

Abstract
In mitosis the kinetochores of chromosomes that lack full microtubule attachments and/or mechanical tension activate a signaling pathway called the mitotic spindle checkpoint that blocks progression into anaphase and prevents premature segregation of the chromatids until chromosomes become aligned at the metaphase plate (1). The spindle checkpoint is responsible for arresting cells in mitosis in response to chemotherapeutic spindle poisons such as paclitaxel or vinblastine. Some cancer cells show a weakened checkpoint signaling system that may contribute to chromosome instability in tumors. Since complete absence of the spindle checkpoint leads to catastrophic cell division, we reasoned that drugs targeting the checkpoint might provide a therapeutic window in which the checkpoint would be eliminated in cancer cells but sufficiently preserved in normal cells. We developed an assay to identify lead compounds that inhibit the spindle checkpoint. Most cells respond to microtubule drugs by activating the spindle checkpoint and arresting in mitosis with a rounded morphology. Our assay depended on the ability of checkpoint inhibitor compounds to drive mitotic exit and cause cells to flatten onto the substrate in the continuous presence of microtubule drugs. In this study we characterize one of the compounds, OM137, as an inhibitor of Aurora kinases. We find that this compound is growth inhibitory to cultured cells when applied at high concentration and potentiates the growth inhibitory effects of subnanomolar concentrations of paclitaxel.
Keywords: Mitosis, Cancer, Microtubule, Chromosome Instability, Paclitaxel
Introduction
During mitosis in metazoans the duplicated chromosomes, composed of two identical chromatids joined at the centromere, move as individuals to align at the metaphase plate. The mitotic spindle checkpoint is a cell signaling pathway that blocks the premature onset of chromatid separation until all the kinetochores of chromosomes are fully occupied by microtubule and/or under appropriate mechanical tension. Spontaneous or chemically induced defects in the mitotic spindle can lead to long term activation of the spindle checkpoint, whereupon cells arrest at the pre-anaphase stage for many hours.
Many cancers show abnormal chromosome content, often being hyperdiploid. Certain lines derived from tumors have abnormal but relatively stable chromosome content while others exhibit substantial chromosome instability (CIN), a condition in which chromosomes frequently missegregate. The role of CIN as a causative factor in oncogenesis is both complex and controversial (2, 3). However, it is likely that CIN contributes to the production of variant populations with the potential for increased malignancy or resistance to therapy. In some instances, tumor lines exhibiting CIN have been shown to have partially defective spindle checkpoint signaling, and these checkpoint defects have been shown to reflect checkpoint gene mutation or altered levels of checkpoint protein expression (4–6). Whether the partially inactive checkpoint that is characteristic of many tumor cells renders the cells more or less sensitive to treatments with microtubule drugs such as paclitaxel remains controversial (7–10). While tumor cells often exhibit a partially defective spindle checkpoint, total inactivation of the checkpoint results in catastrophic cell division that is incompatible with cell survival (11, 12). We reasoned that differences in the potency of checkpoint systems in normal versus tumor cells might provide a therapeutic window for the development of useful drugs that target the mitotic spindle checkpoint. At appropriate doses these drugs could fully eliminate the checkpoint response in tumor cells while sparing sufficient checkpoint activity for survival of normal cells. Checkpoint inhibitor drugs might thus prove preferentially toxic to tumor cells, particularly when used in combination with microtubule drugs that cause checkpoint activation. We devised a cellular assay to test small molecules for compounds that would override the spindle checkpoint. Here we describe the assay used to identify checkpoint inhibitors and characterize one of those lead compounds, OM137, that functions to override the spindle checkpoint primarily through inhibition of the class of mitotic kinases called the Aurora kinases.
Materials and Methods
Cell Culture
Hela cells were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% bovine calf serum or fetal bovine serum, 20mM Hepes, 1x non-essential amino acids, 1 mM sodium pyruvate, 60 µg/ml of penicillin, and 100 µg/ml streptomycin at 37°C under 5% CO2. Ptk1 (rat kangaroo kidney) cells were cultured in Minimal Essential Medium supplemented with 10% fetal bovine serum, 20 mM Hepes buffer, 1x nonessential amino acids, 1 mM sodium pyruvate, 60 µg/ml penicillin, and 100 µg/ml streptomycin. LLC-Pk (porcine kidney) cells were grown in Dulbecco's Modified Minimal Essential Medium with 20 mM Hepes, 10% fetal bovine serum, 60 µg/ml penicillin, and 100 µg/ml streptomycin. Xenopus S3 cells were grown at 23°C in 70% Leibovitz’s L-15 medium containing 15% fetal bovine serum, L-glutamine, 60 µg/ml penicillin, and 100 µg/ml streptomycin.
High Throughput Assay for Chemical Inhibitors of the Spindle Checkpoint
Hela cells in ten 150 mm tissue culture dishes were blocked for 16 to 17 h with nocodazole at 100 ng/ml. The rounded mitotic cells were released from the substratum by gentle agitation and collected. Assays were conducted in duplicate in 384 well plates. A commercial library of 10,000 diverse small molecules (Chembridge Corp.) was stored at −20°C in 96 well plates at an approximate concentration of 2.5 mM in DMSO. Ten microliters of medium were distributed to each well of the 384 well plates. A 0.5 µl 96 pin transfer device (V.P Scientific) was used to transfer an initial aliquot of the test compounds to the upper left well (well A) of a four well quadrant in the 384 well plate. A 2 µl 96 pin transfer device was used to make serial dilutions to the other 3 wells (wells B, C, D) of the quadrant. An additional 10 ul of medium containing approximately 104 cells was added to each well. Assuming a uniform molecular weight of 500 for the compounds, each test compound is tested at concentrations of 55, 10, 1.7, and 0.3 µM in the four wells of the quadrant. The nocodazole concentration was maintained at 20 ng/ml. Negative controls were included in each plate including wells with only medium or cells tested with carrier (DMSO). As a positive control, RO-31-8220 at 10 µM was added. RO-31-8220 is an inhibitor of cyclin-dependent kinase 1 (13) and elicits mitotic exit and flattening onto the substrate for cells in nocodazole.
