

Abstract
Sensory experience is crucial for shaping the cortical microcircuit during development and is thought to modify network function through several forms of Hebbian and homeostatic plasticity. Where and when these different forms of plasticity are expressed at particular synapse types within cortical microcircuits, and how they interact, is poorly understood. Here we investigated how two different visual deprivation paradigms, lid suture (LS) and intraocular TTX, affect the local microcircuit within layer 2/3 of rat visual cortex during the classical critical period for visual system plasticity. Both forms of visual deprivation produced a compensatory increase in the spontaneous firing of layer 2/3 pyramidal neurons in acute slices derived from monocular visual cortex. TTX increased spontaneous activity through an increase in the excitation/inhibition (E/I) balance within layer 2/3. In contrast, LS decreased the E/I balance by strongly depressing excitatory transmission, and the homeostatic increase in spontaneous activity was instead achieved through an increase in the intrinsic excitability of layer 2/3 pyramidal neurons. The microcircuit in layer 2/3 can thus use different forms of homeostatic plasticity to compensate for the loss of visual drive, depending on the specific demands produced by visual experience. The existence of multiple, partially redundant forms of homeostatic plasticity may ensure that network compensation can be achieved in response to a wide range of sensory perturbations.
Keywords: visual cortex, synaptic plasticity, homeostatic plasticity, intrinsic plasticity, visual deprivation, microcircuitry
Introduction
Cortical microcircuitry can be modified in an activity-dependent manner in response to changes in sensory experience (Katz and Shatz, 1996; Fox, 2002; Feller and Scanziani, 2005; Hofer et al., 2006). This experience-dependent circuit refinement has been studied extensively in visual cortex and is thought to require both synapse-specific forms of plasticity such as long-term potentiation (LTP) and long-term depression (LTD) (Katz and Shatz, 1996; Kirkwood et al., 1996; Rittenhouse et al., 1999; Malenka and Bear, 2004) and homeostatic forms of plasticity such as synaptic scaling that stabilize the activity of neurons and networks (Turrigiano et al., 1998; Desai et al., 2002; Turrigiano and Nelson, 2004; Goel and Lee, 2007). How these different forms of plasticity interact within complex cortical microcircuits is poorly understood.
The homeostatic maintenance of an optimal level of network excitability (“gain”) is likely important for allowing cortical neurons to efficiently detect incoming signals and produce the appropriate output (Davis and Bezprozvanny, 2001; Turrigiano and Nelson, 2004). Interestingly, homeostatic plasticity in visual cortex is strongly developmentally regulated and in layer 4 is present only during the first week after eye opening [up to postnatal day 18 (P18)] (Desai et al., 2002; Maffei et al., 2004). Beginning around P19 (after the opening of the classical critical period in rodents) (Fagiolini et al., 1994; Gordon and Stryker, 1996), synaptic scaling can no longer be induced in layer 4 by brief visual deprivation (Maffei et al., 2004, 2006) and instead migrates to layer 2/3, where it can be induced into adulthood (Desai et al., 2002; Goel and Lee, 2007). Visual cortex therefore is endowed with layer-specific critical periods for homeostatic plasticity (Desai et al., 2002).
In addition to synaptic scaling, visual deprivation during the critical period can induce prominent LTD at excitatory synapses (Kirkwood et al., 1996; Rittenhouse et al., 1999), raising the question of how these two opposing forms of excitatory synaptic plasticity interact. To begin to address this, we studied the cellular and synaptic changes induced in layer 2/3 by two modes of visual deprivation: intraocular TTX injection and lid suture (LS). Whereas LS reduces visual drive and decorrelates visual inputs (Wiesel and Hubel, 1965; Van Sluyters and Levitt, 1980), TTX blocks any discharge from the optic nerve and has been shown to rapidly and persistently reduce activation of lateral geniculate nucleus neurons driven by the deprived eye (Stryker and Harris, 1986; Rittenhouse et al., 1999). LS and TTX differentially engage LTD-like synaptic changes (Frenkel and Bear, 2004), but whether they also differentially engage homeostatic plasticity has not been explored. Here we show that both forms of sensory deprivation increase the spontaneous firing of layer 2/3 pyramidal neurons, suggesting a general network-level compensation for reduced sensory drive. TTX increased spontaneous activity through an increase in the excitation/inhibition (E/I) balance within layer 2/3. In contrast, LS decreased the E/I balance but increased the intrinsic excitability of layer 2/3 pyramidal neurons. The microcircuit in layer 2/3 can thus use different forms of homeostatic plasticity to compensate for the loss of visual drive, depending on the specific demands produced by visual experience.
Materials and Methods
Monocular deprivation.
Monocular eyelid suture and intraocular TTX injections were performed on P18 Long–Evans rats and were maintained until P21. All experimental procedures were approved by the Brandeis Animal Use Committee and followed the guidelines of the National Institutes of Health. Animals were anesthetized with a mixture containing 70 mg/kg ketamine, 3.5 mg/kg xylazine hydrochloride, and 0.7 mg/kg acepromazine maleate, injected intraperitoneally. The area surrounding the eye was cleaned with isopropanol, eye drops were administered to maintain a good level of eye moisture, and the eye was covered with a thin layer of xylocaine gel. Lids were closed with four mattress sutures using 6-0 polyester (Ethicon, Somerville, NJ). The closure of the eye was checked every day. Intraocular TTX injection was repeated every 24 h by inserting a micropipette tip of <30 μm in diameter at the ora serrata. A volume of 2 μl of 0.1 mm TTX was injected at a rate of 1 μl/min with a microinjection pump (Harvard Apparatus, Holliston, MA). Dilation of the pupil was observed after every injection to confirm the effectiveness of the TTX injection and was checked again after 24 h to verify that the block was maintained.
Electrophysiology.
