

Abstract
A region along chromosome 7q was recently linked to components of the metabolic syndrome (MetS) in several genome-wide linkage studies. Within this region, the CD36 gene, which encodes a membrane receptor for long-chain fatty acids and lipoproteins, is a potentially important candidate. CD36 has been documented to play an important role in fatty acid metabolism in vivo and subsequently may be involved in the etiology of the MetS. The protein also impacts survival to malaria and the influence of natural selection has resulted in high CD36 genetic variability in populations of African descent. We evaluated 36 tag SNPs across CD36 in the HyperGen population sample of 2020 African-Americans for impact on the MetS and its quantitative traits. Five SNPs associated with increased odds for the MetS [P = 0.0027–0.03, odds ratio (OR) = 1.3–1.4]. Coding SNP, rs3211938, previously shown to influence malaria susceptibility, is documented to result in CD36 deficiency in a homozygous subject. This SNP conferred protection against the MetS (P = 0.0012, OR = 0.61, 95%CI: 0.46–0.82), increased high-density lipoprotein cholesterol, HDL-C (P = 0.00018) and decreased triglycerides (P = 0.0059). Fifteen additional SNPs associated with HDL-C (P = 0.0028–0.044). We conclude that CD36 variants may impact MetS pathophysiology and HDL metabolism, both predictors of the risk of heart disease and type 2 diabetes.
INTRODUCTION
The metabolic syndrome (MetS) is a cluster of risk factors that increase susceptibility to cardiovascular disease (∼4-fold) and type 2 diabetes (T2DM) (∼7–34-fold) (1,2). Its components, which include hypertension, obesity, insulin resistance and dyslipidemia, are influenced by both environmental and genetic factors. Current estimates indicate a prevalence (age-adjusted) of ∼25% (3,4) for the MetS in the USA and UK which continues to increase at alarming rates in parallel with obesity. Thus, understanding genetic variations that increase susceptibility to the MetS and its risk factors is important.
A region of chromosome 7 (7q11.2–7q21.11) has been linked recently to components of the MetS in several genome-wide linkage scans (5–8). This region contains the CD36 gene, which encodes for a membrane protein that facilitates fatty acid (FA) uptake and utilization by key metabolic tissues (9,10). FAs could impact MetS susceptibility since they have been shown to induce insulin resistance, obesity and inflammation (11). In addition to FAs, CD36 binds native lipoproteins (12,13) and functions in the uptake of cholesteryl esters although less efficiently than its family member SR-B1. CD36 also facilitates uptake of oxidized low (14), and high-density lipoproteins (15), likely recognizing the modified lipids in these particles. Recent studies also implicate CD36 in cholesterol uptake by enterocytes (16). As a result of its many ligands and functions, CD36 could impact a variety of conditions linked with the MetS, including insulin resistance, inflammation and atherosclerosis (17,18).
The CD36 gene spans 36 kb and is comprised of 15 alternatively spliced exons that are differentially regulated by several upstream promoters (19). Single nucleotide polymorphisms (SNPs) in CD36 gene are frequent in humans (http://pga.mbt.washington.edu/). Physiological significance has been examined in a number of studies. In Caucasians from Italy and the USA, common haplotypes in CD36 associated with abnormalities of serum FA, triglyceride (TG) levels and increased risk of coronary artery disease (20). In a French population, a rare mutation (21) was linked with insulin resistance while a promoter variant was associated with low adiponectin in diabetes subjects (22). In a Dutch population, a promoter SNP was also more common in subjects with diabetes (23).
CD36 deficiency, rare in Caucasians (<0.3%), is at least 10 times more frequent in Asians and African-Americans (3–6%) (24,25). Polymorphisms in the gene are also more common in these populations when compared with Caucasians, likely reflecting the influence of natural selection (26,27) linked to malaria susceptibility (28,29) since CD36 is a receptor for Plasmodium falciparum infected erythrocytes. Analysis of genes influenced by natural selection may facilitate identification of variants involved in the etiology of complex diseases. To our knowledge, there are no large-scale population studies regarding the metabolic impact of CD36 deficiency. Reports with a small number of subjects suggested that it was associated with abnormal glucose metabolism and altered serum lipids (17,30). Findings of insulin resistance, mild hypertension and low HDL-C levels lead to the suggestion that CD36 deficiency is a risk factor for the MetS (25). In rodents, the metabolic phenotype of CD36 deficiency includes high blood TG and FA due to slow lipid clearance (31,32) and high HDL-C in mice (33). The CD36 null mouse exhibits peripheral insulin sensitivity (34) and hepatic insulin resistance (35), and also the spontaneously hypertensive rat with CD36 deficiency is insulin resistant (31).
This report is the first large-scale study to investigate the role of CD36 genetic variations in the etiology of the MetS. Our findings identify significant associations between relatively common variants in the gene, the MetS and also serum HDL-C levels.
RESULTS
Characteristics of the sample population
Thirty-six CD36 tag SNPs with minor allele frequencies (MAFs) ≥5% (Table 1) were analyzed in 2020 African-Americans representing 490 families and 429 singletons of the HyperGEN study. Demographic and clinical information for this population are shown in Supplementary Material, Table S1. The average age of the population was 46.1 ± 13.1 years with 36.5% males. The MetS was identified in 26.8% of the sample with hypertension and obesity as the major contributors. Mean body mass index (BMI) of 32 ± 7.8 kg/m2 and waist girth of 102.3 cm indicated prevalence of abdominal adiposity. The average systolic BP was slightly elevated (129.1 ± 22.1 mmHg) and the mean diastolic pressure was normal (74.2 ± 11.9 mmHg). HDL-C (53.5 ± 15.4), triglyceride (108.9 ± 113.6) and fasting glucose (95.8 ± 21.4) concentrations (mg/dl) were well within the normal range.
Table 1.
