.6 × 106 | 2.6 × 105* | 4.7 × 103** |
= pfu;
= pfu/lung;
n = 5 mice per group;
one mouse succumbed to infection;
two mice succumbed to infection. RacL11BD is parent RacL11 virus derived from BAC.
IR4 is essential for EHV-1 replication in non-equine cell types
EHV-1 can be propagated in a large number of cell types (O’Callaghan and Osterrieder, 2008). The finding that RacL11ΔIR4 was not able to grow in either the mouse lung or in equine ETCC cells prompted the question as to whether the deletion of IR4 prohibited EHV-1 replication in other cell types routinely used to cultivate this alphaherpesvirus. To address this question, assays examining the replication of RacL11BD and RacL11ΔIR4 were performed in mammalian cell cultures representing a diversity of species, including mouse L-M, rabbit RK13, non-human primate Vero, and human HeLa cells. Whereas RacL11ΔIR4 titers were lower than those that of RacL11BD in equine NBL-6 cells as shown above (Fig. 3), the IR4 mutant virus failed to replicate in all other cell types tested (Table 2). Expression of IR4 in RK13 cells by transient transfection resulted in the recovery of viable RacL11ΔIR4 (Table 2), albeit at lower levels than that recovered from NBL-6 cells. These results indicated that replication of EHV-1 is severely restricted in the absence of IR4 in many cell types normally permissive for EHV-1, and that ectopic expression of IR4 in RK13 cells restores the ability of the IR4-null virus to replicate.
Synthesis of some essential viral proteins is altered or abrogated in the absence of IR4
To identify the nature of the block in the replication of the IR4-null virus, RK13 cells infected with RacL11BD and RacL11ΔIR4 (MOI=1) were examined at various times post infection by western blot analysis for the production of the IE protein and representative early and late proteins. Production of the IE protein in cells infected with the IR4-null mutant was indistinguishable from that observed in cells infected with the parent virus (Fig. 5A). The early essential regulatory protein UL5P (Fig. 5A) and the EHV-1 major DNA binding protein (UL53P, data not shown) were likewise produced independently of the presence of IR4 both in terms of kinetics and total protein levels. In the absence of IR4, the synthesis of the early EICP0 protein, a potent transcriptional trans-activator (Bowles et al., 1997), was found to be initially delayed but by 16 hours post-infection reached levels comparable to those in cells infected with the parent virus (Fig 5A). Late viral protein production was examined using antibodies specific for glycoproteins D and K (gD, gK) and ORF12, the EHV-1 trans-inducing factor (ETIF). Production of the essential ETIF protein, which is synthesized at the initiation of viral DNA replication, was both delayed and reduced in the absence of IR4 (Fig. 5A). Production of gD (Fig. 5A) and gK (data not shown), which are both essential envelope proteins, was reduced to undetectable levels in the non-permissive cell line, whereas gD production in NBL-6 cells infected with RacL11ΔIR4 was detected, albeit at reduced levels, after an initial delay (Fig. 5A and B). Examination of the production of the EICP0 protein in infected NBL-6 cells at 2 and 8 hours post infection revealed no difference between RacL11BD and RacL11ΔIR4 (Fig. 5B).
Fig. 5.
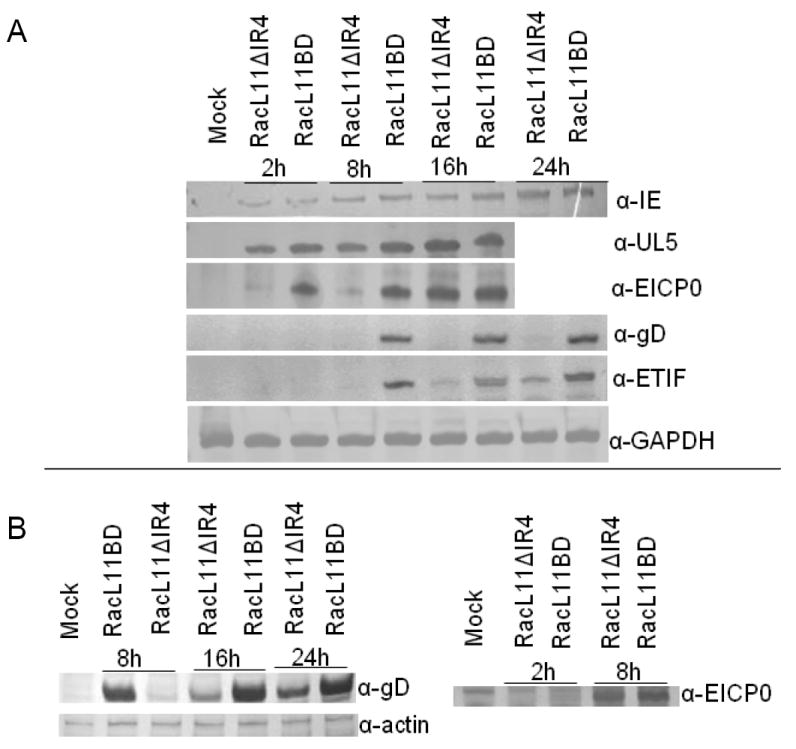
Western blot analysis of RK13 (A) and NBL-6 (B) cells infected with RacL11BD or RacL11ΔIR4. Infected cells (MOI=1) were harvested at indicated times and probed with antisera specific for the IE, UL5, EICP0, ETIF, and gD proteins. Rabbit gapdh or horse actin was detected as loading controls.