For the remainder of the protocol one of the duplicate plates was inverted to counteract processing artifacts such as inhomogeneities in certain channels of the washer or fluorescent plate reader. Plates were incubated for 4 hours at 37°C to allow mitotic exit and attachment of cells in wells where the spindle checkpoint was abrogated. Plates were then washed with 5 cycles in a Tecan PW-384 plate washer using MOPS/Triton/DNAse (0.1% Triton X-100, 150 mM NaCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 10 mM MOPS, pH 7.3 and 10 µg/ml DNAse1 (Sigma)). The DNAse serves to reduce non-specific background due to cells becoming trapped in DNA gel released by dead or dying cells. After the final wash, wells were treated with a fixation/permeablilzation/staining solution consisting of 2% paraformaldehyde, 0.5% Triton X-100, 60 mM Pipes, 25 mM HEPES, 10 mM EGTA, 4 mM MgSO4, pH 6.9 and the fluorescent DNA label Syber Gold used at the manufacturer’s recommended concentration diluted 1:10,000 from the stock (Invitrogen). The plates were then read with a Tecan Genios fluorescent plate reader.
Immunofluorescence
Xenopus S3 cells were grown on glass coverslips and incubated in 25 µM MG132 for 90 minutes to accumulate cells arrested at metaphase. Cells were then incubated in media containing 25 µM MG132 and OM137 ranging from 0.8 to 100 µM for 60 minutes. Cells were treated with fixation-extraction solution (0.5%Triton X-100, 1.5% paraformaldehyde, 60 mM Pipes, 25 mM HEPES, 10 mM EGTA, 4 mM MgSO4, 400 nM Microcystin-LR, pH 6.9) for 15 minutes at room temperature. Mouse anti-phospho-histone H3 (Cell Signaling, 1/2000) and Cy3-conjugated goat anti-mouse (Jackson Immunoresearch, 1/1,000) antibodies were used to detect phosphorylated histone H3. DNA was stained with DAPI. Labeled cells were mounted in Vectashield (Vector Industries) containing 10 mM MgSO4. 3D images were collected using a Ziess Axioplan IIe microscope, 100X objective (Plan-APOCHROMAT, 1.4 NA) and a Hammamatsu C47472-98 CCD camera. Fluorescence images were analyzed using Metamorph software (Molecular Devices).
Chromosome Isolation and Immunofluorescence
HeLa cells were treated with 330 nM nocodazole for 4 hours to accumulate mitotic cells. Mitotic cells were collected by shake-off and their media was exchanged to 330 nM nocodazole and 25 mM MG132. OM137 was added to experimental cultures for a final concentration of 100 uM, while control cultures received an equivalent volume of DMSO. Mitotic cells were washed in 10 mM HEPES (pH 7.4), 40 mM KCl, 5 mM EGTA, 4 mM MgSO4, and 400 nM Microcystin-LR by centrifugation at 200 × g for 4 minutes. Mitotics were lysed in 60 mM Pipes, 25 mM HEPES (pH 6.9), 10 mM EGTA, 4 mM MgSO4 (PHEM), 0.5% Triton X-100, 1 mM DTT, 400 nM Microcystin-LR, and 5 µg/ml protease inhibitor cocktail. The extracts were centrifuged through a cushion of lysis buffer containing 10% glycerol over poly-L-lysine-treated glass coverslips at 1500 × g for 10 min at 4°C in order to collect chromosomes for immunofluorescence labeling. The chromosome-coated coverslips were then fixed in PHEM and 1.5% formaldehyde for 15 min and processed for immunofluorescence analysis as described previously (14). Mouse anti-Aurora B (BD Biosciences), Rabbit anti-MAD2 (generously provided by Dr. Rey Chen, Academia Sinica, Taiwan), and Rabbit anti-BUBR1 (generously provided by Dr. Todd Stukenberg, University of Virginia) were used at 1.25 ug/ml, 0.5 ug/ml, and 1:100 respectively. Chromosome fluorescence image capture and analysis was performed with a Zeiss Axioplan II microscope equipped with a 100× (NA 1.4) objective, a Hamamatsu Orca 2 camera (Hamamatsu Photonics), and Metamorph imaging software (Molecular Devices).
Kinase Assays
All kinase assays were carried out in 30 µl. Reaction mixes contained 50 µM ATP, 1 mM DTT, 1 mM Na3VO4, 5 µCi [γ−32P]-ATP, 1 µg of the appropriate substrate (see below), 1 µl DMSO or drugs dissolved in DMSO, and 50 to 100 ng kinase. Reaction mixes were incubated for 1 h at 30°C, quenched with SDS loading buffer and resolved on 14 % SDS-PAGE. Incorporation of 32P was visualized by autoradiography. Densitometry analysis was performed using ImageJ software (W. Rasband, NIH). IC50 values were calculated from log-dose response curves using Prism 4 software (Graph Pad). Aurora A:TPX2, Aurora B:INCENP and CDK5:p25 purification protocols and kinase assay conditions have been described previously (15–17). Plk1 and CDK1:Cyclin B were kind gifts of Dr. Aldo Tarricone. Bub1:Bub3 complex and Mps1 kinase were expressed in, and purified from, Sf9 insect cells infected with recombinant baculoviruses. The complex was isolated on Ni-NTA beads and further purified by size-exclusion chromatography. Bub1:Bub3 kinase reaction buffer contained 50 mM Tris-HCl pH 7.6, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA and histone H3 was used as substrate. Human Mps1 was expressed and purified in Sf9 cells. Mps1 was assayed in 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10 mM MnCl2, and Mad1:Mad2 complex as a substrate (18). Human Nek2A (residues 1–291) was expressed in Escerichia coli as a fusion to GST. The protein was purified on GSH Sepharose Fast Flow (GE Healthcare) and the GST tag cleaved using PreScission Protease (GE Healthcare). The cleaved product was further purified by size exclusion chromatography. Nek2A assays were carried out in 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10 mM MnCl2 with α-casein as a substrate. Human Plk1 (a kind gift from Nerviano Medical Sciences) was tested in 50 mM Tris-HCl pH 7.6, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA with α-casein as a substrate. Human Tao1 (residues 1–356) cDNA was a kind gift of Dario Alessi (University of Dundee, Dundee, UK). Tao1 was expressed as an N-terminal GST fusion in Escherichia coli and isolated on GSH Sepharose Fast Flow (GE Healthcare). GST-tagged TAO1 immobilized on GSH Sepharose beads was direclty used in kinase assay in 40 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM EDTA and myelin basic protein as a substrate. CDK1:cyclin B was assayed under the same conditions previously described for CDK5:p25 (15–17).