36 CD36 tag SNPs evaluated for association analysis
| SNP ID | NCBI dbSNP | Positiona | Major/minor allele | MAFb | Location |
|---|---|---|---|---|---|
| 1 | rs10499859 | 80096746 | A/G | 0.326 | 5′utr |
| 2 | rs17263407 | 80097844 | G/C | 0.229 | 5′utr |
| 3 | rs13438282 | 80100160 | C/T | 0.283 | 5′utr |
| 4 | rs9784998 | 80100937 | C/T | 0.216 | 5′utr |
| 5 | rs1334511 | 80104004 | A/G | 0.238 | 5′utr |
| 6 | rs1049654 | 80113391 | A/C | 0.426 | 5′utr |
| 7 | rs3211805 | 80113512 | G/T | 0.058 | 5′utr |
| 8 | rs1527463 | 80114267 | T/C | 0.059 | Intron |
| 9 | rs3211810 | 80114953 | T/G | 0.105 | Intron |
| 10 | rs3211812 | 80115568 | A/G | 0.054 | Intron |
| 11 | rs3211813 | 80115679 | T/G | 0.051 | Intron |
| 12 | rs3211822 | 80116562 | G/A | 0.328 | Intron |
| 13 | rs997906 | 80117771 | A/T | 0.171 | Intron |
| 14 | rs3211834 | 80118053 | C/A | 0.262 | Intron |
| 15 | rs3211842 | 80120572 | G/A | 0.352 | Intron |
| 16 | rs3211849 | 80121259 | G/A | 0.460 | Intron |
| 17 | rs3211850 | 80121352 | G/A | 0.065 | Intron |
| 18 | rs1054516 | 80122878 | T/C | 0.316 | Intron |
| 19 | rs3173798 | 80123786 | T/C | 0.209 | Intron |
| 20 | rs3211868 | 80124958 | T/C | 0.219 | Intron |
| 21 | rs3211870 | 80125145 | C/T | 0.423 | Intron |
| 22 | rs1358337 | 80126321 | A/G | 0.460 | Intron |
| 23 | rs3211885 | 80127338 | C/A | 0.360 | Intron |
| 24 | rs3211886 | 80127375 | G/A | 0.132 | Intron |
| 25 | rs3211890 | 80127478 | T/G | 0.047 | Intron |
| 26 | rs3211892 | 80127884 | G/A | 0.205 | Intron |
| 27 | rs3173801 | 80128305 | T/A | 0.049 | Intron |
| 28 | rs3211909 | 80132051 | T/C | 0.222 | Intron |
| 29 | rs3211913 | 80132540 | A/G | 0.374 | Intron |
| 30 | rs3211917 | 80133237 | T/C | 0.058 | Intron |
| 31 | rs3173804 | 80137786 | T/A | 0.236 | Intron |
| 32 | rs3211938 | 80138385 | T/G | 0.094 | Coding |
| 33 | rs3211944 | 80139449 | G/T | 0.071 | Intron |
| 34 | rs7755 | 80144207 | G/A | 0.202 | 3′utr |
| 35 | rs13246513 | 80144687 | C/T | 0.200 | 3′Flanking |
| 36 | rs13230419 | 80147221 | C/T | 0.149 | 3′Flanking |
aChromosome 7q, NCBI Build 36, dbSNP Build 127.
bMAF-minor allele frequencies based on founder chromosomes.
The metabolic syndrome
Figure 1A presents the results of the logistic regression model for the effect of CD36 SNPs on the MetS and its components adjusted for age, gender, BMI and field center. Six SNPs (SNP ID: 17, 31, 32, 34–36) with MAFs ranging from 6.5 to 23.6% showed significant association with the MetS (P < 0.05), four of which (31,32,35,36) had P-values <0.01, corresponding to false discovery rate (FDR) ≤0.034 after adjusting for multiple tests. SNPs 17, 31, 34, 35 and 36 increased the odds for the MetS by 29–40% (Table 2). Linkage disequilibrium between SNPs 34 and 35 was strong (D' = 0.98, r2 = 0.86) suggesting the associations may not be independent (Fig. 1B). In contrast to the above SNPs, the minor allele for SNP 32 decreased the odds of MetS by 39% (Table 2). The original analysis was performed using the NCEP 2001 guidelines for fasting glucose (>110 mg/dl); however, the analysis was repeated using the current fasting glucose criteria (>100 mg/dl), which resulted in similar findings. Additionally, the associations with the MetS were similar when adjusted and unadjusted for BMI (data not shown) as well as when adjusted for waist circumference (Supplementary Material, Table S3). SNPs 17 and 31 are located in introns 3 and 9, respectively, while SNPs 34, 35 and 36 are located in the 3′ untranslated and the 3′ flanking locus region. SNP 32 is a non-synonymous polymorphism located in exon 10 of CD36 and is predicted to result in premature truncation of the protein (28).
Figure 1.
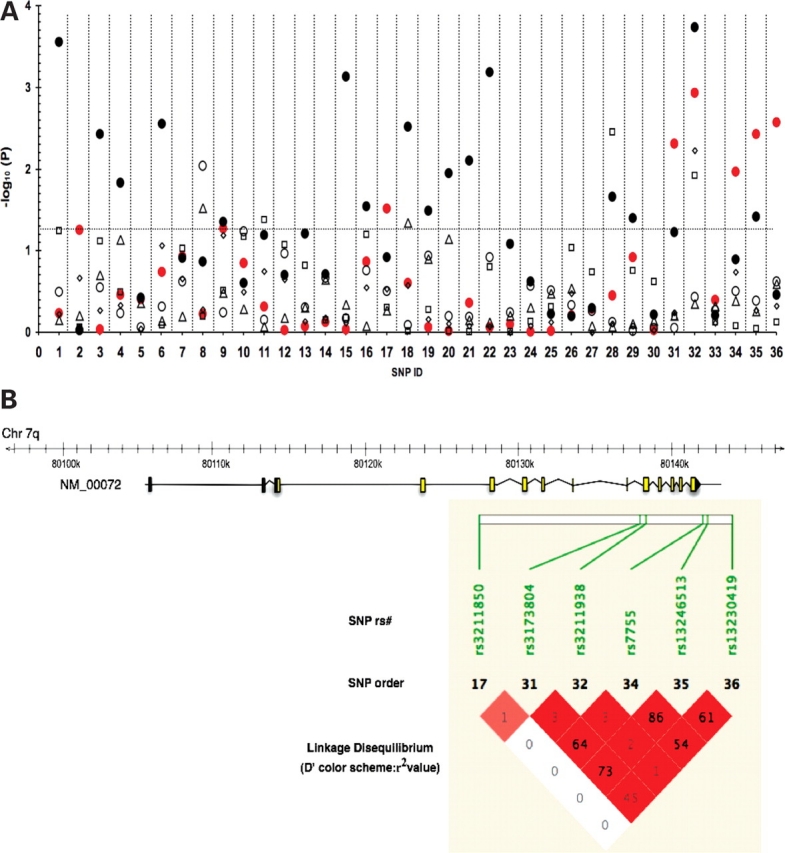
Plot of association analysis between 36 CD36 tag SNPs, the metabolic syndrome and its components. The horizontal dotted line indicates a P-value of 0.05 with significant associations above the line. SNPs (X-axis) are displayed according to the order on chromosome 7q (corresponding dbSNP ID shown in Table 1). (A) Regression analysis identified the following associations: 6 SNPs and the metabolic syndrome (filled circle, red) 16 SNPs and HDL-C (filled circle), 1 SNP and waist circumference (open circle), 2 SNPs and fasting glucose (triangle), 3 SNPs and hypertension (square), 1 SNP and triglycerides (diamond). (B) In the CD36 gene schematic, yellow boxes indicate coding exons, gray boxes indicate untranslated exonic regions. Pairwise measures of linkage disequilibrium (LD) between CD36 tag SNPs associated with the MetS. LD measures presented as D′ standard color scheme (bright red indicates D' = 1 with a LOD score ≥2) and values represent r2, correlation coefficients generated using default settings in Haploview.