Coronal slices of 300 μm were obtained from the hemisphere contralateral to the open eye (control condition) and contralateral to the injected or closed eye (deprived condition). Visualized patch-clamp recordings were performed by targeting layer 2/3 pyramidal neurons in the monocular portion of primary visual cortex (V1). The identity of the cell type was further confirmed by testing their firing pattern in response to depolarizing current steps and by post hoc biocytin staining and morphological reconstruction.
Spontaneous firing was measured as described previously (Maffei et al., 2004). Briefly, slices were incubated in a modified artificial CSF (ACSF) 1 h before recording, and spontaneous spiking activity was recorded in current clamp at a bath temperature of 35°C. Spontaneous firing rates were recorded by injecting a small amount of bias current to bring the resting membrane potential to −60 mV and were then allowed to fluctuate freely in response to spontaneous synaptic events. Spontaneous excitatory and inhibitory currents were recorded in voltage clamp after incubation in the modified ACSF as described previously (Maffei et al., 2004). To isolate combined AMPA–NMDA currents, neurons were held at the E rev for GABAA-mediated currents, and vice versa. The E rev for each was determined by holding neurons between −50 and −30 mV in 5 mV increments (GABAA) and between −5 and +15 mV (AMPA–NMDA). When corrected for the liquid junction potential, the average E rev for GABAA was −49.3 ± 1.1 mV and for AMPA–NMDA was 9.6 ± 0.9 mV, close to the predicted values for our internal solution. The spontaneous excitatory and inhibitory synaptic charge was obtained by integrating the current traces at the E rev for inhibition and excitation, respectively.
Paired recording techniques were used to measure strength and dynamics of monosynaptic recurrent connections within layer 2/3 (Maffei et al., 2004, 2006). Patch-clamp recordings were obtained from quadruplets of visually identified pyramidal neurons. Presynaptic neurons were activated to fire trains of five action potentials precisely timed at 20 Hz, whereas postsynaptic neurons were voltage clamped at −70 mV. Each connected pair was tested by recording 40 repetitions. Off-line average traces were obtained for each pair, and the amplitudes of the five EPSCs in the train were measured. Differences in the strength of the connection were obtained by comparing the amplitudes of the first EPSC in the train across conditions. The paired-pulse ratio (PPR) was calculated as the ratio of EPSC 2/EPSC 1 for each pair, and average values were compared across conditions. The coefficient of variation (CV) of EPSC amplitude was calculated as the SD/mean amplitude (for the first EPSC in the train) for each pair.
Evoked feedforward EPSCs and IPSCs were obtained with extracellular stimulation in layer 4. A bipolar tungsten electrode was positioned in layer 4, and patch-clamp recordings were obtained from pyramidal neurons in layer 2/3. EPSCs were recorded by holding the neurons at the E rev for GABAA-mediated currents, and IPSC were recorded at E rev for AMPA–NMDA-mediated currents. The distance between stimulating and recording electrodes was generally 300 μm (325 ± 25 μm), and we used electrodes with a distance between the poles that did not exceed 50 ± 15 μm with a 1 MΩ resistance, to obtain very focal and reliable stimulation across slices. Evoked responses were successfully induced in pyramidal neurons positioned vertically directly above the stimulating electrode. In recordings where two pyramidal neurons were recorded simultaneously, synaptic currents were always successfully evoked in the one positioned directly above the stimulating electrode, but not in neurons positioned, on average, 50 ± 10 μm lateral. For each neuron, we recorded evoked EPSCs and IPSCs in response to increasing intensity of stimulation by holding the neuron sequentially at the E rev for inhibition and excitation as described above. We compared minimal amplitude EPSCs and IPSCs across experimental conditions, by slowly increasing the stimulus intensity until synaptic responses were evoked ∼50% of the time; the minimal nature of these responses was confirmed by the presence of an amplitude plateau as the stimulus was increased further (see Fig. 5 B, arrows indicate plateau).
Figure 5.
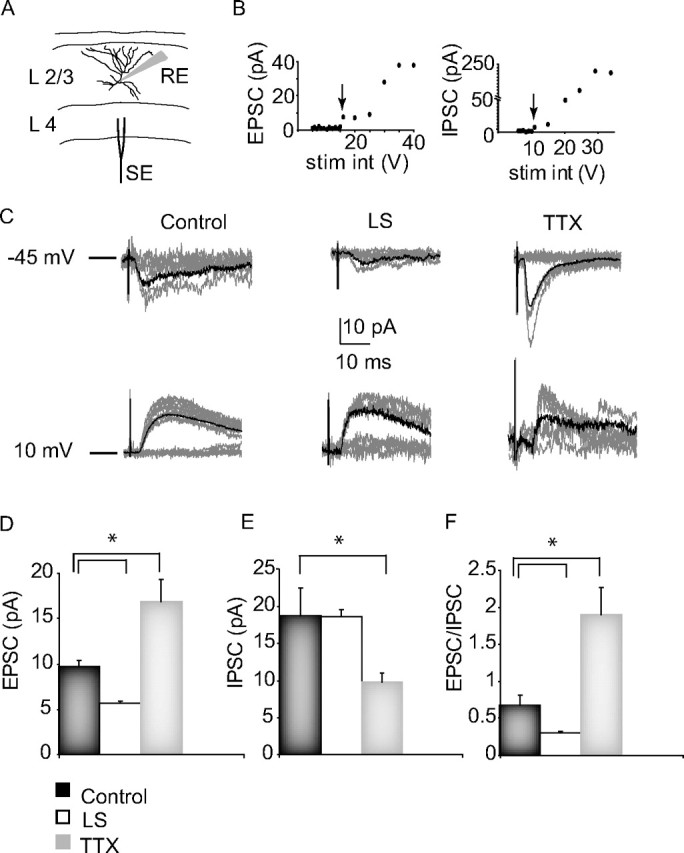
The effects of visual deprivation on evoked minimal synaptic transmission onto layer 2/3 pyramidal neurons from lower layers. A, Diagram of the experimental setup, showing the laminar and lateral positions of the whole-cell recording (RE) and the bipolar stimulating electrode (SE), and a morphological reconstruction of a layer 2/3 pyramidal neuron. B, Example EPSC and IPSC amplitude versus stimulus intensity (stim int) plots. The arrows indicate the minimal EPSC and IPSC for this neuron. C, Example minimal evoked EPSCs (−45 mV) and IPSCs (+10 mV) from control, LS, and TTX conditions. Each plot shows 10 responses for each condition (gray traces). The average of 30 events for each condition is overlaid in black. D, Average amplitudes (EPSC), paired-pulse ratio (PP ratio), and CV of minimal EPSC for control (black bar), LS (white bar), and TTX (gray bar) conditions. The same color code will be used also for the plots in E and F. E, Average amplitude (IPSC), PP ratio, and CV of minimal IPSC for each condition. F, Average EPSC/IPSC ratio (computed for each neuron and then averaged for each condition). *Significantly different from control.