Viral DNA synthesis is inhibited in RK13 cells in the absence of IR4
While analysis of non-permissive RK13 cells infected with RacL11ΔIR4 revealed impaired synthesis of a number of important viral proteins, the ability of the mutant virus to replicate DNA remained in question. To address whether IR4 production is required for viral DNA synthesis, real-time PCR was performed on samples containing total DNA harvested from RK13 cells infected with either RacL11BD or RacL11ΔIR4 at 2, 8, 12, 24, and 48 hours post-infection. The results were normalized to the cellular gapdh gene as an internal control and expressed as the relative change in viral genomic copy number after an initial 2 hour time point. Compared to that of RacL11BD, the replication of RacL11ΔIR4 DNA was inhibited in excess of 99.9%, and the earliest detection of viral DNA replication was delayed by 16 hours compared to RacL11BD (Fig. 6A). Interestingly, there was a 50% reduction in RacL11ΔIR4 DNA copies detected at 8 hours post-infection which may indicate degradation of the mutant viral genome following entry. While RacL11ΔIR4 also exhibited a lag in the synthesis of viral DNA and, ultimately, viral DNA failed to reach levels comparable to RacL11BD in permissive NBL-6 cells, the inhibition of viral DNA synthesis was far less pronounced at late times (Fig 6B). Severe cytopathology of cells infected with RacL11BD made reliable analysis of RacL11BD DNA replication at the 48h time point impractical.
Fig. 6.
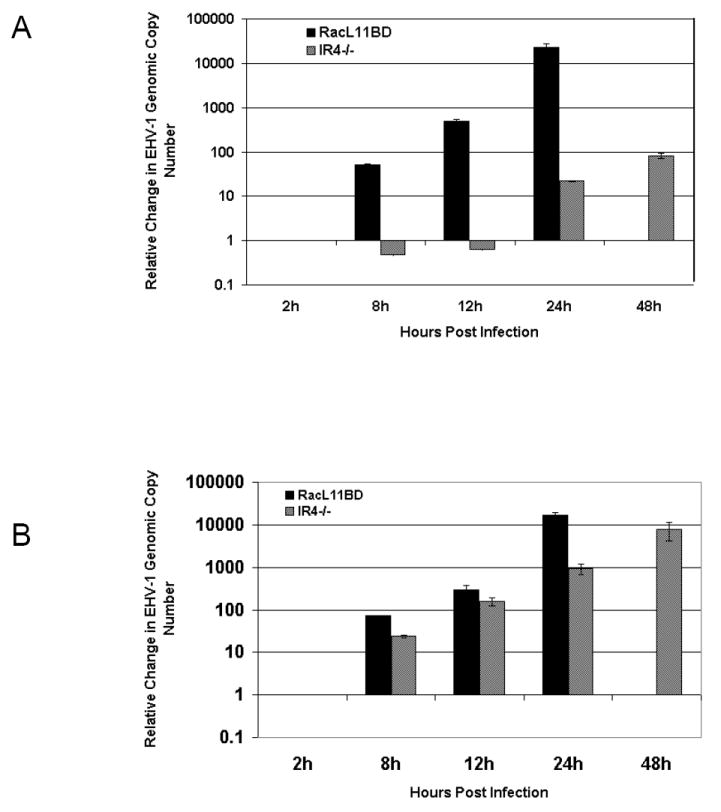
Comparison of viral DNA synthesis. RK13 (A) or NBL-6 (B) cells were infected with RacL11BD or RacL11ΔIR4 and harvested at indicated times post-infection. Cell pellets were prepared as described in Materials and Methods, and the relative change in viral DNA copy number after two hours was measured using real-time PCR. Samples were tested in triplicate, and the results were normalized to total sample DNA. Extensive cytopathology made testing of RacL11BD infected cells at 48h impractical. Error bars indicate standard deviation.