Table 2.
Tag SNPs associated with the metabolic syndrome
| SNP | OR | 95%CI | P-value | FDR |
|---|---|---|---|---|
| 32 | 0.61 | 0.46–0.82 | 1.2 × 10−3 | 3.3 × 10−2 |
| 36 | 1.36 | 1.11–1.67 | 2.7 × 10−3 | 3.4 × 10−2 |
| 35 | 1.32 | 1.09–1.58 | 3.7 × 10−3 | 3.4 × 10−2 |
| 31 | 1.29 | 1.08–1.54 | 4.9 × 10−3 | 3.4 × 10−2 |
| 34 | 1.35 | 1.07–1.69 | 1.1 × 10−2 | 6.0 × 10−2 |
| 17 | 1.4 | 1.0 3–1.90 | 3.0 × 10−2 | 1.4 × 10−1 |
Associations from logistic regression (additive model) were adjusted for age, gender, BMI and recruitment center. SNPs listed by ascending P-values. SNP ID in Table 1.
Components of the metabolic syndrome
HDL-C and TG
We further examined whether any of the 36 tag SNPs were associated with components of the MetS (Fig. 1A), using an additive regression model adjusted for age, gender, BMI and recruitment center. Association with triglyceride concentrations was identified only at coding SNP 32, which decreased TG levels by 8% (β = −8.0% per allele, P = 0.006). Adjustment for multiple testing yielded an FDR of 0.09.
Table 3 displays parameter estimates for 16 SNPs that were found to associate with HDL-C levels (P≤0.05). The MAF for these SNPs ranged between 9.4 and 46%. Minor alleles at 9 of the 16 SNPs were associated with increased HDL-C concentrations as opposed to decreased HDL-C for the remaining 7. Coding SNP 32 (MAF 9.4%) showed the strongest effect with a predicted increase in HDL-C of 5.5% for the minor allele (‘G’), (P < 0.0002, FDR 0.02). FDRs were <0.05 for SNPs 1, 15 and 22 (MAF of 23, 35 and 46%, respectively).
Table 3.
CD36 tag SNPs associated with HDL-C
| SNP | βa | P | FDR | Adjustment for TG | |
|---|---|---|---|---|---|
| βa | P | ||||
| 32 | 0.055 | 1.8 × 10−4 | 0.021 | 0.035 | 1.2 × 10−2 |
| 1 | 0.042 | 2.8 × 10−4 | 0.021 | 0.043 | <1.0 × 10−4 |
| 3 | 0.039 | 3.8 × 10−3 | 0.062 | 0.041 | 7.0 × 10−4 |
| 22 | 0.031 | 6.6 × 10−4 | 0.028 | 0.032 | 2.0 × 10−4 |
| 18 | 0.028 | 3.0 × 10−3 | 0.062 | 0.032 | 4.0 × 10−4 |
| 6 | 0.027 | 2.8 × 10−3 | 0.062 | 0.032 | 2.0 × 10−4 |
| 28 | 0.025 | 2.2 × 10−2 | 0.186 | 0.023 | 2.1 × 10−2 |
| 16 | 0.019 | 2.9 × 10−2 | 0.221 | 0.024 | 2.7 × 10−3 |
| 29 | 0.019 | 3.9 × 10−2 | 0.254 | 0.018 | 3.9 × 10−2 |
| 35 | −0.022 | 3.8 × 10−2 | 0.254 | −0.017 | 8.6 × 10−2 |
| 19 | −0.023 | 3.3 × 10−2 | 0.230 | −0.028 | 6.2 × 10−3 |
| 21 | −0.024 | 7.9 × 10−3 | 0.106 | −0.024 | 2.9 × 10−3 |
| 15 | −0.029 | 7.4 × 10−4 | 0.028 | −0.031 | <1.0 × 10−4 |
| 4 | −0.030 | 1.5 × 10−2 | 0.137 | −0.028 | 1.9 × 10−2 |
| 9 | −0.030 | 4.4 × 10−2 | 0.259 | −0.018 | 2.0 × 10−1 |
| 20 | −0.032 | 1.1 × 10−2 | 0.117 | −0.031 | 6.2 × 10−3 |
Linear-regression analysis of SNP association with log HDL-C. SNPs are listed by descending estimate (β). Estimates represent percent change in HDL-C. Adjustment for TG means that associations were corrected for an effect of triglyceride levels. FDR, false discovery rate.
a±SE <0.01 for all estimates with the exception of SNP 9 where ± SE = 0.02.
In a previous HyperGEN analysis, HDL-C showed a negative correlation with TG concentration (36). It has also been shown that TG levels impact HDL-C concentrations as a result of lipid transfer between blood lipoproteins (37). Thus, all HDL-C-associated SNPs were adjusted for TG. After this adjustment (Table 3), the associations with HDL-C remained significant for 14/16 SNPs indicating that the effects on HDL were largely independent of TG. Consistent with this observation, the parameter estimates remained the same in magnitude and direction except for coding SNP 32 and for SNPs 9 and 35. For SNP 32, the association was slightly attenuated (β = 0.035, P = 0.012). For SNPs 9 and 35, the association appeared to be confounded by TG (β = −0.018, P = 0.2 and β = −0.017, P = 0.086, respectively).
Although HDL-C distribution levels differ between men and women, we found no significant interactions between gender and SNPs associated with HDL-C and TG (data not shown).
To examine direct impact of significant SNPs on levels of HDL-C and TG, pairwise comparisons across genotype groups adjusted for age, gender, BMI and recruitment center were performed. Figure 2 shows significant differences between genotypes in mean HDL-C levels at SNPs 22 (rs1358337), 1 (rs10499859) and 6 (rs109654). In each case, the magnitude of the increase in HDL-C was positively associated with the number of minor alleles present. Mean HDL-C concentrations were increased between 1.5 and 2.5 mg/dl per allele.
Figure 2.

Impact of SNPs 22, 1 and 6 on HDL-C levels (mg/dl ± SE) from pairwise genotype comparisons. Mean HDL-C for non-carriers (black bars), for heterozygous (gray bars) and subjects homozygous for the minor allele (white bars). Sample sizes (n) for non-carriers, heterozygous and homozygous subjects (in this order) for SNPs 22 (rs13583337) n = 574, 819, 430, for SNPs 1 (rs10499859) n = 564, 500, 142 and for SNP 6 (rs1049654) n = 630, 959, 318. Resulting P-values for SNP 22: *0.04, ***0.0007; SNP 1: *0.02, **0.032, ***0.0004; SNP 6: *0.035, **0.002.