Frequency versus current intensity curves were plotted by measuring the average rate of action potentials in current clamp during 500-ms-long depolarizing steps of increasing intensity in the presence of synaptic blockers (in μm: 50 APV, 20 DNQX, and 20 bicuculline). The spike threshold was measured as the interpolated membrane potential at which dV/dt equaled 20 V/s. All neurons included in the analysis showed less than a 20% change in R in, V m, and R s.
Statistical analysis.
All data are presented as mean ± SEM for the number of neurons indicated. To determine statistical significance, single-factor ANOVAs were run for each data set; if significant, this was followed by two-tailed Student's t tests corrected for multiple comparisons using a Bonferroni correction. In some cases, the nonparametric Mann–Whitney test (also corrected for multiple comparisons) was used for underlying distributions with large deviations from normality. The statistical significance for connection probability was assessed using Pearson χ2 for contingency test. Corrected p values ≤0.05 were considered significant.
Solutions.
Regular ACSF contained (in mm) 126 NaCl, 3 KCl, 2 MgSO4, 1 NaHPO4, 25 NaHCO3, 2 CaCl2, and 25 dextrose. For paired recordings, the Mg2+ concentration was 1 mm. Modified ACSF for inducing spontaneous firing contained (in mm) 124 NaCl, 3.5 KCl, 0.5 MgCl2, 1.25 NaHPO4, 26 NaHCO3, 1 CaCl2, and 25 dextrose. The internal solution for current-clamp and paired recordings contained 20 mm KCl, 100 mm K-gluconate, 10 mm HEPES, 0.2% biocytin, 4 mm Mg-ATP, 0.3 mm Na-GTP, and 10 mm Na-phosphocreatine. For measuring spontaneous currents and feedforward EPSC and IPSC from layer 4, the internal solution contained (in mm) 20 KCl, 100 Cs-methyl-sulfonate, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 Na-phosphocreatine, and 3 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide].
Results
We investigated the plasticity mechanisms underlying changes in layer 2/3 circuit excitability in response to two different paradigms of sensory deprivation: intraocular TTX injection and LS. One eye was visually deprived, and all recordings were performed in monocular cortex from the deprived and the spared hemispheres as described previously (Maffei et al., 2004, 2006). After 2 d of visual deprivation, we prepared coronal slices containing primary visual cortex from P21 rats. Patch-clamp recordings were obtained to measure excitatory and inhibitory synaptic connections and intrinsic excitability from pyramidal neurons in layer 2/3.
Both LS and TTX increase circuit excitability in layer 2/3
Reduced visual drive can induce compensatory homeostatic changes in layer 4 circuit excitability during early postnatal life, but not later during the classical rodent critical period for visual system plasticity (after P19). We began by asking whether layer 2/3 exhibits compensatory changes in excitability during the classical rodent visual system critical period. One probe for changes in microcircuit excitability is the spontaneous firing of pyramidal neurons, which is strongly affected by changes in excitation and inhibition within the local microcircuit (Maffei et al., 2004, 2006; Dani et al., 2005). We therefore obtained current-clamp recordings from visually identified layer 2/3 pyramidal neurons and measured spontaneous activity in slices from the control and deprived hemispheres.
Both LS and TTX increased the spontaneous firing rates of layer 2/3 pyramidal neurons by approximately fourfold over the control hemisphere (control: 0.78 ± 0.24, n = 10; LS: 3.57 ± 0.54 Hz, n = 8, p < 0.004; TTX: 3.08, n = 8, p < 0.006) (Fig. 1 A, B). In control hemispheres from both LS- and TTX-treated animals, spontaneous firing was comparable (control LS: 0.78 ± 0.21 Hz, n = 5; control TTX: 0.55 ± 0.15 Hz, n = 5; p = 0.2; ANOVA, p = 0.45) (Fig. 1 B). This was also the case for every parameter we measured; therefore, data from the control hemispheres for the two visual deprivation conditions were combined throughout this manuscript. These data indicate that reduced visual drive induces a compensatory readjustment in microcircuit excitability within layer 2/3. Layer 2/3 thus becomes a locus for homeostatic plasticity during the critical period, when layer 4 has lost the ability to produce any compensatory response to visual deprivation (Desai et al., 2002; Maffei et al., 2006). Furthermore, this homeostatic increase in circuit excitability is a robust response that does not depend on the mode of visual deprivation.
Figure 1.

Sensory deprivation increased layer 2/3 pyramidal neuron spontaneous firing. A, Examples of spontaneous firing from layer 2/3 pyramidal neurons from control (black), LS (dark gray), and TTX (light gray) conditions. B, Bar plot showing average spontaneous firing frequency for each condition. Data from slices ipsilateral to the sutured eye (control LS; black bar), contralateral to the sutured eye (LS; white bar), ipsilateral to the TTX-injected eye (control TTX; dark gray bar), and contralateral to the TTX-injected eye (TTX; light gray bar) are shown. *Significantly different from control.