Discussion
IR4 homologues have regulatory roles in many alphaherpesviruses. The HSV-1 homologue, ICP22, is thought to direct transcription away from cellular promoters to viral promoters via hyper-phosphorylation of RNA polymerase II (Rice et al., 1994, 1995; Leopardi et al., 1997). Likewise, the ORF63/70 protein is thought to have a role in regulating varicella-zoster virus (VZV) promoters, but its function is less clear (Jackers et al., 1992; Kost et al., 1995). Discerning the role of the IR4 protein of EHV-1 based on characterized functions of its homologues may be problematic, however, as the similarity among the different proteins is, in many cases, quite low. Indeed, ICP22 of HSV-1 is a 420aa immediate-early protein, whereas IR4 is 293aa and is an early regulatory protein. Additionally, IR4 is diploid in EHV-1, while its counterpart is haploid in HSV-1.
In HSV-1, ICP22 is not essential for virus growth in some cell types (Post and Roizman 1981; Poffenberger et al., 1993, 1994, Orlando et al., 2006). The VZV genome is structured more closely to that of EHV-1, and in VZV one copy of the diploid IR4 homologue ORF63/70 is required for virus replication (Sommer et al., 2001). EHV-1 deleted for both copies of IR4 was able to replicate in non-immortalized equine cells, although its growth kinetics were significantly retarded in comparison to those of the parent virus. It should be noted that while the diploid nature of IR4 in EHV-1 made the generation of full IR4 revertants unfeasible, the analysis of both BAC DNA and the properties of the progeny viruses demonstrated that any unintended alterations of the EHV-1 genome were unlikely, in agreement with the demonstration that IR4 produced in trans restored RacL11ΔIR4 replication in RK13 cells. Additionally, recent experiments succeeded in generating an EHV-1 mutant virus devoid of the entire internal repeat of the S region and indicated that one copy of IR4 is sufficient to support viral replication in RK13 cells that are non-permissive for RacL11ΔIR4 (Ahn, Breitenbach, Zhang, and O’Callaghan, unpublished results).
The findings that infection of mice with the IR4-null virus failed to cause clinical signs and to result in virus replication in the mouse lung were not unexpected given the observation that the IR4-null virus fails to replicate in murine cells in culture. The facts that the IR4 mutant virus can be propagated in equine NBL-6 cells and is non pathogenic in the mouse model that mimics EHV-1 infection in the equine may be observations that could be exploited in future work to develop vaccines and gene delivery vehicles for the equine. In permissive NBL-6 cells, plaque morphology was indistinguishable in the absence of IR4, and average plaque size was not significantly different in the absence of IR4. It is likely that the production of late protein species, such as glycoprotein D, that mediate cell fusion and influence plaque morphology is sufficient by 72 hours post-infection in NBL-6 cells to yield similar plaque characteristics, even though overall virus production is reduced in this cell type.
A recent report (Orlando et al., 2006) showed that deletion of ICP22 results in HSV-1 particles that contain greatly reduced quantities of the late proteins US11 and gC, produce increased amounts of ICP0 and ICP4, and exhibit biochemical and physical properties that are distinct from wt virus, resulting in a 500-fold decrease in infectivity. In the case of EHV-1, it remains to be determined whether non-infectious particles are produced in cells non-permissive for the IR4-null virus. However, in these cells, the IEP protein and early proteins UL5P and UL53P were produced in nearly identical amounts and with similar kinetics in the presence or absence of IR4, indicating that viral entry and the initiation of the viral replication cycle were not impaired. Also, the inhibition of the synthesis of ETIF, recently shown to be essential for EHV-1 secondary envelopment and virion maturation (von Einem et al., 2006), and the complete inhibition of synthesis of gD and gK, which are known to play essential roles in maturation, egress, and infectivity, argue that EHV-1 replication in the absence of IR4 is blocked at a stage prior to that of particle formation and release. Since EHV-1 DNA replication is impaired in excess of three logs in the absence of IR4 in non-permissive RK13 cells, future work will be required to determine whether IR4 plays a direct role in DNA replication and/or whether the IR4 protein is essential for transcribing early genes required for DNA synthesis
Materials and Methods
Virus and cells
Pathogenic EHV-1 strain RacL11 was propagated in equine NBL-6 cells. Mouse L-M, rabbit RK-13, equine NBL-6, equine ETCC (Allen and Bryans, 1974), primate Vero, and human HeLa cells were propagated in Dulbecco’s minimum essential medium (DMEM) supplemented with 5% or 10% fetal bovine serum (FBS), and selected cells were used in transfections and infections as indicated.