Subjects heterozygous for coding SNP 32 (Fig. 3A) had higher (by ∼4 mg/dl) mean HDL-C when compared with non-carriers, P < 0.0001. Mean TG (Fig. 3B) was lower (by ∼2.5 mg/dl), P = 0.0015. These data were consistent with a protective metabolic effect for subjects heterozygous for this allele. In contrast, an opposite trend was observed for subjects homozygous for the minor allele (i.e. G/G) as they had significantly lower mean HDL-C levels when compared with heterozygous subjects (47.8 ± 3.3 mg/dl versus 56.1 ± 0.8 mg/dl, respectively), P = 0.008. However, when compared to the non-carrier group (i.e. T/T), the difference was not significant (47.8 versus 52.9 ± 0.4 mg/dl, G/G versus T/T, respectively), P = 0.096. In Figure 3B, a similar trend for homozygous subjects was observed for TG levels although the differences were not significant. The lack of significance likely reflects the small sample of homozygous (G/G) subjects (n = 9) in addition to large variability in TG levels (standard deviations were ± 15.4 for T/T, ± 15.0 for T/G, and ± 9.9 for the G/G subjects).
Figure 3.

Impact of coding SNP 32 on HDL-C and TG levels (mg/dl ± SE) from pairwise genotype comparisons. (A) Mean HDL-C levels: non-carriers for SNP 32 (rs3111938) (T/T) = 52.94 ± 0.39; heterozygous (T/G) = 56.10 ± 0.82; homozygous (G/G) 47.78 ± 3.29. (B) Mean TG levels: T/T = 111.75 ± 3.10; T/G 99.29 ± 3.9; G/G 127.33 ± 22.9. Sample sizes are: n = 1576 (T/T), 334 (T/G) and 9 (G/G). Resulting P-values the pairwise comparisons: ***<0.0001, **00.008, *0.0015. (C) Absence of CD36 expression on monocytes and platelets from a subject homozygous for coding SNP 32 (rs3211938). Shown are representative histograms of flow cytometric analysis from a non-carrier and heterozygote (carrier) for SNP 32. (D) Representative western blots and densitometry analysis of total CD36 protein in monocytes and platelets from a non-carrier (−/−), a homozygote (+/+) and a heterozygote (−/+) for SNP 32. The RAN (ras-related nuclear protein) was used as the loading control.
SNP 32 is predicted to result in a truncated protein and to alter CD36 expression. To determine the impact of this SNP on CD36 level, we genotyped an independent sample of 350 unrelated African-American subjects. The frequency of SNP 32 in this cohort was 9.7% (n = 280 non-carriers, 65 carriers and 1 subject homozygous for the minor allele). Cell surface CD36 expression was evaluated by flow cytometry on monocytes and platelets isolated from subjects identified as non-carriers, heterozygous and homozygous for SNP 32. The subjects were matched for age, BMI and had no history of diabetes mellitus or cardiovascular disease (n = 7, age = 46.0 ± 1.5, BMI = 26.7 ± 3.43). In Figure 3C, we demonstrate that a subject homozygous for this allele is CD36 deficient as determined from the lack of CD36 expression on both monocytes and platelets by flow cytometry. A subject heterozygous for SNP 32 had reduced levels of CD36 on both cell types. These findings were confirmed by western blotting (Fig. 3D). Thus, the above difference in serum lipid concentrations between heterozygous and homozygous subjects (Fig. 3A) may reflect the differing impact of partial versus complete CD36 deficiency on HDL-C and TG.
Fasting plasma glucose
SNPs 18 and 8 showed modest association with plasma glucose (P = 0.05 and P = 0.03, respectively). The minor allele of SNP 8 also associated with decreased waist circumference (P = 0.009). However, neither of these associations remained significant after correcting for multiple tests (FDR >0.05).
Hypertension
SNPs 11, 28 and 32 associated with decreased hypertension risk (OR = 0.70, P = 0.004; OR = 0.63, P = 0.012, respectively). Adjusting for multiple testing yielded FDRs of 0.06 and 0.12, respectively.
DISCUSSION
The region of chromosome 7 (7q11.2–7q21.11) where the CD36 gene is located has been linked to components of the MetS in several genome-wide scans (5–8). We evaluated the potential of variations in CD36 to impact susceptibility to the MetS, its components and documented the following: five common CD36 SNPs increased risk for the MetS, whereas a non-synonymous SNP, which results in haploinsufficiency of CD36, associated with lower TG and higher HDL-C levels. The protective effect of this SNP on MetS and serum lipids was not observed in subjects homozygous for the minor allele, suggesting different effects of partial versus total CD36 deficiency. Additionally, strong associations were observed between multiple SNPs and different components of the MetS. The majority of these associations were with serum HDL-C and most remained significant after adjusting for TG levels, suggesting an important role of CD36 in HDL metabolism in humans.
CD36 variants associate with the MetS
Five intronic SNPs, shown in Table 2, significantly increased odds (29–40%) for the MetS. Based on the strength of the associations and the frequency of the minor alleles involved (6.5–23.6%), the risk contributed has potential significance. This risk may also be impacted by environmental factors, which have been shown to increase the effects of genetic variants (38,39). Additionally, the MetS strongly increases risk of subsequent T2D and cardiovascular disease. However, the influence of the identified SNPs on these outcomes will need to be examined in large populations. In this context, a common haplotype in CD36 was previously reported to associate with the increased cardiovascular disease in diabetic Caucasian subjects (20). The above findings together with the key role of CD36 in lipid metabolism suggest that CD36 contributes to individual susceptibility to the MetS and variants in the gene could provide biomarkers for targeted preventive strategies.
FASTSNP, a functional analysis tool (40), was used to predict how the SNPs that associated with the MetS might impact CD36 expression or function. SNP 31 (rs3173804, located in intron 9) was identified as lying in a predicted intronic enhancer sequence homologous to a binding site for CDX1, a caudal-type homeobox transcription factor. CDX1 is restricted to the intestine in humans and involved in regulation of intestine-specific gene transcription and in maintenance of intestinal cell phenotype (41). Thus, SNP 31 may alter intestinal CD36 expression, which is important for the proximal uptake of FA and cholesterol and for their export into chylomicron size particles (16). In addition, CD36 deficiency results in postprandial hyperlipidemia consequent to significantly delayed clearance of blood lipids in both humans (30) and rodents (16).