Video Microscopy
S3, Ptk1, or Hela cells were grown on 25 mm round coverslips. The coverslips were sealed into Sykes Moore Chambers (Bellco) and medium containing test compounds were added using a syringe. Cells were cultured at 37°C on the stage of a Zeiss Axiovert 200 microscope or a Nikon Eclipse TE2000-E microscope. Images were collected at intervals using phase contrast or Nomarski DIC optics with Roper Coolsnap-HQ2 or Hamamtsu Orca-ERG cameras using Metamorph software (Molecular Devices) or NIS-Elements software (Nikon).
Cell Proliferation Assay
Hela cells at 80 cells/well were seeded in 96 well plates and permitted to adhere to the substratum for 6 hours while incubating at 37°C under 5% CO2. Test compounds were then added; paclitaxel at 0.25 nM and OM137 ranging from 6.25 uM to 100 uM. Controls received equivalent levels of DMSO (the diluent for both compounds). All conditions were assayed in quadruplicate. Cells were incubated for 4 days under these conditions. At the end of the 4th day, the media was exchanged with fresh media containing OM137 at the same concentrations, but paclitaxel was increased to 0.75 nM. Cells were incubated for an additional 4 days. The amount of cell proliferation was measured using the CellTiter 96®AQueous One Solution Cell Proliferation Assay (Promega Corporation, G3580). Absorbance measurements were obtained using a Tecan Genios plate reader. Data from cells treated solely with OM137 were normalized to untreated cell values. Values obtained from cells exposed to taxol and OM137 were normalized to data from cells treated with taxol alone.
Results
High Throughput Screening Identifies Chemical Inhibitors of the Mitotic Spindle Checkpoint
Many cultured cells that are well attached during interphase become rounded during mitosis and maintain only weak attachment to the substratum. Upon division and exit from mitosis they reattach and reflatten (Fig 1A). Cells treated with microtubule drugs such as nocodozole arrest in mitosis through the action of the spindle checkpoint and remain arrested in this rounded state for several hours. They can be dislodged easily with gentle agitation of the medium. However, if the spindle checkpoint is inactivated these cells will flatten and reattach without division (Fig 1A). We transferred nocodazole-arrested mitotic cells to wells of 384 well dishes and tested a library of small molecules for their ability to induce mitotic exit in the arrested cells. Compounds that inactivate the checkpoint caused cells to exit mitosis, flatten, and reattach firmly to the substratum. The cells in wells containing inactive compounds remained rounded and were easily washed from the dishes (Fig 1B). After fixation in a solution containing a fluorescent DNA label, we used a fluorescence plate reader to rapidly assess which test compounds could induce mitotic exit and cell reattachment. Because the assay requires cells to actively flatten onto the substrate it selects against compounds that are merely cytotoxic.
Figure 1.
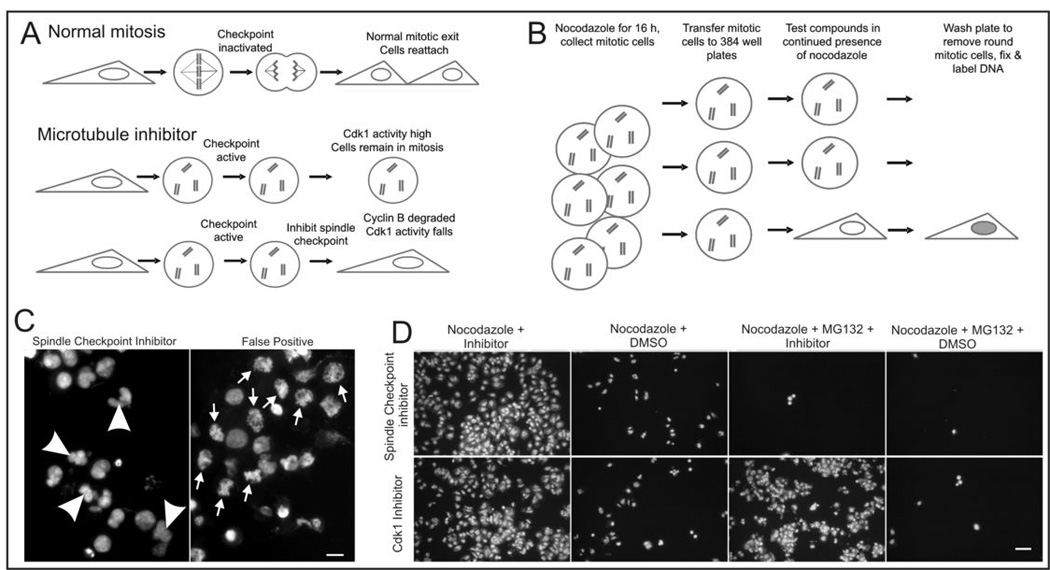
A high throughput, whole cell assay for small molecule inhibitors of the mitotic spindle checkpoint. A, In normal cell division cells become rounded during mitosis and flatten onto the substrate following cytokinesis. In response to microtubule drugs the spindle checkpoint is activated and cells remain arrested as rounded cells while the checkpoint is active. If the checkpoint fails, cells flatten and reattach often in the absence of cytokinesis. B, Schematic for checkpoint inhibitor assay. Cells forced to exit mitosis will flatten and reattach to the substratum. Mitotic cells remain rounded and are washed from the assay plates. C, DNA labeling of cells in assay plates allows immediate visualization of wells in which cells exit M phase with decondensed chromosomes and reformed nuclei that are often abnormal in shape (arrowheads in left panel). In contrast, drugs that induce cells to remain stuck to plates in the absence of mitotic exit show condensed chromosomes (arrows in right panel). These are scored as false positives. Bar = 10 µm. E, Secondary screening with proteasome inhibitor-treated cells distinguishes drugs that inhibit the spindle checkpoint from those that induce mitotic exit through other mechanisms such as inhibition of cyclin-dependent kinases. Checkpoint inhibitors induce exit from nocodazole arrest but do not induce exit in the presence of proteasome inhibitors. Bar = 50 µm.