Plasmids
pGexEuS4 containing full length EUs4 that encodes gp2 of EHV-1 (RacL11 strain) was generated by PCR amplification of EUs4 and surrounding sequences with primers incorporating BamHI and EcoRI restriction sites. pSVIR4 (formerly pSVEICP22) has been previously described. (Derbigny et al., 2000).
Generation and confirmation of IR4-null BAC
An EHV-1 RacL11 BAC (Rudolph et al., 2002) was deleted for a single copy of IR4 by RED recombination as previously described (Yao et al., 2003). Briefly, a kanamycin (KAN) resistance marker generated by PCR using primers containing approximately 50bp of IR4 flanking sequences in their 5’ ends was electroporated into E. coli DY380 (containing the EHV-1 BAC) previously grown at 42°C to induce RED recombinases. Electroporated cultures were inoculated onto medium containing kanamycin (30μg/mL) to select for transformants. The second copy of IR4 was deleted in the same manner employing a zeocin (ZEO) marker. Putative transformants were screened by: 1) PCR amplification of both EHV-1/antibiotic resistance marker junction sequences and IR4 internal sequences; 2) Southern blot analysis using probes specific for IR4 sequences, kanamycin (KAN) marker sequences, or zeocin (ZEO) marker sequences; and 3) sequencing of EHV-1/antibiotic resistance marker junctions.
Generation of IR4 null virus
Purified BAC constructs were electroporated (Nucleofector, Amaxa Corp, Germany) into NBL-6 cells that were then incubated at 37°C for 5-7 days. Supernatant was transferred to fresh NBL-6 monolayers, and cells were observed for cytopathic effects. Green fluorescent plaques were selected and purified three times. EUs4 revertant viruses were generated by co-electroporating BAC constructs with pGexEUs4. In the case of RacL11ΔIR4, cells transfected with the EUs4 restoration plasmid were subsequently infected with the RacL11ΔIR4ΔEUs4 virus. In both cases, non-fluorescing plaques were isolated and purified three times. DNA was isolated from each putative revertant and screened by PCR targeting full length EUs4.
Western blot analysis
RK13 or NBL-6 cells were seeded onto 60mm tissue culture dishes (BD Biosciences, Durham, NC) and infected the subsequent day at a confluency of 80-90% with either RacL11BD or RacL11ΔIR4 viruses. Protein extracts of infected cells were separated by SDS-PAGE, electrophoretically transferred to a nitrocellulose membrane (Bio-Rad LSG, Hercules, CA), blocked in 1% skim milk, rinsed in Tris-Buffered Saline (pH 7.4) with 0.5% Tween-20 (TBST), and incubated with the indicated primary antibodies at a concentration of 1:2500 to 1:10,000. The membranes were washed again and incubated with a 1:10,000 dilution of the secondary antibody (alkaline phosphatase-conjugated goat anti-rabbit or anti-mouse antiserum; Sigma, St Louis, MO). Protein-antibody complexes were visualized by incubating the membranes in AP color reagent (Bio-Rad LSG, Hercules, CA) per the manufacturer’s instructions.
Growth kinetics and plaque morphology
NBL-6, RK-13, L-M, Vero, HeLa, ETCC, and pSVIR4 tranfected RK13 cells were seeded to 80% confluence in 60mm dishes and infected at an MOI=1. Cells and supernatants were harvested at the indicated times post-infection. Serial dilutions of each sample were used to inoculate fresh NBL-6 monolayers. Monolayers were incubated in medium containing 1.5% methylcellulose, and plaques were quantitated after 3 days by fixing with 10% formalin and staining with 0.5% crystal violet.