CD36 variants associate with HDL-C
The finding of strong associations between multiple CD36 SNPs and HDL-C suggests an important role for CD36 in HDL-C metabolism in humans. Furthermore, the most significant SNPs had MAF between 23 and 46%. In vitro evidence previously documented the ability of CD36 to bind HDL with high affinity (12) but the role of the protein in cholesteryl ester uptake from these particles was proposed to be less important than that of SR-B1 (13). Recent evidence obtained with enterocytes isolated from CD36 null mice supported a role of CD36 in cholesterol transport in the proximal intestine (16). Such a role was consistent with the defect in cholesterol secretion in the lymph of these mice (42). However, it is also possible that the effect of CD36 variants on HDL-C is exerted indirectly. For example, it could be mediated by abnormal regulation of PPAR transcription factors. In this context, PPAR delta has been shown to have significant effects on blood HDL-C, as recently reviewed (43) and activity of this PPAR is regulated by CD36-mediated FA uptake (Nahlé Z, Hsieh M, Pietka T, Coburn CC, Grimaldi PA, Das D, Abumrad NA, manuscript in preparation).
Low HDL-C is a major determinant of the MetS and an independent risk factor for cardiovascular disease. Plasma HDL-C levels have a strong hereditary component accounting for ∼70% of individual variability (38). Multiple genes related to cellular reverse cholesterol transport and to lipid transfer between various lipoproteins affect HDL-C levels. However, the cumulative effects of variants identified so far are reported to explain less than 20% of the inter-individual HDL-C variance (44).
Small increases in HDL-C have a significant protective effect for numerous cardiovascular outcomes. It is estimated that increasing HDL-C by 1 mg/dl decreases cardiovascular risk by 2–3%. Individuals with 6–7 mg/dl higher than average HDL-C have a 20–27% lower risk of coronary disease (45). Thus, the effects (2–5 mg/dl) we identified are suggestive of physiological significance.
The direction of the effects on HDL-C (increase versus decrease) likely reflects the underlying alterations in CD36 expression/function or tissue distribution. FASTSNP identified SNP 19 (rs3173798, intron 3) to be located in a potential splice site while SNP 22 (rs1358337) was within a predicted intronic enhancer sequence similar to an Oct-1 and/or Retinoid-related orphan nuclear receptor alpha (RORα), binding site. RORα is involved in the regulation of FA catabolism and of HDL-C and attenuating RORα is associated with repression of CD36 expression in muscle cells (46). Our data may suggest that either regulation of CD36 by RORα influences HDL-C or alternatively that CD36 function modulates this nuclear receptor. Additional studies are needed to assess the true biological function of these SNPs. Possible LD between these SNPs and potentially unidentified causative variants will also need to be considered by extensive re-sequencing of the CD36 gene.
CD36 deficiency
The observation that carriers of SNP 32 (rs3211938) who are haploinsufficient for CD36 are protected against the MetS and exhibit increased HDL-C and decreased TG is a significant finding. This polymorphism results in a T to G substitution at nucleotide 1264 in the mRNA sequence (Accession Number: NM_000072.2) and leads to the insertion of a stop codon at amino acid 325 (UniProtKB Entry: P16671). As shown in Figure 3C and D, this mutation results in CD36 deficiency in the homozygous state. The data with heterozygote subjects would indicate that down-regulation of CD36 expression has a beneficial effect on blood lipids and is protective against the MetS. Although limited by the small number of individuals involved (n = 9), the data with subjects homozygous for the minor allele at SNP 32 suggest that the beneficial effects of haploinsufficiency are reversed in total CD36 deficiency. Homozygote subjects deficient in CD36 have a deleterious plasma lipid profile (Fig. 3) consistent with the previous reports from the studies conducted in a small number of CD36 deficient Japanese subjects (25). Opposite outcomes of partial versus total CD36 on lipid metabolism and susceptibility to the MetS may reflect the effect of both conditions on lipid utilization. For example, complete lack of CD36 may impair the adaptive ability of muscle tissues to transition between utilization of glucose and fatty acids during the stress of fasting or high FA flux, which chronically would promote metabolic pathology (Nahlé Z, Hsieh M, Pietka T, Coburn CC, Grimaldi PA, Das D, Abumrad NA, manuscript in preparation). In contrast, reduced levels of CD36 would allow metabolic transitions and may be beneficial by limiting FA intake under conditions of high FA supply possibly alleviating the associated negative cellular effects.
In summary, we present significant evidence for the association between common variants in the CD36 gene and the MetS and its components, particularly HDL-C. Regions of linkage with components of the MetS have been identified on chromosome 7 by whole genome studies across different populations (5–8) consistent with common genetic influences. Our findings suggest that CD36 may contribute to the underlying linkage signal previously reported. Together with the established role of CD36 in tissue lipid utilization and prior knowledge of the influence of natural selection on CD36 variants, our data provide evidence suggesting that CD36 polymorphisms contribute to individual and population variability in blood lipids and susceptibility to the MetS. Finally, examination of genetic variants known to be impacted by natural selection may expedite the identification of genetic influences involved in the etiology of common diseases.
METHODS AND MATERIALS
Subjects
Recruitment and phenotyping of African-American subjects by the Hypertension Genetic Epidemiology Network (HyperGEN) have been previously described (47). For these analyses, subjects with fasting times <8 h, plasma glucose <18 mg/dl or >450 mg/dl, insulin <0.144 or >316.78 uIU/ml) were excluded. Washington University Institutional Review Board and the HyperGEN Investigators approved the use of the genomic DNA samples and clinical data.
Because materials to assess the impact of CD36 variants on CD36 expression were not available from the HyperGEN study, we recruited an additional cohort for genotype and expression studies. To evaluate the function of the non-synonymous SNP 32 (rs3211938) on CD36 expression, 350 unrelated subjects of African-American descent (20.9% male, age 41.8 ± 12.03 years, BMI 32.4 ± 8.60 kg/m2) were recruited via the Volunteer for Health Program at Washington University. Medical and family histories were ascertained by self-reporting. Height (cm) and weight measurements (kg) were collected in duplicate to determine BMI. The institutional review board of Washington University School of Medicine approved the protocols for this study.
SNP genotyping and data cleaning
Thirty-six tag SNPs across a 50.5 kb region encompassing CD36 were selected based on the predicted function, allele frequencies (MAF > 5%) identified from the public database SeattleSNP and the Yoruba (Ibadan, Nigeria) data from the HapMap Project, and functionality as a tag SNP as determined using Tagger (Haploview) and TAMAL (48).
SNPs were genotyped (Washington University, Department of Human Genetics) using the high-performance Sequenom MassARRAY system, which relies on matrix-assisted laser desorption/ionization-Time-of-Flight (MALDI-TOF) mass spectrometry (49). SNP rs997906 was represented in two multiplexes yielding duplicate genotypes with 99.6% accuracy.
SNP 32 was genotyped in the cohort of 350 unrelated African-American subjects by pyrosequencing technology and confirmed by PCR-RFLP analysis of genomic DNA extracted from peripheral blood with the Puregene DNA purification Kit (Gentra Systems, Minneapolis, MN, USA).