The screen was also designed to eliminate a number of false positives. Because a fluorescent DNA label was used it was simple matter to examine microscopically all the wells scored as positive from the plate reader analysis and confirm that they contained live cells that had been induced to exit mitosis (Fig. 1C). In wells were cells exited mitosis, the chromatin was decondensed and assembled into one or more rounded nuclei within the attached cells. In a few instances however, we noted that positive wells contained a high proportion of attached cells in which chromatin remained condensed in mitotic chromosomes. These false positives were excluded from further analysis.
The spindle checkpoint functions by inhibiting the ubiquitylation pathway that targets cyclin B and other proteins for degradation by the proteasome. Thus proteasome activity is downstream of the checkpoint and is absolutely required for mitotic exit induced by chemical inhibitors of the spindle checkpoint. As a secondary screen potential spindle checkpoint inhibitors were tested for the ability to override a mitotic block imposed by a combination of nocodazole and the proteasome inhibitor MG132 (Fig 1D). Thus those compounds that induced mitotic exit of nocodazole treated cells in the absence of MG132 but failed to induce mitotic exit in its presence were scored as positive inhibitors of the mitotic spindle checkpoint. As controls for this assay we used chemical inhibitors of cyclin-dependent kinase 1, which are able to induce mitotic exit in the presence of proteasome inhibitors (13).
We screened a commercial library of 10,000 small molecules (Chembridge Corp.) From these, we identified 11 compounds of distinct structure that inhibited the spindle checkpoint at micromolar concentrations. Most often hits scored at the highest concentration tested. Here we report in detail the characterization of the cell biological effects and the molecular targets of one compound, OM137, that scored as a hit at the highest concentration tested in the initial screen. Characterization of other lead compounds and identification of cellular targets for other compounds identified in the screen is underway and will be reported subsequently.
Compound OM137 is an inhibitor of Aurora kinases
To obtain clues to the potential molecular targets of lead compounds, we assayed their effects on phosphorylation of serine 10 in histone H3 using an antibody that specifically binds this site when phosphorylated. Serine 10 phosphorylation of histone H3 is catalyzed by Aurora B kinase, which becomes activated in mitosis. We and others have shown that Aurora B activity is required for maintenance of the spindle checkpoint (19–21). To ensure that loss of phosphorylation of histone H3 was a direct consequence of inhibition of Aurora B and not an indirect effect of mitotic exit, we carried out the assay using cells cultured in the presence of the proteasome inhibitor, MG132. As shown in Fig. 2A and Fig. 2B, amongst the lead compounds, OM137 showed the most potent inhibition of expression of the serine 10 phosphoepitope on histone H3. Certain other lead compounds, particularly F and K, showed somewhat weaker inhibitory activity. When tested at a range of concentrations for inhibition of histone H3 phosphorylation in mitotic cells, OM137 showed an IC50 of approximately 15 µM (Fig 3A and 3B).
Figure 2.
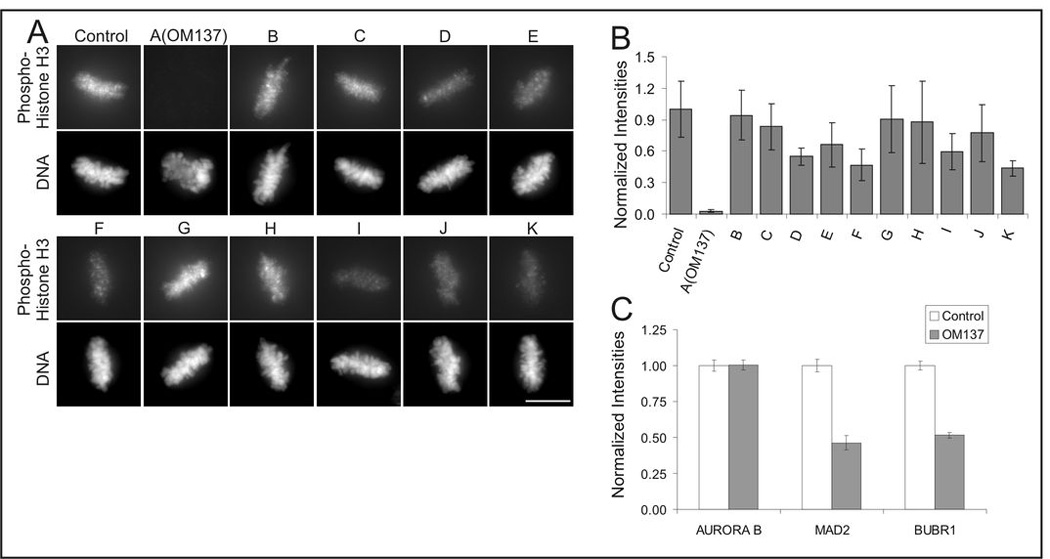
Comparison of checkpoint inhibitors for their ability to inhibit expression of phosphorylated serine 10 of histone H3, a target of Aurora B kinase. A, Xenopus S3 cell cultures were incubated in the proteasome inhibitor MG132 in combination with the set of spindle checkpoint inhibitors identified in the screen and then labeled with antibody to phospho-H3 (upper rows) and with DAPI to label DNA (lower rows). Bar = 10 µm. B, Quantitation of immunolabeling with anti-phospho-H3. Error bars indicate S.D. C, OM137 induces loss of spindle checkpoint proteins from kinetochores. Hela cells were treated with nocodazole plus the proteasome inhibitor, MG132, were treated with OM137 and then mitotic cells were washed off and used to prepare isolated chromosomes that were then antibody labeled for the indicated spindle checkpoint protein. Error bars indicate S.E.M.