Animal experiments
Groups (n=10) of three-week-old CBA mice were infected intranasally with 5 × 105 pfu of RacL11ΔIR4 or RacL11BD, or were mock-infected with sterile medium. Mice were weighed prior to inoculation and every 24h post-inoculation for five days. Differences in weight loss or gain over time were measured by comparing individual mouse weight at each interval to weight prior to inoculation. Virus replication in mice was measured by infecting groups of 20 three-week-old CBA mice as described above. Five mice from each group were sacrificed at 4, 48, 96, and 144h post-infection, and lungs were harvested. Whole lungs were combined in diluent and homogenized, and the virus concentration in each sample was quantitated as previously described (Smith et al., 2005).
Real-time PCR analysis of viral DNA replication
RK13 cells were infected with RacL11ΔIR4 or RacL11BD as described, and monolayers were harvested at 2, 8, 12, 24, and 48 hours post-infection. Cell pellets were resuspended in 500μL of DNAzol (MRC, Inc., Cincinnati, OH) and incubated at 100°C for five minutes to ensure release of DNA. The samples were then diluted by a factor of 100 and stored at 20°C until ready for use. Primers specific for gapdh (forward) 5’ - TGC CCC CAT GTT TGT GAT G - 3’ and (reverse) 5’ - TGT GGT CAT GAG CCC TTC C - 3’ and primers specific for EHV-1 UL5 (forward) 5’ - CAA CTG GAA GCA GCA ACA GC - 3’ and (reverse) 5’ - GCG GTG AAC TCT GGC CAC GC - 3’ were utilized to amplify viral and cellular DNA using an IQ5 real-time PCR instrument and IQ SYBR Green real-time PCR supermix (Bio-Rad LSG, Hercules, CA). The relative change in genomic copy number was extrapolated by determining threshold detection cycle differences in viral DNA after two hours and normalized against the cellular gapdh gene as an internal control for total sample DNA.
Acknowledgments
The authors thank Ms. Suzanne Zavecz, Ms. Shannon Kahan, and Mr. Kyle Coffin for their technical assistance in these experiments and members of the laboratory for helpful suggestions. This study was conducted with the support of NIH research grant AI-22001 and NIH center grant P20-RR018724 from the National Center for Research Resources. PDE was supported by NIH fellowship grant F32 AI-060113. We dedicate this paper to the memory of the late Dr. Patrick M. Smith who contributed to the early phase of this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn BC, Breitenbach JE, Kim SK, O’Callaghan DJ. The equine herpesvirus-1 IR3 gene that lies antisense to the sole immediate-early (IE) gene is trans-activated by the IE protein, and is poorly expressed to a protein. Virology. 2007;316:15–25. doi: 10.1016/j.virol.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht RA, Jang HK, Kim SK, O’Callaghan DJ. Direct interaction of TFIIB and the IE protein of equine herpesvirus 1 is required for maximal trans-activation function. Virology. 2003;316:302–312. doi: 10.1016/j.virol.2003.08.017. [DOI] [PubMed] [Google Scholar]
- Albrecht RA, Kim SK, O’Callaghan DJ. The EICP27 protein of equine herpesvirus 1 is recruited to viral promoters by its interaction with the immediate-early protein. Virology. 2005;333:74–87. doi: 10.1016/j.virol.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Albrecht RA, Kim SK, Zhang Y, Zhao Y, O’Callaghan DJ. The equine herpesvirus 1 EICP27 protein enhances gene expression via an interaction with TATA box-binding protein. Virology. 2004;324:311–326. doi: 10.1016/j.virol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- Allen GP, Bryans JT. Studies of an established equine cell line derived from a transitional cell carcinoma. Am J Vet Res. 1974;35:1153–1160. [PubMed] [Google Scholar]
- Baumann RP, Staczek J, O’Callaghan DJ. Equine herpesvirus type 1 defective interfering (DI) particle DNA structure: The central region of the inverted repeat is deleted from DI DNA. Virology. 1987;159:137–146. doi: 10.1016/0042-6822(87)90356-4. [DOI] [PubMed] [Google Scholar]