Visual deprivation alters the E/I balance
Increased spontaneous activity could be achieved through several different mechanisms, among them increased excitatory drive, decreased inhibitory drive, or increased intrinsic excitability. To determine which mechanism(s) is used within layer 2/3, we began by examining the balance between total excitatory and inhibitory synaptic charge onto layer 2/3 pyramidal neurons after TTX and LS, by holding layer 2/3 pyramidal neurons at the reversal potential for inhibitory and excitatory currents, respectively, and quantifying the spontaneous synaptic charge (Maffei et al., 2004) (see Materials and Methods). TTX increased total excitatory charge to 166.7 ± 38.6% of control (n = 6; p < 0.02) (Fig. 2, AMPA–NMDA) and decreased inhibitory charge to 47.9 ± 7.1% of control (n = 6; p < 0.004) (Fig. 2, GABAA). The E/I ratio was increased to 255.3 ± 25.9% control (n = 6; p < 0.006) (Fig. 2). This increase in the E/I ratio is likely a major contributor to the increased spontaneous firing of pyramidal neurons after TTX treatment. In contrast to TTX, LS reduced the excitatory drive onto pyramidal neurons to 35.3 ± 8.4% of control (control, n = 13; LS, n = 6; p < 0.02) (Fig. 2), whereas the total inhibitory charge was unchanged (9.5 ± 16.2% change; p = 0.61) (Fig. 3, LS). The E/I ratio was therefore reduced to 32.4 ± 4.9% of control in this condition (p < 0.02) (Fig. 2). These data indicate that although TTX and LS produce a similar increase in spontaneous firing, the mechanism by which this is achieved is fundamentally different.
Figure 2.

TTX increases, whereas LS decreases, the E/I balance. A, Examples of spontaneous excitatory and inhibitory synaptic currents from layer 2/3 pyramidal neurons in control, LS, and TTX conditions; neurons were held sequentially at the E rev for GABAA currents (−45 mV) to isolate spontaneous excitatory currents and AMPA–NMDA (+10 mV) to isolate spontaneous inhibitory currents. B, Average excitatory and inhibitory synaptic charge in control (black), LS (white), and TTX (gray) conditions. C, Average excitation (Exc)/inhibition (Inh) ratio (computed for individual neurons and then averaged for each condition) for control (black), LS (white), and TTX (gray) conditions. *Significantly different from control.
Figure 3.

TTX increases, whereas LS decreases, mEPSC amplitude. A, Example traces showing raw mEPSC recordings (top) and average mEPSCs (bottom) for control (black), LS (dark gray), and TTX (light gray) conditions. B, Scaled average mEPSCs for each condition; control scaled to peak of LS (top) and to peak of TTX (bottom). Note similar kinetics in each condition. C, Average mEPSC amplitude (Amp) and frequency (Freq), computed for each neuron and then averaged for each condition. Data are presented as percentage of control. Black bar, Control; white bar, LS; gray bar, TTX. D, Cumulative distribution of mEPSC amplitudes in control (black), LS (dark gray), and TTX (light gray). E, Cumulative distributions of intereven intervals (IEI) between mEPSC for control (black), LS (dark gray), and TTX (light gray) conditions. For cumulative histograms, 50 events/neuron were included. *Significantly different from control.
Experience-dependent plasticity at excitatory layer 2/3 synapses
Previous experiments showed that intraocular TTX injection (Desai et al., 2002) and dark rearing (Desai et al., 2002; Goel and Lee, 2007) during the critical period induce synaptic scaling of AMPA miniature EPSCs (mEPSCs) onto layer 2/3 pyramidal neurons. Because TTX and LS have different effects on the E/I ratio, we compared their abilities to induce synaptic scaling. We measured AMPA mEPSCs from deprived and control hemispheres after TTX and LS and compared their amplitude and frequency. TTX increased mEPSC amplitude to 131.4 ± 11.8% of control (n = 10; p < 0.05) and shifted the entire amplitude distribution toward larger values, without affecting mEPSC frequency (Fig. 3, TTX) (p = 0.75). In contrast, LS depressed mEPSC amplitude to 89.4 ± 5.6% of control (n = 27; p < 0.012) (Fig. 3, LS) and also significantly decreased mEPSC frequency (control: 2.1 ± 0.2 Hz, n = 27; LS: 1.4 ± 0.1 Hz, n = 27; p < 0.02) (Fig. 3, LS). There was no significant effect on mEPSC rise times (control, 2.04 ± 0.26 ms; LS, 2.45 ± 0.31 ms; TTX, 2.19 ± 0.32 ms; ANOVA, p = 0.73) and decay time constants (control, 4.16 ± 0.52 ms; LS, 4.14 ± 0.31 ms; TTX, 4.32 ± 0.39 ms; ANOVA, p = 0.53) for either condition. These data confirm that TTX induces synaptic scaling at layer 2/3 pyramidal synapses and are consistent with the increase in total excitatory drive onto layer 2/3 elicited by TTX treatment (Fig. 2 B). Consistent with the reduction in total excitatory synaptic drive induced by LS (Fig. 2 B), LS depressed mEPSCs at these same synapses, possibly through an LTD-like mechanism.
mEPSC recordings sample from all excitatory inputs onto layer 2/3 pyramidal neurons, which arise from several sources. One prominent source is the recurrent connections from other layer 2/3 pyramidal neurons. To determine how evoked transmission at this recurrent synapse is affected by sensory deprivation, we compared the connection probability, synaptic strength, and short-term plasticity in control, TTX, and LS conditions using paired recording techniques. With both visual deprivation protocols, there was a large and significant decrease in EPSC amplitude (TTX, p < 0.01; LS, p < 0.046; n = 10 from each condition) (Fig. 4) and a significant increase in the PPR (TTX, p < 0.01; LS, p < 0.04) (Fig. 4), the CV (TTX, p < 0.05; LS, p < 0.014) (Fig. 4), and the rate of failures (TTX, p < 0.038; LS, p < 0.048) (Fig. 4). No significant differences were observed in connection probability. These changes indicate a major presynaptic contribution to the reduction in unitary EPSCs and suggest that both TTX and LS induce a presynaptic form of synaptic depression at this synapse.