Figure 3.
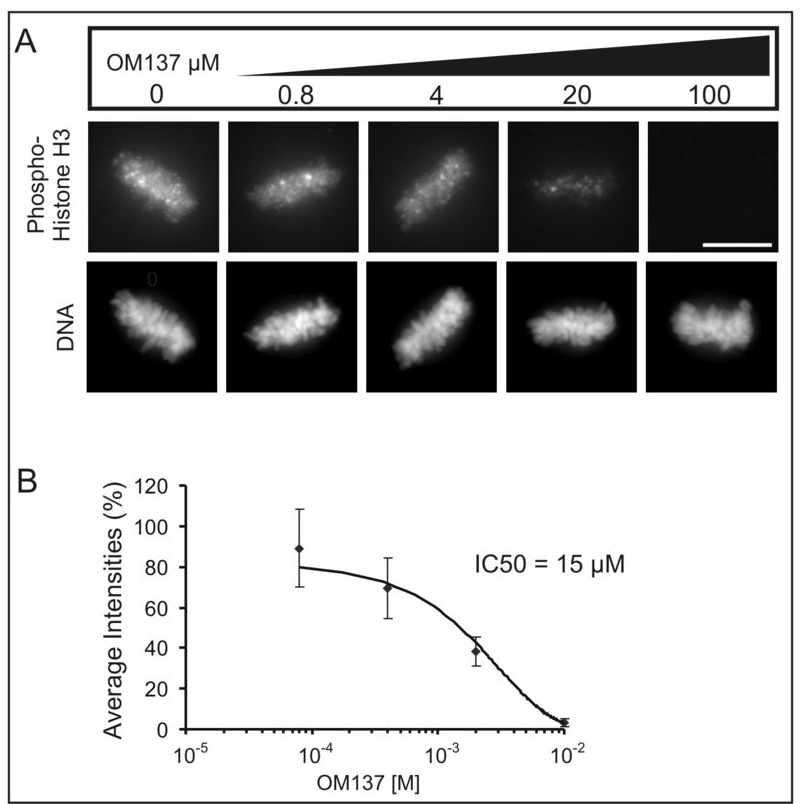
OM137 inhibits expression of phospho-H3 in mitotic cells. A, Increasing concentrations of OM137 leads to progressively increased inhibition of phospho-H3 expression from mitotic Xenopus S3 cells. Bar = 10 µm. B, Plot of phospho-H3 levels with increasing concentrations of OM137.
We tested OM137 for direct inhibition of Aurora A and Aurora B kinase as well as with a variety of other mitotic kinases. We found that OM137 inhibited Aurora A kinase (IC50 = 21.7 µM) and Aurora B kinase (IC50 = 2.4 µM) (Fig 4A). When tested with other mitotic kinases Mps1, Bub1, Plk1, Nek2A, and Tao1 that have been implicated in spindle checkpoint signaling, OM137 showed no significant inhibition (Fig. 4B). We did observe that OM137 showed in vitro activity in inhibiting cyclin-dependent kinases, Cdk1/cyclinB and Cdk5/p25 with an approximate IC50 of 20 µM.
Figure 4.
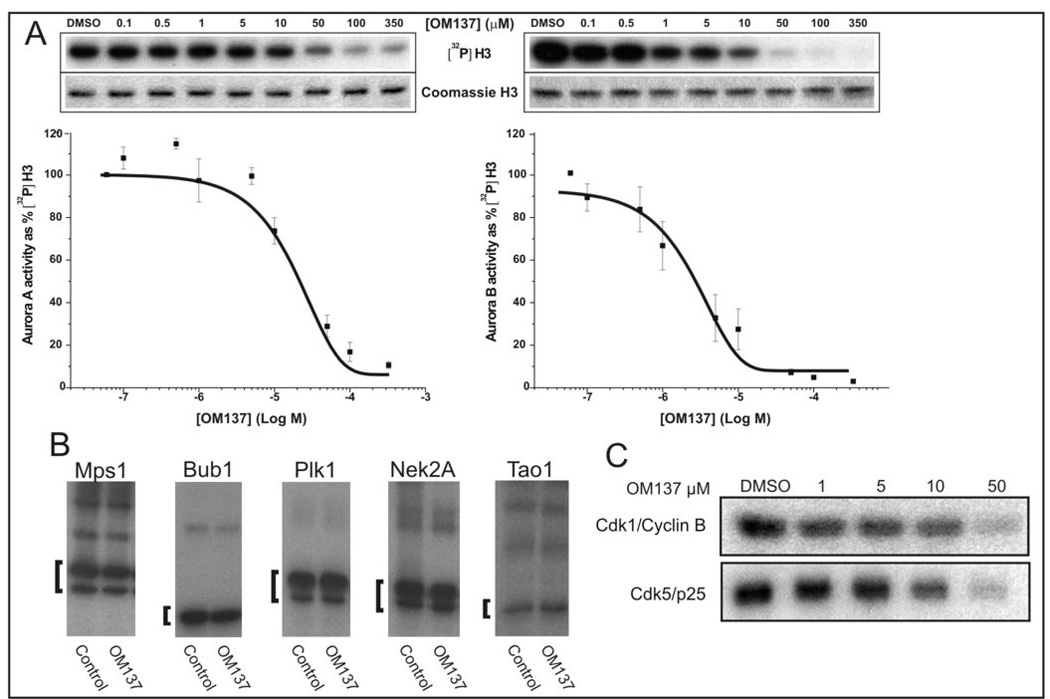
OM137 inhibits Aurora kinases. (A) Assay for OM137 inhibition of Aurora A (left) and Aurora B (right) kinases. For Aurora A IC50 = 21.7 µM and for Aurora B IC50 = 2.4 µM. B, OM137 at 10 µM does not inhibit other mitotic kinases: Mps1, Bub1, Plk1, Nek2A and Tao1. Brackets denote test substrates for each kinase (see Methods). C, OM137 shows inhibition of Cdk1/CyclinB and Cdk5/p25 in vitro. Error bars indicate S.D.