- Bowles DE, Holden VR, Zhao Y, O’Callaghan DJ. The ICP0 protein of equine herpesvirus 1 is an early protein that independently transactivates expression of all classes of viral promoters. J Virol. 1997;71:4904–4914. doi: 10.1128/jvi.71.7.4904-4914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczynski KA, Kim SK, O’Callaghan DJ. Characterization of the transactivation domain of the equine herpesvirus type 1 immediate-early protein. Virus Res. 1999;65:131–140. doi: 10.1016/s0168-1702(99)00116-1. [DOI] [PubMed] [Google Scholar]
- Buczynski KA, Kim SK, O’Callaghan DJ. Initial characterization of 17 viruses harboring mutant forms of the immediate early gene of equine herpesvirus 1. Virus Genes. 2005;31:229–239. doi: 10.1007/s11262-005-1801-2. [DOI] [PubMed] [Google Scholar]
- Caughman GB, Lewis JB, Smith RH, Harty RN, O’Callaghan DJ. Detection and intracellular localization of equine herpesvirus 1 IR1 and IR2 gene products by using monoclonal antibodies. J Virol. 1995;69:3024–3032. doi: 10.1128/jvi.69.5.3024-3032.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughman GB, Staczek J, O’Callaghan DJ. Equine herpesvirus type l infected cell polypeptides: Evidence for immediate early/early/late regulation of viral gene expression. Virology. 1985;145:49–61. doi: 10.1016/0042-6822(85)90200-4. [DOI] [PubMed] [Google Scholar]
- Chen M, Garko KA, Zhang Y, O’Callaghan DJ. The defective interfering particles of equine herpesvirus 1 encode an ICP22/ICP27 hybrid protein that alters viral gene regulation. Virus Res. 1999;59:149–164. doi: 10.1016/s0168-1702(98)00128-2. [DOI] [PubMed] [Google Scholar]
- Chen M, Harty RN, Zhao Y, Holden VR, O’Callaghan DJ. Expression of an equine herpesvirus 1 ICP22/CP27 hybrid protein encoded by defective interfering particles associated with persistent infection. J Virol. 1996;70:313–320. doi: 10.1128/jvi.70.1.313-320.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbigny WA, Kim SK, Caughman GB, O’Callaghan DJ. The EICP22 protein of equine herpesvirus 1 physically interacts with the immediate-early protein and with itself to form dimers or higher-ordered complexes. J Virol. 2000;74:1425–1435. doi: 10.1128/jvi.74.3.1425-1435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbigny WA, Kim SK, Jang HK, O’Callaghan DJ. EHV-1 EICP22 protein sequences that mediate its physical interaction with the immediate-early protein are not sufficient to enhance the trans-activation activity of the IE protein. Virus Res. 2002;84:1–15. doi: 10.1016/s0168-1702(01)00377-x. [DOI] [PubMed] [Google Scholar]
- Ebner PD, Kim SK, O’Callaghan DJ. Biological and genotypic properties of defective interfering particles of equine herpesvirus 1 that mediate persistent infection. Virology. 2008 doi: 10.1016/j.virol.2008.08.024. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner PD, O’Callaghan DJ. Genetic complexity of EHV-1 defective interfering particles and identification of novel IR4/UL5 hybrid proteins produced during persistent infection. Virus Genes. 2006;32:313–320. doi: 10.1007/s11262-005-6916-y. [DOI] [PubMed] [Google Scholar]
- Harty RN, Colle CF, O’Callaghan DJ. Equine herpesvirus type 1 gene regulation: Characterization of transcription from the immediate early gene region in productive infection. In: Wagner EK, editor. Herpesvirus Transcriptional and Its Regulation. Chapter 16. CRC Press, Inc; Boca Raton, FL: 1990. pp. 319–338. [Google Scholar]
- Harty RN, O’Callaghan DJ. An early gene maps within and is 3’ co-terminal with the immediate-early gene of equine herpesvirus 1. J Virol. 1991;65:3829–3838. doi: 10.1128/jvi.65.7.3829-3838.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden VR, Caughman GB, Zhao Y, Harty RN, O’Callaghan DJ. Identification and characterization of the ICP22 protein of equine herpesvirus 1. J Virol. 1994;68:4329–4340. doi: 10.1128/jvi.68.7.4329-4340.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden VR, Harty RN, Yalamanchili RR, O’Callaghan DJ. The IR3 gene of equine herpesvirus type 1: a unique gene regulated by sequences within the intron of the immediate-early gene. DNA Sequence. 1992a;3:143–152. doi: 10.3109/10425179209034010. [DOI] [PubMed] [Google Scholar]