Figure 4.

Both TTX and LS depressed the amplitude of recurrent monosynaptic connections between layer 2/3 pyramidal neurons. A, Examples of evoked recurrent EPSCs in response to the firing of presynaptic neurons at 20 Hz in control, LS, and TTX conditions. B, Average EPSC amplitude (Amp; in picoamperes), paired-pulse ratio (PP ratio), CV, and failure rates (% failures) for control (black bar), LS (white bar), and TTX (gray bar) conditions. *Significantly different from control.
Feedforward excitatory and inhibitory synaptic transmission
Although TTX and LS have opposing effects on mEPSC amplitude and on total excitatory synaptic drive, they both reduce the amplitude of recurrent EPSCs. Together, the data on LS and TTX suggest that several forms of synaptic plasticity coexist at layer 2/3 synapses and are induced in different combinations by TTX and LS. Our data suggest that LS produces a generalized depression of excitatory transmission onto layer 2/3 neurons, whereas TTX produces a net potentiation of excitatory transmission coupled to a selective depression of recurrent excitatory synapses. To support this, we wanted to compare recurrent layer 2/3 connections to a second input pathway onto layer 2/3 pyramidal neurons. A major source of excitatory drive to layer 2/3 neurons comes from excitatory neurons in layer 4; layer 2/3 neurons also receive excitatory input from layer 5 (Burkhalter, 1999). The relative contribution to experience-dependent plasticity of recurrent inputs and inputs external to layer 2/3 is not known.
To activate inputs from lower layers to layer 2/3, we positioned a bipolar tungsten electrode in layer 4 and performed patch-clamp recordings from pyramidal neurons in layer 2/3 (Fig. 5 A). Our aim was to compare putative single axon evoked EPSCs and IPSCs from axons in layer 4 onto layer 2/3 pyramidal neurons. For each neuron, we recorded evoked EPSCs and IPSCs by holding the postsynaptic neuron at the reversal potential for IPSCs and EPSCs, respectively (Maffei et al., 2004). By slowly increasing the stimulus intensity, we were able to identify minimal responses (Fig. 5 B) and compare the amplitude of these minimal evoked currents in the control, TTX, and LS conditions (Fig. 5 C). The monosynaptic nature of these evoked responses was confirmed by the short and constant delay between the stimulus and the onset of synaptic currents (Fig. 5 C) (control: EPSC, 3.3 ± 0.3 ms; IPSC, 3.8 ± 0.5; LS, 2.9 ± 0.3 ms; TTX: EPSC, 3.5 ± 0.5; IPSC, 4.4 ± 0.4 ms; ANOVA, p = 0.83).
TTX increased the minimal feedforward EPSC to 174.2 ± 14.4% of control (p < 0.04), without affecting the PPR or CV (Fig. 5 D), consistent with a postsynaptic increase in synaptic transmission. In contrast, TTX decreased the minimal feedforward IPSC to 48.4 ± 13.4% of control, and this change was accompanied by a significant increase in CV, consistent with a presynaptic locus of change (control, n = 8; TTX, n = 7; p < 0.047) (Fig. 5 E). The E/I balance of the feedforward input to layer 2/3 was thus shifted to favor excitation, consistent in both direction and magnitude with our data on total synaptic drive onto these neurons (Fig. 5 D, EPSC/IPSC; compare also Fig. 2, TTX). These data strongly suggest that intraocular TTX induces a selective depression of layer 2/3 recurrent connections, while at the same time boosting other excitatory inputs onto layer 2/3 pyramidal neurons.
A very different picture arose from the LS experiments. Consistent with the reduction in total excitatory synaptic drive induced by LS (Fig. 2), LS decreased the feedforward minimal EPSC amplitude to 40.9 ± 4.2% of control, while increasing the CV and PPR (Fig. 5 C,D, LS) (control, n = 8; LS, n = 6; p < 0.002). Also consistent with the lack of change in total inhibitory input after LS (Fig. 2), the feedforward minimal IPSCs were unaffected by LS (Fig. 5 C,E) (1.2 ± 5.1%; p = 0.94). Together, these data show that LS and TTX act differently on the cortical microcircuit in layer 2/3. LS produces a generalized decrease in excitatory drive onto layer 2/3 pyramidal neurons without affecting inhibitory synaptic transmission, through a net decrease in the E/I ratio (Fig. 5 F) (p < 0.05). In contrast, TTX shifts the E/I ratio to favor excitation, through reciprocal changes in net excitation (an increase) and inhibition (a decrease).
Increased intrinsic excitability after eyelid suture
The shift in the E/I balance produced by TTX can account nicely for the increase in spontaneous firing rates of layer 2/3 pyramidal neurons. However, LS shifts this balance to favor inhibition, which would be expected to decrease, rather than increase, layer 2/3 pyramidal neuron firing rates. In addition to synaptic changes, activity deprivation can induce homeostatic changes in intrinsic neuronal excitability by modulating voltage-dependent conductance (Desai et al., 1999a; Aizenman et al., 2003). There is therefore the possibility that the increase in spontaneous firing of layer 2/3 pyramidal neurons after LS could be driven by changes in intrinsic excitability, which might compensate for (and overcome) the effects of decreased excitatory synaptic drive.