Analogs of OM137
A number of compounds with alternative substitutions on the aryl ring were available commercially. We tested several in our checkpoint assay. As shown in Figure 5 we found several analogs with activities in the spindle checkpoint assay similar to (compounds 3 and 4) or even stronger than (compound 2) OM137 and we noted certain substitutions (compounds 5 – 8) led to loss of activity. These structure-activity relationship data highlight the importance of the amino group on the thiazole moiety and the presence and position of the hydroxyl group on the aryl moiety as important determinants for checkpoint inhibition.
Figure 5.
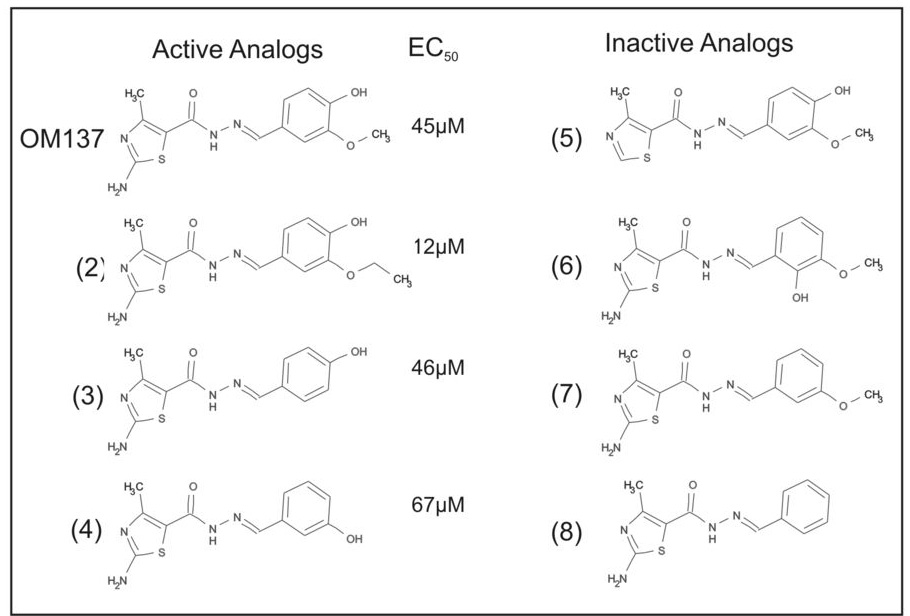
Active and inactive analogs of OM137. Analogs 1–3 showed similar activity to OM137 in inducing mitotic exit. EC50’s for the checkpoint assay are depicted. Analogs 4–7 showed no activity in the checkpoint assay.
Compound OM137 induces mitotic exit in cells arrested by the spindle checkpoint
With video microscopy we studied cellular responses to abrogation of the spindle checkpoint by OM137 using cells that remain relatively flat in mitosis. In cultured Xenopus S3 cells treated with OM137 prior to nuclear envelope breakdown, many chromosomes failed to align at the metaphase plate. Cells then entered anaphase with massive chromosome mis-segregation; cytokinesis failed, and mitotic exit resulted in the formation of a misshapen and multi-lobed nucleus (Fig 6A and Supplemental Video 1). Similarly, when cells were treated with OM137 in the early stages of prometaphase after nuclear envelope breakdown, premature mitotic exit mitotic exit occurred accompanied by chromosome decondensation and reformation of a misshapen interphase nucleus. OM137 treatment of mitotic cells also caused restructuring of the microtubule network from the mitotic spindle array to the interphase pattern (Fig 6B and Supplemental Video 2). As expected OM137 also overrode chronic checkpoint activation induced by treatment of cells with microtubule poisons. Ptk1 cells treated with nocodazole remained arrested with condensed mitotic chromosomes for several hours (Fig 6C left column and Supplemental Video 3). In contrast when nocodazole-arrested cells were co-treated with OM137, the chromosomes rapidly decondensed and an interphase nucleus reformed around the undivided chromosomes (Fig 6C right column and Supplemental Video 4).
Figure 6.