- Holden VR, Yalamanchili RR, Harty RN, O’Callaghan DJ. The ICP22 homologue of equine herpesvirus 1: Expression from early and late promoters. J Virol. 1992b;66:664–673. doi: 10.1128/jvi.66.2.664-673.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden VR, Zhao Y, Thompson Y, Caughman GB, Smith RH, O’Callaghan DJ. Characterization of the regulatory function of the ICP22 protein of equine herpesvirus type 1. Virology. 1995;210:273–282. doi: 10.1006/viro.1995.1344. [DOI] [PubMed] [Google Scholar]
- Jackers P, Defechereux PL, Baudoux C, Lambert M, Massaer M, Merville-Louis MP, Rentier B, Piette J. Characterization of regulatory functions of the varicella-zoster virus gene 63-encoded protein. J Virol. 1992;66:3899–3903. doi: 10.1128/jvi.66.6.3899-3903.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HK, Albrecht RA, Buczynski KA, Kim SK, Derbigny WA, O’Callaghan DJ. Mapping the sequences that mediate interaction of the equine herpesvirus 1 immediate-early protein and human TFIIB. J Virol. 2001;75:10219–10230. doi: 10.1128/JVI.75.21.10219-10230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Ahn BC, Albrecht RA, O’Callaghan DJ. The unique IR2 protein of equine herpesvirus 1 (EHV-1) negatively regulates viral gene expression. J Virol. 2006;80:5041–5049. doi: 10.1128/JVI.80.10.5041-5049.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Bowles DE, O’Callaghan DJ. The γ2 late glycoprotein K promoter of equine herpesvirus 1 is differentially regulated by the IE and EICP0 proteins. Virology. 1999;256:173–179. doi: 10.1006/viro.1999.9608. [DOI] [PubMed] [Google Scholar]
- Kim SK, Buczynski KA, Caughman GB, O’Callaghan DJ. The equine herpesvirus 1 immediate-early protein interacts with EAP, a nucleolar-ribosomal protein. Virology. 2001;279:173–184. doi: 10.1006/viro.2000.0725. [DOI] [PubMed] [Google Scholar]
- Kim SK, Holden VR, O’Callaghan DJ. The ICP22 protein of equine herpesvirus 1 cooperates with the IE protein to regulate viral gene expression. J Virol. 1997;71:1004–1012. doi: 10.1128/jvi.71.2.1004-1012.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Jang HK, Albrecht RA, Derbigny WA, Zhang Y, O’Callaghan DJ. Interaction of the equine herpesvirus 1 EICP0 protein with the immediate-early (IE) protein, TFIIB, and TBP may mediate the antagonism between the IE and EICP0 proteins. J Virol. 2003;77:2675–2685. doi: 10.1128/JVI.77.4.2675-2685.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Smith RH, O’Callaghan DJ. Characterization of DNA binding properties of the immediate-early gene product of equine herpesvirus type 1. Virology. 1995;213:46–56. doi: 10.1006/viro.1995.1545. [DOI] [PubMed] [Google Scholar]
- Kost RG, Kupinsky H, Straus SE. Varicella-zoster virus gene 63: transcript mapping and regulatory activity. Virology. 1995;209:218–224. doi: 10.1006/viro.1995.1246. [DOI] [PubMed] [Google Scholar]
- Leopardi R, Ward PL, Ogle WO, Roizman B. Association of herpes simplex virus regulatory protein ICP22 with transcriptional complexes containing EAP, ICP4, RNA polymerase II, viral DNA requires posttranslational modification by the UL13 protein kinase. J Virol. 1997;71:1133–1139. doi: 10.1128/jvi.71.2.1133-1139.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan DJ, Osterrieder N. Herpesviruses of Horses. In: Mahy B, Van Regenmortel M, editors. Encyclopedia of Virology. 3. Elsevier Ltd.; Oxford, UK: 2008. pp. 411–420. [Google Scholar]
- Orlando JS, Balliet JW, Kushnir AS, Astor TL, Kosz-Vnenchak M, Rice SA, Knipe DM, Schaffer PA. ICP22 is required for wild-type composition and infectivity of herpes simplex virus type 1 virions. J Virol. 2006;8:9381–9390. doi: 10.1128/JVI.01061-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poffenberger KL, Idowu AD, Fraser-Smith EB, Raichlen PE, Herman RC. A herpes simplex virus type 1 ICP22 deletion mutant is altered for virulence and latency in vivo. Arch Virol. 1994;139:111–119. doi: 10.1007/BF01309458. [DOI] [PubMed] [Google Scholar]