To probe for changes in intrinsic excitability, we measured the frequency of action potential firing to different levels of depolarizing current injection (FI curve), in the presence of blockers of synaptic transmission. There was no significant change in the FI curve after TTX (Fig. 6). However, there was a significant shift of the FI curve to the left after LS, indicating an increase in intrinsic excitability (Fig. 6, LS). This shift was accompanied by a significant increase in firing rate for each depolarizing current step tested (Fig. 6 A,B) (p < 0.02). LS also produced a large and significant decrease in the threshold voltage for action potential generation (Fig. 6 C) (action potential threshold decreased by 4.9 ± 0.5 mV; control, n = 11; LS, n = 9; p < 0.0006), whereas resting membrane potential, input resistance, and membrane time constant were not significantly affected by either treatment (Fig. 6 C). These data suggest that the major factor driving the increase in layer 2/3 pyramidal neuron firing after LS is an increase in intrinsic excitability.
Figure 6.

Plasticity of intrinsic excitability. A, Example firing in response to DC depolarizing current steps for control, LS, and TTX conditions. For each neuron, the response to the same three current injection amplitudes are shown from low (black) to middle (dark gray) to high (light gray). B, Frequency versus current curves for control (black circles), LS (white circles), and TTX (gray circles) conditions. Asterisks highlight significant differences. C, Summary plots for action potential threshold (AP thr), resting membrane potential (V m), resting input resistance (R in), and membrane time constant (tau) for control (black bar), LS (white bar), and TTX (gray bar). Data are mean ± SEM. *Different from control.
Discussion
Here we show that brief periods of visual deprivation ending at P21 induce a compensatory increase in the excitability of the layer 2/3 microcircuit that manifests as an increase in the spontaneous firing of pyramidal neurons. This network compensation is robust and independent of the mode of visual deprivation, because it can be induced by both LS and blockade of retinal activity with TTX. The forms of plasticity that underlie this increased excitability, however, are strongly dependent on the sensory deprivation paradigm (Fig. 7). Our data suggest that a palette of homeostatic mechanisms (changes in excitation, inhibition, and intrinsic excitability) can be differentially recruited depending on which forms of synapse-specific plasticity are engaged by a particular deprivation paradigm.
Figure 7.

Summary diagram of microcircuit changes induced by LS and intraocular TTX. Pyramidal neurons (black) and inhibitory interneurons (gray) are show in layers 4 and 2/3, under control conditions (top) and after LS (bottom left) or intraocular TTX injection (bottom right). Both LS and TTX increased the spontaneous firing rate of layer 2/3 pyramidal neurons, but via distinct mechanisms. LS reduced excitatory synaptic drive onto layer 2/3 pyramidal neurons from multiple sources through a mixture of presynaptic and postsynaptic mechanisms (denoted by thin axons) but increased the intrinsic excitability of layer 2/3 pyramidal neurons to compensate (denoted by somatic arrows). In contrast, TTX produced a selective presynaptic depression of recurrent layer 2/3 excitatory connections but scaled up mEPSCs as well as evoked excitatory drive from layer 2/3 (denoted by thick axons), while simultaneously reducing inhibitory drive (denoted by thin axons). The net effect of these changes was to shift the E/I balance toward increased excitation.
Recent experiments have shown that both Hebbian and homeostatic forms of plasticity contribute to experience-dependent changes in visual cortex, and multiple synaptic plasticity mechanisms can coexist at particular neocortical excitatory and inhibitory synapses. At excitatory recurrent layer 5 synapses, presynaptic and postsynaptic forms of LTP coexist with a presynaptic form of LTD, and all three can be activated to different degrees by correlated presynaptic and postsynaptic firing (Markram et al., 1997; Sjostrom et al., 2001, 2003, 2007). Feedforward connections onto layer 2/3 pyramidal neurons also exhibit a presynaptic form of LTD in both visual and somatosensory cortex, and this LTD can be partially occluded by previous sensory deprivation (Kirkwood et al., 1996; Allen et al., 2003; Bender et al., 2006; Crozier et al., 2007). Synaptic scaling has been demonstrated at excitatory layer 4 synapses early in postnatal development and at layer 2/3 synapses later in development, in response to both intraocular TTX injection and dark rearing (Desai et al., 2002; Goel and Lee, 2007). Finally, in response to visual deprivation, inhibitory synapses onto layer 4 pyramidal neurons exhibit homeostatic plasticity early in development (Maffei et al., 2004) and a form of anticorrelation induced inhibitory LTP during the critical period (Maffei et al., 2006). Our data show that, during the critical period, intraocular TTX and LS activate different subsets of these available plasticity mechanisms within layer 2/3.
Visual deprivation via intraocular TTX injection shifted the balance between excitation and inhibition in layer 2/3 to favor excitation, through a combination of increased excitatory and decreased inhibitory synaptic drive. Quantal excitatory currents onto layer 2/3 pyramids were increased in amplitude (but not frequency), consistent with the induction of synaptic scaling (Turrigiano et al., 1998; Desai et al., 2002). Interestingly, although feedforward excitatory connections from layers 4 and 5 were enhanced, there was a selective presynaptic depression of recurrent layer 2/3 excitatory synapses. This suggests that blocking retinal activity modifies layer 2/3 pyramidal neuron firing in such a way as to drive presynaptic LTD at this synapse. This depression is more than compensated for by the combined effects of global postsynaptic enhancement of excitation and a reduction in inhibition, so that the net effect of the synaptic changes in the local microcircuit is to enhance drive from layer 4/5 and to render layer 2/3 more excitable (Fig. 7, TTX). This shift in the E/I balance closely resembles that seen in layer 4 in response to visual deprivation during the precritical period (Maffei et al., 2004), suggesting that homeostatic regulation of the E/I balance is a common compensatory mechanism used across cortical layers and developmental time periods.