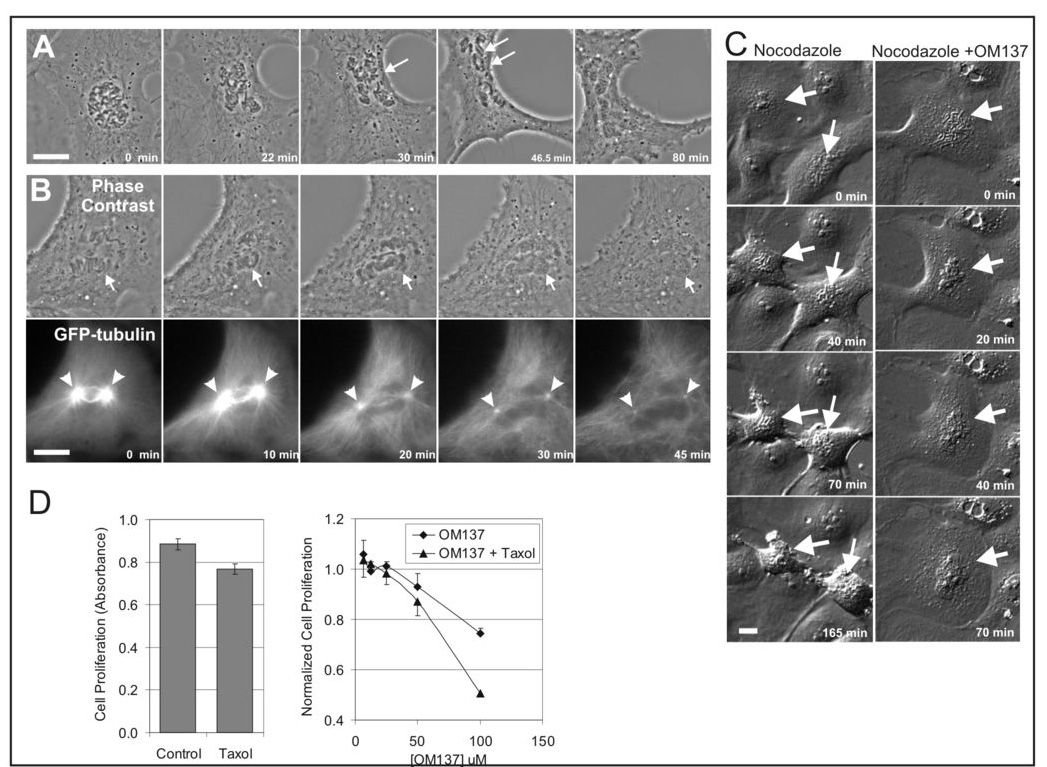
OM137 inhibits chromosome alignment at metaphase, induces premature anaphase, overrides the mitotic spindle checkpoint promotes the growth inhibitory activity of paclitaxel. A, A Xenopus S3 cell was treated prior to nuclear envelope breakdown with OM137 at 100 µM and then imaged by time lapse phase contras microscopy. A minority of the chromosomes aligned at the metaphase plate (single arrow) and these underwent chromatid separation when the cell entered premature anaphase (double arrow). As the cell exits M phase, the chromosomes decondensed and a large multi-lobed nucleus formed. The time-lapse sequence for this cell is shown in Supplemental Video 1. B, A Xenopus S3 cells stably expressing GFP-tubulin was treated with OM137 at 50 µM and imaged by phase contrast and fluorescence microscopy. Shortly after treatment at time 0 chromosomes (arrows) begin to decondense eventually forming an abnormally shaped interphase nucleus by 30 min. In the GFP-tubulin image, the cell at time 0 has bright mitotic spindle poles (arrowheads) and a nascent mitotic spindle. Treatment with OM137 leads to loss of the spindle structure and reversion to the interphase microtubule array. The time-lapse sequence for this cell is shown in Supplemental Video 2. B, Cultures of flat epithelial Ptk1 cells were treated with 100ng nocodazole for 20 min before DIC imaging. Control cultures that did not receive OM137 (left column) remain arrested in mitosis. At time 0 one cell is already arrested at prometaphase (upper arrow) while another cell is in prophase (lower arrow). A few minutes later, both cells are arrested at prometaphase and remain arrested for the duration of imaging (165 min). As they remain in mitosis, cells tend to become more rounded. The time-lapse sequence for this sequence is shown in Supplemental Video 3. In experimental cultures that were treated with nocodazole plus 80 µM OM137 (lower row) cells escape spindle checkpoint arrest. The arrow points to a prometaphase cell. By 20 min, the chromosomes have begun to decondense. Decondensation and nuclear envelope reformation take place over the ensuing time such that by 70 min a characteristic interphase nucleus has reformed showing that the cell has exited M phase without chromosome segregation and cytokinesis. The time-lapse sequence for this cell is shown in Supplemental Video 4. Bars = 10 µm. D, Subnanomolar concentrations of paclitaxel caused only minor inhibition of Hela cell growth over 8 days (left panel). OM137 at higher concentrations inhibited cell growth. When used in combination with subnanomalar concentrations of paclitaxel, growth inhibition was enhanced (right panel).
Compound OM137 inhibits cell growth and enhances growth inhibitory effects of paclitaxel
Paclitaxel is a commonly used anti-tumor drug. We tested whether OM137 would inhibit Hela cell growth when used alone or in combination with paclitaxel. At higher concentrations, OM137 showed growth inhibition and inhibition was significantly increased when OM137 was applied with subnanomlar concentrations of paclitaxel (Fig 6D, right panel). Subnanomolar concentrations of paclitaxel showed only minimal growth inhibition when used alone (Fig 6D, left panel).
Discussion
Human tumors have also been reported to show altered spindle checkpoint signaling characteristics that, in some instances, are due to mutations or altered levels of checkpoint signaling proteins. Aurora kinases are often misregulated in human tumors (reviewed in (22, 23). These changes may lead to alterations in events of mitosis, e.g. malfunctions in spindle assembly and chromosome segregation. Aurora B is required for normal function of the mitotic spindle checkpoint (19–21). Mitotic defects may contribute to chromosome mis-segregation and aneuploidy in human cancers and these chromosomal abnormalities may contribute to tumor malignancy (reviewed in (2, 10). However, altered checkpoint activity due to improper expression of Aurora kinases in tumor cells may also present a target for tumor-specific anti-cancer therapeutics. A number of other Aurora kinase inhibitors have been reported and several of these are currently in clinical trial (reviewed in (23).
Here we show that a screen to detect compounds that inhibit the spindle checkpoint identified an inhibitor of Aurora kinases termed OM137. OM137 is an aminothiazole derivative. Thiazole derivatives have previously been identified as Aurora kinase inhibitors (24). Recently a large scale screen was carried out assaying compounds for inhibition of Aurora A kinase in vitro that identified and characterized a large number of small molecule aminothiazole compounds related to but distinct from OM137 (25). Although many of the compounds analyzed in that study were more potent inhibitors of Aurora A kinase in vitro, the authors reported that obtaining responses consistent with Aurora kinase inhibition in living cells required concentrations many fold higher than that required in vitro, attributing the difference in potency to problems with cell permeability of the compounds.