Mendelian errors for all SNPs were identified using PEDCHECK (50). Genotypes of individuals with fewer than 90% of SNPs successfully genotyped were considered missing. SNPs were excluded based on call rates <90%, MAF <5% or Hardy–Weinberg P < 0.001. As a result, a total of 36 tag SNPs (Table 1) were analyzed with an average density of 1 SNP/1.4 kb.
Statistical analysis
Association between the MetS, its components and SNPs in the CD36 gene were analyzed using regression models implemented in GENMOD (SAS v9.1), which corrects for genotypic and phenotypic correlations among family members. Logistic regression models were used for dichotomous traits (MetS and hypertension) and linear models for quantitative traits. Each SNP was coded using a variable that indicated the number of minor alleles (0, 1 or 2) in each subject. All analyses were adjusted for age, gender, field center and BMI.
The MetS was designated as a dichotomous trait based on its most widely accepted definition by the National Cholesterol Education Program (NCEP) Adult Treatment Panel III (ATP III) 2001 (51), which requires presence of three or more of the following traits: waist circumference >102 cm in men and 88 cm in women, fasting glucose ≥110 mg/dl, hypertension, TG ≥150 mg/dl and HDL-C <40 mg/dl in men and <50 mg/dl in women). Serum glucose was excluded as a potential MetS criterion for individuals on glucose lowering or insulin sensitizing medications, but they could be considered positive for the MetS if they met three of the remaining criteria. Quantitative traits were log-transformed where necessary to better approximate a normal distribution. Adjustment for tests of association was performed using the FDR in the Q-Value software (52). FDRs were computed for MetS association tests (N = 36) and for the five MetS component tests (N = 36 × 5 = 190). FDRs ≤ 0.05 were considered as significant.
Determination of CD36 expression
Fasting venous blood was collected in K2-EDTA tubes and evaluated for CD36 expression on monocytes and platelets (53) by Fluorescence Activated Cell Sorting (FACS) using Phycoerythrin (PE)-conjugated monoclonal antibody against CD36 (SMϕ-Sc7309: Santa Cruz Biotechnology, Santa Cruz, CA, USA) and gating with Fluorescein isothiocyanate (FITC)-conjugated monoclonal antibody against CD14 (BD Pharmigen) for monocytes, and FITC conjugated CD42 (BD Pharmigen) for platelets.
For western blot analysis, whole blood (2 ml) was used for isolation of monocytes and platelets. Monocytes isolated by ficoll separation (Ficoll-Paque PLUS, GE Health), washed in calcium/magnesium free PBS were lysed in SDS buffer (0.5 M Tris–HCl, pH 6.8, Glycerol, 10% SDS w/v and 2-mercaptoethanol with 1% Triton X-100). Platelets were prepared as described (54) and lysed in 250 mm NaCl, 25 mm Tris–HCl, pH 7.5, 5 mm EDTA, 2 ug/ml Aprotinin, 100 ug/ml PMSF, with 1% Triton X-100. Twenty micrograms of protein was resolved on a 10% SDS-polyacrylamide gel. Blotted proteins were blocked in 5% milk in PBS with 0.1% Tween 20, incubated overnight at 4°C with primary anti-human monoclonal CD36 antibody 1:1000 (Abcam Inc.; Cat No. ab17044) then washed in PBS with 0.1% Tween 20, before incubation with anti-mouse HRP-conjugated (Pierce; Cat No. 1858413) antibody. SuperSignal was used for immunodetection (Pierce; Cat. No. 34096). Ras-related nuclear (RAN) protein was used as a loading control (Santa Cruz; Cat No. sc-1156), bound by an anti-goat HRP-conjugated (Santa Cruz, Cat No. sc-2350) and detected with ECL reagent (Amersham Biosciences; Cat No. RPN2106). Blots were exposed to Kodak BioMax XAR film.
SUPPLEMENTARY MATERIAL
FUNDING
National Institutes of Health (R01 DK60022, R37 DK016746); Washington University Clinical Nutrition Research Unit (DK56351). The HyperGEN network of the NHLBI Family Blood Pressure Program was funded by cooperative agreements (U10): HL54471, HL54472, HL54473, HL54495, HL54496, HL54497, HL54509, HL54515.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to Terri Pietka in the Center for Human Nutrition at Washington University for valuable help with the flow cytometry. We gratefully acknowledge Jennifer McRae (Center for Human Nutrition) and the Volunteer for Health program at Washington University for help with study coordination and subject recruitment.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Klein B.E., Klein R., Lee K.E. Components of the metabolic syndrome and risk of cardiovascular disease and diabetes in Beaver Dam. Diabetes Care. 2002;25:1790–1794. doi: 10.2337/diacare.25.10.1790. [DOI] [PubMed] [Google Scholar]
- 2.Malik S., Wong N.D., Franklin S.S., Kamath T.V., L'Italien G.J., Pio J.R., Williams G.R. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110:1245–1250. doi: 10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- 3.Ford E.S., Giles W.H., Dietz W.H. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 4.Tonkin R. The X factor: obesity and the metabolic syndrome in. The Science and Public Affairs Forum. 2003 [Google Scholar]
- 5.An P., Freedman B.I., Hanis C.L., Chen Y.D., Weder A.B., Schork N.J., Boerwinkle E., Province M.A., Hsiung C.A., Wu X., et al. Genome-wide linkage scans for fasting glucose, insulin, and insulin resistance in the National Heart, Lung, and Blood Institute Family Blood Pressure Program: evidence of linkages to chromosome 7q36 and 19q13 from meta-analysis. Diabetes. 2005;54:909–914. doi: 10.2337/diabetes.54.3.909. [DOI] [PubMed] [Google Scholar]
- 6.Arya R., Blangero J., Williams K., Almasy L., Dyer T.D., Leach R.J., O'Connell P., Stern M.P., Duggirala R. Factors of insulin resistance syndrome-related phenotypes are linked to genetic locations on chromosomes 6 and 7 in nondiabetic Mexican-Americans. Diabetes. 2002;51:841–847. doi: 10.2337/diabetes.51.3.841. [DOI] [PubMed] [Google Scholar]