It has been suggested that LS drives cortical LTD more effectively than intraocular TTX because of higher levels of uncorrelated activity from the periphery (Rittenhouse et al., 1999). Consistent with this view, LS induced a strong and pervasive depression of excitatory synapses onto layer 2/3 pyramidal neurons. Recurrent excitatory layer 2/3 connections were strongly depressed, as were feedforward excitatory inputs from layer 4. This depression had both a presynaptic component (suggested by changes in CV and PPR) and a postsynaptic component (suggested by a reduction in mEPSC amplitude). It is unclear why LS did not produce a net scaling up of the amplitude of mEPSCs onto layer 2/3 neurons. Synaptic scaling can be induced by intraocular TTX injection (Desai et al., 2002; present study) and by brief periods of dark rearing (Desai et al., 2002; Goel and Lee, 2007) at this developmental stage, so the inability of LS to scale up excitatory synaptic strengths is not attributable to the absence of synaptic scaling at this synapse. One explanation may be that LS induces such strong postsynaptic LTD at layer 2/3 synapses that synaptic scaling cannot overcome it; the effect of LS on mEPSCs may thus be the net effect of opposing changes produced by LTD and synaptic scaling, with LTD winning.
LS increased spontaneous layer 2/3 pyramidal neuron firing to the same degree as intraocular TTX injection, but rather than changing the E/I balance, this increase was driven by changes in intrinsic excitability (Fig. 7, LS). Plasticity of intrinsic excitability has now been demonstrated in several systems and, like synaptic plasticity, can take a variety of forms, some of which are destabilizing and some of which are homeostatic (Turrigiano et al., 1994; Desai et al., 1999a,b; Aizenman et al., 2003; Marder and Prinz, 2003; Zhang and Linden, 2003; Fan et al., 2005; Frick and Johnston, 2005). Several recent studies have shown that changes in intrinsic excitability contribute to network-level homeostatic compensation in the visual system of lower vertebrates and can function to increase the signal-to-noise ratio (Aizenman et al., 2003; Pratt and Aizenman, 2007), but homeostatic intrinsic excitability has not previously been demonstrated in the intact mammalian visual system. Here we show that after LS, layer 2/3 pyramidal neurons became more intrinsically excitable, as measured by a shift to the left of the FI curve and a decrease in action potential threshold. These effects were large enough to overcome the depression of excitatory drive and generate a net increase in spontaneous firing rates in layer 2/3 pyramidal neurons. Intrinsic changes were specific to LS, because intraocular TTX had no effect on intrinsic excitability. In neocortical cultures (Desai et al., 1999a) and hippocampal circuits (Karmarkar and Buonomano, 2006; Echegoyen et al., 2007), complete blockade of spikes with TTX induces homeostatic intrinsic and synaptic changes in parallel, but in vivo manipulations of sensory input (which lower drive but do not block spiking) can induce synaptic scaling in layer 4 without affecting intrinsic excitability (Maffei et al., 2004). Together with the data presented here, these studies suggest that in neocortical circuits intrinsic homeostatic plasticity is recruited only when synaptic homeostatic mechanisms are not sufficient to fully compensate for a drop in sensory drive, either because they are opposed by strong destabilizing forms of synaptic plasticity or because the feedback signal (firing) is absent (Desai et al., 1999a).
In invertebrate preparations, it has been shown that similar rhythmic patterns can be achieved through a combination of different synaptic and intrinsic properties (Prinz et al., 2004). Our data suggest that cortical microcircuits are endowed with the ability to reach a target level of excitability using at least two fundamentally different homeostatic mechanisms: changes in the E/I balance and changes in the intrinsic excitability of principal neurons. Different modes of visual deprivation (LS, intraocular TTX, and dark rearing) are likely to have very different effects on correlated firing within cortical networks and to differentially engage correlation-based and/or spike-timing-dependent plasticity mechanisms. This may, in turn, necessitate the use of different forms of homeostatic plasticity to reach a similar level of excitability. Interestingly, whereas the output of layer 2/3 (spontaneous firing rates) is similar after TTX and LS, the microcircuit is configured very differently in terms of synaptic strengths, and the consequences for visual function may be quite different. One possible effect of the increase in intrinsic excitability of layer 2/3 pyramidal neurons after LS is to increase their sensitivity to strong sensory stimuli that still manage to propagate through layer 4. Decreased excitatory transmission coupled to increased intrinsic excitability may ensure that small noisy signals are ignored, while salient signals are still effective.
The ability to compensate for decreased sensory input by increasing circuit excitability may be a critical feature of cortical networks that allows them to maintain sensitivity in the face of fluctuations in drive. Our data argue that the effects of sensory deprivation cannot be explained by a single form of synaptic plasticity, but rather arise through a complex interplay between many forms of excitatory, inhibitory, and intrinsic mechanisms occurring at many sites within the microcircuit. Most of these forms of plasticity have been independently proposed as mechanisms for experience-dependent changes in visual response properties (Kirkwood et al., 1996; Maffei et al., 2004, 2006; Hensch, 2005; Crozier et al., 2007; Mrsic-Flogel et al., 2007), but understanding experience-dependent plasticity will likely require an integrated understanding of how these various forms of plasticity cooperate to modify microcircuit function. The existence of such a rich palette of plasticity mechanisms suggests that cortical microcircuits can respond in a very flexible manner to changes in sensory input. In particular, the existence of redundant forms of homeostatic plasticity may ensure that network compensation can be achieved in response to a wide range of sensory perturbations.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant EY 014429 (G.G.T.) and NIH Training Grant T32 NS07292 (A.M.). We thank Roman Pavlyuk for help with histology.