- Poffenberger KL, Raichlen PE, Herman RC. In vitro characterization of a herpes simplex virus type 1 ICP22 deletion mutant. Virus Genes. 1993;7:171–186. doi: 10.1007/BF01702397. [DOI] [PubMed] [Google Scholar]
- Post LE, Roizman B. A generalized technique for deletion of specific genes in large genomes: alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell. 1981;25:227–232. doi: 10.1016/0092-8674(81)90247-6. [DOI] [PubMed] [Google Scholar]
- Rice SA, Long MC, Lam V, Schaffer PA, Spencer CA. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J Virol. 1995;69:5550–5559. doi: 10.1128/jvi.69.9.5550-5559.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice SA, Long MC, Lam V, Spencer CA. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J Virol. 1994;68:988–1001. doi: 10.1128/jvi.68.2.988-1001.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph J, O’Callaghan DJ, Osterrieder N. Cloning of the genomes of equine herpesvirus type 1 (EHV-1) strains KyA and RacL11 as bacterial artificial chromosomes (BAC) J Vet Med B. 2002;49:31–36. doi: 10.1046/j.1439-0450.2002.00534.x. [DOI] [PubMed] [Google Scholar]
- Smith PM, Kahan SM, Rorex CB, von Einem J, Osterrieder N, O’Callaghan DJ. Expression of the full length form of gp2 of equine herpesvirus 1 (EHV-1) completely restores respiratory virulence to the attenuated EHV-1 strain KyA in CBA mice. J Virol. 2005;79:5105–5115. doi: 10.1128/JVI.79.8.5105-5115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH, Caughman GB, O’Callaghan DJ. Characterization of the regulatory functions of the equine herpesvirus 1 immediate-early gene product. J Virol. 1992;66:936–945. doi: 10.1128/jvi.66.2.936-945.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH, Holden VR, O’Callaghan DJ. Nuclear localization and transcriptional activation activities of truncated versions of the immediate-early gene product of equine herpesvirus 1. J Virol. 1995;69:3857–3862. doi: 10.1128/jvi.69.6.3857-3862.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH, Zhao Y, O’Callaghan DJ. The equine herpesvirus 1 (EHV-1) UL3 gene, an ICP27 homologue, is necessary for full activation of gene expression directed by an EHV-1 late promoter. J Virol. 1993;67:1105–1109. doi: 10.1128/jvi.67.2.1105-1109.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH, Zhao Y, O’Callaghan DJ. The equine herpesvirus type 1 immediate-early gene product contains an acidic transcriptional activation domain. Virology. 1994;202:760–770. doi: 10.1006/viro.1994.1398. [DOI] [PubMed] [Google Scholar]
- Sommer MH, Zagha E, Serrano OK, Ku CC, Zerboni L, Baiker A, Santos R, Spengler M, Lynch J, Grose C, Ruyechan W, Hay J, Arvin AM. Mutational analysis of the repeated open reading frames, ORFs 63 and 70 and ORFs 64 and 69, of varicella-zoster virus. J Virol. 2001;75:8224–8239. doi: 10.1128/JVI.75.17.8224-8239.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Einem J, Schumacher D, O’Callaghan DJ, Osterrieder N. The α-TIF (VP16) homologue (ETIF) of equine herpesvirus 1 (EHV-1) is essential for secondary envelopment and virus egress. J Virol. 2006;80:2609–2620. doi: 10.1128/JVI.80.6.2609-2620.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalamanchili RR, Raengsakulrach B, Baumann RP, O’Callaghan DJ. Identification of the site of recombination in the generation of the genome of DI particles of equine herpesvirus type 1. Virology. 1990;175:448–455. doi: 10.1016/0042-6822(90)90429-u. [DOI] [PubMed] [Google Scholar]
- Yao H, Osterrieder N, O’Callaghan DJ. Generation and characterization of an EICP0 null mutant of equine herpesvirus 1. Virus Res. 2003;98:163–172. doi: 10.1016/j.virusres.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Holden VR, Smith RH, O’Callaghan DJ. Regulatory function of the equine herpesvirus 1 ICP27 gene product. J Virol. 1995;69:2786–2793. doi: 10.1128/jvi.69.5.2786-2793.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]