- 7.Duggirala R., Blangero J., Almasy L., Dyer T.D., Williams K.L., Leach R.J., O'Connell P., Stern M.P. A major susceptibility locus influencing plasma triglyceride concentrations is located on chromosome 15q in Mexican Americans. Am. J. Hum. Genet. 2000;66:1237–1245. doi: 10.1086/302849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malhotra A., Elbein S.C., Ng M.C., Duggirala R., Arya R., Imperatore G., Adeyemo A., Pollin T.I., Hsueh W.C., Chan J.C., et al. Meta-analysis of genome-wide linkage studies of quantitative lipid traits in families ascertained for type 2 diabetes. Diabetes. 2007;56:890–896. doi: 10.2337/db06-1057. [DOI] [PubMed] [Google Scholar]
- 9.Coburn C.T., Knapp F.F., Jr, Febbraio M., Beets A.L., Silverstein R.L., Abumrad N.A. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem. 2000;275:32523–32529. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka T., Nakata T., Oka T., Ogawa T., Okamoto F., Kusaka Y., Sohmiya K., Shimamoto K., Itakura K. Defect in human myocardial long-chain fatty acid uptake is caused by FAT/CD36 mutations. J. Lipid. Res. 2001;42:751–759. [PubMed] [Google Scholar]
- 11.Roden M. Blocking fatty acids' mystery tour: a therapy for metabolic syndrome? Cell. Metab. 2007;6:89–91. doi: 10.1016/j.cmet.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 12.Calvo D., Gomez-Coronado D., Suarez Y., Lasuncion M.A., Vega M.A. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J. Lipid. Res. 1998;39:777–788. [PubMed] [Google Scholar]
- 13.Connelly M.A., Klein S.M., Azhar S., Abumrad N.A., Williams D.L. Comparison of class B scavenger receptors, CD36 and scavenger receptor BI (SR-BI), shows that both receptors mediate high density lipoprotein-cholesteryl ester selective uptake but SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J. Biol. Chem. 1999;274:41–47. doi: 10.1074/jbc.274.1.41. [DOI] [PubMed] [Google Scholar]
- 14.Nicholson A.C., Hajjar D.P. CD36, oxidized LDL and PPAR gamma: pathological interactions in macrophages and atherosclerosis. Vascul. Pharmacol. 2004;41:139–146. doi: 10.1016/j.vph.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Thorne R.F., Mhaidat N.M., Ralston K.J., Burns G.F. CD36 is a receptor for oxidized high density lipoprotein: implications for the development of atherosclerosis. FEBS Lett. 2007;581:1227–1232. doi: 10.1016/j.febslet.2007.02.043. [DOI] [PubMed] [Google Scholar]
- 16.Nassir F., Wilson B., Han X., Gross R.W., Abumrad N.A. CD36 is important for fatty acid and cholesterol uptake by the proximal but not distal intestine. J. Biol. Chem. 2007;282:19493–19501. doi: 10.1074/jbc.M703330200. [DOI] [PubMed] [Google Scholar]
- 17.Hirano K., Kuwasako T., Nakagawa-Toyama Y., Janabi M., Yamashita S., Matsuzawa Y. Pathophysiology of human genetic CD36 deficiency. Trends Cardiovasc. Med. 2003;13:136–141. doi: 10.1016/s1050-1738(03)00026-4. [DOI] [PubMed] [Google Scholar]
- 18.Pravenec M., Kurtz T.W. Molecular genetics of experimental hypertension and the metabolic syndrome: from gene pathways to new therapies. Hypertension. 2007;49:941–952. doi: 10.1161/HYPERTENSIONAHA.107.086900. [DOI] [PubMed] [Google Scholar]
- 19.Andersen M., Lenhard B., Whatling C., Eriksson P., Odeberg J. Alternative promoter usage of the membrane glycoprotein CD36. BMC Mol. Biol. 2006;7:8. doi: 10.1186/1471-2199-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma X., Bacci S., Mlynarski W., Gottardo L., Soccio T., Menzaghi C., Iori E., Lager R.A., Shroff A.R., Gervino E.V., et al. A common haplotype at the CD36 locus is associated with high free fatty acid levels and increased cardiovascular risk in Caucasians. Hum. Mol. Genet. 2004;13:2197–2205. doi: 10.1093/hmg/ddh233. [DOI] [PubMed] [Google Scholar]
- 21.Leprêtre F., Vasseur F., Vaxillaire M., Scherer P.E., Ali S., Linton K., Aitman T., Froguel P. A CD36 nonsense mutation associated with insulin resistance and familial type 2 diabetes. Hum. Mutat. 2004;24:104. doi: 10.1002/humu.9256. [DOI] [PubMed] [Google Scholar]
- 22.Leprêtre F., Linton K.J., Lacquemant C., Vatin V., Samson C., Dina C., Chikri M., Ali S., Scherer P., Séron K., et al. Genetic study of the CD36 gene in a French diabetic population. Diabetes Metab. 2004;30:459–463. doi: 10.1016/s1262-3636(07)70143-x. [DOI] [PubMed] [Google Scholar]
- 23.Corpeleijn E., van der Kallen C.J., Kruijshoop M., Magagnin M.G., de Bruin T.W., Feskens E.J., Saris W.H., Blaak E.E. Direct association of a promoter polymorphism in the CD36/FAT fatty acid transporter gene with Type 2 diabetes mellitus and insulin resistance. Diabet. Med. 2006;23:907–911. doi: 10.1111/j.1464-5491.2006.01888.x. [DOI] [PubMed] [Google Scholar]
- 24.Curtis B.R., Aster R.H. Incidence of the Nak(a)-negative platelet phenotype in African Americans is similar to that of Asians. Transfusion. 1996;36:331–334. doi: 10.1046/j.1537-2995.1996.36496226147.x. [DOI] [PubMed] [Google Scholar]
- 25.Yamashita S., Hirano K., Kuwasako T., Janabi M., Toyama Y., Ishigami M., Sakai N. Physiological and pathological roles of a multi-ligand receptor CD36 in atherogenesis; insights from CD36-deficient patients. Mol. Cell Biochem. 2007;299:19–22. doi: 10.1007/s11010-005-9031-4. [DOI] [PubMed] [Google Scholar]
- 26.Ayodo G., Price A.L., Keinan A., Ajwang A., Otieno M.F., Orago A.S., Patterson N., Reich D. Combining evidence of natural selection with association analysis increases power to detect malaria-resistance variants. Am. J. Hum. Genet. 2007;81:234–242. doi: 10.1086/519221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabeti P.C., Schaffner S.F., Fry B., Lohmueller J., Varilly P., Shamovsky O., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 28.Aitman T.J., Cooper L.D., Norsworthy P.J., Wahid F.N., Gray J.K., Curtis B.R., McKeigue P.M., Kwiatkowski D., Greenwood B.M., Snow R.W., et al. Malaria susceptibility and CD36 mutation. Nature. 2000;405:1015–1016. doi: 10.1038/35016636. [DOI] [PubMed] [Google Scholar]
- 29.Omi K., Ohashi J., Patarapotikul J., Hananantachai H., Naka I., Looareesuwan S., Tokunaga K. CD36 polymorphism is associated with protection from cerebral malaria. Am. J. Hum. Genet. 2003;72:364–374. doi: 10.1086/346091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furuhashi M., Ura N., Nakata T., Shimamoto K. Insulin sensitivity and lipid metabolism in human CD36 deficiency. Diabetes Care. 2003;26:471–474. doi: 10.2337/diacare.26.2.471. [DOI] [PubMed] [Google Scholar]