References
- Aizenman C, Akerman C, Jensen K, Cline H. Visually driven regulation of intrinsic neuronal excitability improves stimulus detection in vivo. Neuron. 2003;39:831–842. doi: 10.1016/s0896-6273(03)00527-0. [DOI] [PubMed] [Google Scholar]
- Allen CB, Celikel T, Feldman DE. Long-term depression induced by sensory deprivation during cortical map plasticity in vivo. Nat Neurosci. 2003;6:291–299. doi: 10.1038/nn1012. [DOI] [PubMed] [Google Scholar]
- Bender KJ, Allen CB, Bender VA, Feldman DE. Synaptic basis for whisker deprivation-induced synaptic depression in rat somatosensory cortex. J Neurosci. 2006;26:4155–4165. doi: 10.1523/JNEUROSCI.0175-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhalter A. Intrinsic connections of rat primary visual cortex: laminar organization of axonal projections. J Comp Neurol. 1999;279:171–186. doi: 10.1002/cne.902790202. [DOI] [PubMed] [Google Scholar]
- Crozier RA, Wang Y, Liu CH, Bear MF. Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc Natl Acad Sci USA. 2007;104:1383–1388. doi: 10.1073/pnas.0609596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani V, Chang Q, Maffei A, Turrigiano G, Jaenisch R, Nelson S. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci USA. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I. Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol. 2001;63:847–869. doi: 10.1146/annurev.physiol.63.1.847. [DOI] [PubMed] [Google Scholar]
- Desai N, Cudmore R, Nelson S, Turrigiano G. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002;5:783–789. doi: 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci. 1999a;2:515–520. doi: 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. 1999b;6:284–291. [PMC free article] [PubMed] [Google Scholar]
- Echegoyen J, Neu A, Graber KD, Soltesz I. Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PLoS ONE. 2007;2:e700. doi: 10.1371/journal.pone.0000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Pizzorusso T, Berardi N, Domenici L, Maffei L. Functional postnatal development of the rat primary visual cortex and the role of visual experience: dark rearing and monocular deprivation. Vision Res. 1994;34:709–720. doi: 10.1016/0042-6989(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager D, Chen X, Lu H, Chitwood R, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I (h) Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- Feller MB, Scanziani M. A precritical period for plasticity in visual cortex. Curr Opin Neurobiol. 2005;15:94–100. doi: 10.1016/j.conb.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Fox K. Anatomical pathways and molecular mechanisms for plasticity in the barrel cortex. Neuroscience. 2002;111:799–814. doi: 10.1016/s0306-4522(02)00027-1. [DOI] [PubMed] [Google Scholar]
- Frenkel M, Bear M. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron. 2004;44:917–923. doi: 10.1016/j.neuron.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Frick A, Johnston D. Plasticity of dendritic excitability. J Neurobiol. 2005;64:100–115. doi: 10.1002/neu.20148. [DOI] [PubMed] [Google Scholar]
- Goel A, Lee H. Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J Neurosci. 2007;27:6692–6700. doi: 10.1523/JNEUROSCI.5038-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JA, Stryker MP. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci. 1996;16:3274–3286. doi: 10.1523/JNEUROSCI.16-10-03274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hübener M. Lifelong learning: ocular dominance plasticity in mouse visual cortex. Curr Opin Neurobiol. 2006;16:451–459. doi: 10.1016/j.conb.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Karmarkar UR, Buonomano DV. Different forms of homeostatic plasticity are engaged with distinct temporal profiles. Eur J Neurosci. 2006;23:1575–1584. doi: 10.1111/j.1460-9568.2006.04692.x. [DOI] [PubMed] [Google Scholar]
- Katz L, Shatz C. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Rioult M, Bear M. Experience-dependent modification of synaptic plasticity in visual cortex. Nature. 1996;381:526–528. doi: 10.1038/381526a0. [DOI] [PubMed] [Google Scholar]
- Maffei A, Nelson S, Turrigiano G. Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci. 2004;7:1353–1359. doi: 10.1038/nn1351. [DOI] [PubMed] [Google Scholar]
- Maffei A, Nataraj K, Nelson S, Turrigiano G. Potentiation of cortical inhibition by visual deprivation. Nature. 2006;443:81–84. doi: 10.1038/nature05079. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Marder E, Prinz AA. Current compensation in neuronal homeostasis. Neuron. 2003;37:2–4. doi: 10.1016/s0896-6273(02)01173-x. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Mrsic-Flogel T, Hofer S, Ohki K, Reid R, Bonhoeffer T, Hübener M. Homeostatic regulation of eye-specific responses in visual cortex during ocular dominance plasticity. Neuron. 2007;54:961–972. doi: 10.1016/j.neuron.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Pratt KG, Aizenman CD. Homeostatic regulation of intrinsic excitability and synaptic transmission in a developing visual circuit. J Neurosci. 2007;27:8268–8277. doi: 10.1523/JNEUROSCI.1738-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz A, Bucher D, Marder E. Similar network activity from disparate circuit parameters. Nat Neurosci. 2004;7:1345–1352. doi: 10.1038/nn1352. [DOI] [PubMed] [Google Scholar]
- Rittenhouse C, Shouval H, Paradiso M, Bear M. Monocular deprivation induces homosynaptic long-term depression in visual cortex. Nature. 1999;397:347–350. doi: 10.1038/16922. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Multiple forms of long-term plasticity at unitary neocortical layer 5 synapses. Neuropharmacology. 2007;52:176–184. doi: 10.1016/j.neuropharm.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Stryker M, Harris W. Binocular impulse blockade prevents the formation of ocular dominance columns in cat visual cortex. J Neurosci. 1986;6:2117–2133. doi: 10.1523/JNEUROSCI.06-08-02117.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G, Abbott LF, Marder E. Activity-dependent changes in the intrinsic properties of cultured neurons. Science. 1994;264:974–977. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Van Sluyters R, Levitt F. Experimental strabismus in the kitten. J Neurophysiol. 1980;43:686–699. doi: 10.1152/jn.1980.43.3.686. [DOI] [PubMed] [Google Scholar]
- Wiesel T, Hubel D. Comparison of the effects of unilateral and bilateral eye closure on cortical unit responses in kittens. J Neurophysiol. 1965;28:1029–1040. doi: 10.1152/jn.1965.28.6.1029. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]