- 31.Aitman T.J., Glazier A.M., Wallace C.A., Cooper L.D., Norsworthy P.J., Wahid F.N., Al-Majali K.M., Trembling P.M., Mann C.J., Shoulders C.C., et al. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat. Genet. 1999;21:76–83. doi: 10.1038/5013. [DOI] [PubMed] [Google Scholar]
- 32.Hajri T., Abumrad N.A. Fatty acid transport across membranes: relevance to nutrition and metabolic pathology. Annu. Rev. Nutr. 2002;22:383–415. doi: 10.1146/annurev.nutr.22.020402.130846. [DOI] [PubMed] [Google Scholar]
- 33.Febbraio M., Abumrad N.A., Hajjar D.P., Sharma K., Cheng W., Pearce S.F., Silverstein R.L. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J. Biol. Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 34.Hajri T., Han X.X., Bonen A., Abumrad N.A. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J. Clin. Invest. 2002;109:1381–1389. doi: 10.1172/JCI14596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goudriaan J.R., Dahlmans V.E., Teusink B., Ouwens D.M., Febbraio M., Maassen J.A., Romijn J.A., Havekes L.M., Voshol P.J. CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J. Lipid. Res. 2003;44:2270–2277. doi: 10.1194/jlr.M300143-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Kraja A.T., Hunt S.C., Pankow J.S., Myers R.H., Heiss G., Lewis C.E., Rao D., Province M.A. An evaluation of the metabolic syndrome in the HyperGEN study. Nutr. Metab. (Lond.) 2005;2:2. doi: 10.1186/1743-7075-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ginsberg H.N., Zhang Y.L., Hernandez-Ono A. Regulation of plasma triglycerides in insulin resistance and diabetes. Arch. Med. Res. 2005;36:232–240. doi: 10.1016/j.arcmed.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Coon H., Leppert M.F., Eckfeldt J.H., Oberman A., Myers R.H., Peacock J.M., Province M.A., Hopkins P.N., Heiss G. Genome-wide linkage analysis of lipids in the Hypertension Genetic Epidemiology Network (HyperGEN) Blood Pressure Study. Arterioscler. Thromb. Vasc. Biol. 2001;21:1969–1976. doi: 10.1161/hq1201.100228. [DOI] [PubMed] [Google Scholar]
- 39.Ordovas J.M., Corella D., Kaput J. Nutrient-gene interactions in lipoprotein metabolism—an overview. Forum. Nutr. 2007;60:102–109. doi: 10.1159/000107079. [DOI] [PubMed] [Google Scholar]
- 40.Yuan H.Y., Chiou J.J., Tseng W.H., Liu C.H., Liu C.K., Lin Y.J., Wang H.H., Yao A., Chen Y.T., Hsu C.N. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res. 2006;34:W635–W641. doi: 10.1093/nar/gkl236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walters J.R. Recent findings in the cell and molecular biology of the small intestine. Curr. Opin. Gastroenterol. 2005;21:135–140. doi: 10.1097/01.mog.0000153309.13080.8b. [DOI] [PubMed] [Google Scholar]
- 42.Nauli A.M., Nassir F., Zheng S., Yang Q., Lo C.M., Vonlehmden S.B., Lee D., Jandacek R.J., Abumrad N.A., Tso P. CD36 is important for chylomicron formation and secretion and may mediate cholesterol uptake in the proximal intestine. Gastroenterology. 2006;131:1197–1207. doi: 10.1053/j.gastro.2006.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seedorf U., Aberle J. Emerging roles of PPAR delta in metabolism. Biochim. Biophys. Acta. 2007;1771:1125–1131. doi: 10.1016/j.bbalip.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 44.Boekholdt S.M., Souverein O.W., Tanck M.W., Hovingh G.K., Kuivenhoven J.A., Peters R.I., Jansen H., Schiffers P.M., van der Wall E.E., Doevendans P.A., et al. Common variants of multiple genes that control reverse cholesterol transport together explain only a minor part of the variation of HDL cholesterol levels. Clin. Genet. 2006;69:263–270. doi: 10.1111/j.1399-0004.2006.00578.x. [DOI] [PubMed] [Google Scholar]
- 45.Gotto A.M., Jr, Brinton E.A. Assessing low levels of high-density lipoprotein cholesterol as a risk factor in coronary heart disease: a working group report and update. J. Am. Coll. Cardiol. 2004;43:717–724. doi: 10.1016/j.jacc.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 46.Lau P., Nixon S.J., Parton R.G., Muscat G.E. RORalpha regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: caveolin-3 and CPT-1 are direct targets of ROR. J. Biol. Chem. 2004;279:36828–36840. doi: 10.1074/jbc.M404927200. [DOI] [PubMed] [Google Scholar]
- 47.Williams R.R., Rao D.C., Ellison R.C., Arnett D.K., Heiss G., Oberman A., Eckfeldt J.H., Leppert M.F., Province M.A., Mockrin S.C., Hunt S.C. NHLBI family blood pressure program: methodology and recruitment in the HyperGEN network. Hypertension genetic epidemiology network. Ann. Epidemiol. 2000;10:389–400. doi: 10.1016/s1047-2797(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 48.Hemminger B.M., Saelim B., Sullivan P.F. TAMAL: an integrated approach to choosing SNPs for genetic studies of human complex traits. Bioinformatics. 2006;22:626–627. doi: 10.1093/bioinformatics/btk025. [DOI] [PubMed] [Google Scholar]
- 49.Leushner J., Chiu N.H. Automated mass spectrometry: a revolutionary technology for clinical diagnostics. Mol. Diagn. 2000;5:341–348. doi: 10.1007/BF03262095. [DOI] [PubMed] [Google Scholar]
- 50.O'Connell J.R., Weeks D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection Evaluation Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 52.Storey J.D., Taylor J.E., Siegmund D. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. J. Royal Stat. Soc. 2004;66:187–205. [Google Scholar]
- 53.Sampson M.J., Davies I.R., Braschi S., Ivory K., Hughes D.A. Increased expression of a scavenger receptor (CD36) in monocytes from subjects with Type 2 diabetes. Atherosclerosis. 2003;167:129–134. doi: 10.1016/s0021-9150(02)00421-5. [DOI] [PubMed] [Google Scholar]
- 54.Miao W.M., Vasile E., Lane W.S., Lawler J. CD36 associates with CD9 and integrins on human blood platelets. Blood. 2001;97:1689–1696. doi: 10.1182/blood.v97.6.1689. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.