Our screen for checkpoint inhibitor activity were conducted with whole cells and therefore required that effective compounds be cell permeable. In addition we found that OM137 was a more potent inhibitor of Aurora B compared to Aurora A in vitro, consistent with the effects of OM137 on checkpoint function in living cells. We also found that OM137 showed inhibitory activity against cyclin-dependent kinases. Cdk1 inhibitors can drive mitotic exit when applied to cells in culture (13, 26). However, unlike other Cdk1 inhibitors, OM137 was unable to drive mitotic exit when the proteasome was inhibited. Thus it is likely that the major mode by which OM137 drives mitotic exit of cells arrested in M phase via the spindle checkpoint is through its inhibitory activity against Aurora B kinase. Inhibition of Aurora B kinase is known to induce override of the spindle checkpoint (19–21). We hypothesize that OM137 induces catastrophic mitotic exit in cultured cells through its activity on Aurora kinases. This effect may be assisted by other properties of OM137 including its ability to inhibit of cyclin-dependent kinases. Compounds that inhibit the spindle checkpoint may provide a novel approach toward directly targeting tumors that show partially defective checkpoints. Such compounds may also enhance the anti-tumor potency of M phase checkpoint activators such as paclitaxel and other microtubule poisons.
Acknowledgements
We are grateful to Todd Stukenberg, Fabrizio Villa, and Fabio Sessa for helpful discussion. We thank Rey Chen and Todd Stukenberg for providing antibodies. We also thank Aldo Tarricone for providing Plk1 and CDK1:Cyclin B.
Financial Support: JHD, JRD, and GJG were supported by grants R01GM50412 and R21CA091264 from the National Institutes of Health, by grant HR00-094 from the Oklahoma Center for the Advancement of Science and by the McCasland Foundation. SS and AM were supported by grants from the Italian Foundation for Cancer Research (AIRC) and from the Telethon Foundation.
References
- 1.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 2.Weaver BA, Cleveland DW. Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res. 2007;67:10103–10105. doi: 10.1158/0008-5472.CAN-07-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 5.Pinto M, Soares MJ, Cerveira N, et al. Expression changes of the MAD mitotic checkpoint gene family in renal cell carcinomas characterized by numerical chromosome changes. Virchows Arch. 2007;450:379–385. doi: 10.1007/s00428-007-0386-7. [DOI] [PubMed] [Google Scholar]
- 6.Sotillo R, Hernando E, Diaz-Rodriguez E, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee EA, Keutmann MK, Dowling ML, Harris E, Chan G, Kao GD. Inactivation of the mitotic checkpoint as a determinant of the efficacy of microtubule-targeted drugs in killing human cancer cells. Mol Cancer Ther. 2004;3:661–669. [PubMed] [Google Scholar]
- 8.Sudo T, Nitta M, Saya H, Ueno NT. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 2004;64:2502–2508. doi: 10.1158/0008-5472.can-03-2013. [DOI] [PubMed] [Google Scholar]
- 9.Swanton C, Tomlinson I, Downward J. Chromosomal instability, colorectal cancer and taxane resistance. Cell Cycle. 2006;5:818–823. doi: 10.4161/cc.5.8.2682. [DOI] [PubMed] [Google Scholar]
- 10.Yamada HY, Gorbsky GJ. Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol Cancer Ther. 2006;5:2963–2969. doi: 10.1158/1535-7163.MCT-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorbsky GJ, Chen RH, Murray AW. Microinjection of antibody to Mad2 protein into mammalian cells in mitosis induces premature anaphase. J Cell Biol. 1998;141:1193–1205. doi: 10.1083/jcb.141.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci U S A. 2004;101:8699–8704. doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Potapova TA, Daum JR, Pittman BD, et al. The reversibility of mitotic exit in vertebrate cells. Nature. 2006;440:954–958. doi: 10.1038/nature04652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahonen LJ, Kallio MJ, Daum JR, et al. Polo-like kinase 1 creates the tension-sensing 3F3/2 phosphoepitope and modulates the association of spindle-checkpoint proteins at kinetochores. Curr Biol. 2005;15:1078–1089. doi: 10.1016/j.cub.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 15.Bayliss R, Sardon T, Vernos I, Conti E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell. 2003;12:851–862. doi: 10.1016/s1097-2765(03)00392-7. [DOI] [PubMed] [Google Scholar]
- 16.Mapelli M, Massimiliano L, Crovace C, et al. Mechanism of CDK5/p25 binding by CDK inhibitors. J Med Chem. 2005;48:671–679. doi: 10.1021/jm049323m. [DOI] [PubMed] [Google Scholar]
- 17.Sessa F, Mapelli M, Ciferri C, et al. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell. 2005;18:379–391. doi: 10.1016/j.molcel.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 18.Sironi L, Mapelli M, Knapp S, De Antoni A, Jeang KT, Musacchio A. Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of a 'safety belt' binding mechanism for the spindle checkpoint. Embo J. 2002;21:2496–2506. doi: 10.1093/emboj/21.10.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ditchfield C, Johnson VL, Tighe A, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hauf S, Cole RW, LaTerra S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kallio MJ, McCleland ML, Stukenberg PT, Gorbsky GJ. Inhibition of aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr Biol. 2002;12:900–905. doi: 10.1016/s0960-9822(02)00887-4. [DOI] [PubMed] [Google Scholar]
- 22.Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1–10. doi: 10.1158/1541-7786.MCR-06-0208. [DOI] [PubMed] [Google Scholar]
- 23.Mountzios G, Terpos E, Dimopoulos MA. Aurora kinases as targets for cancer therapy. Cancer Treat Rev. 2008;34:175–182. doi: 10.1016/j.ctrv.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Jung FH, Pasquet G, Lambert-van der Brempt C, et al. Discovery of novel and potent thiazoloquinazolines as selective Aurora A and B kinase inhibitors. J Med Chem. 2006;49:955–970. doi: 10.1021/jm050786h. [DOI] [PubMed] [Google Scholar]
- 25.Andersen CB, Wan Y, Chang JW, et al. Discovery of selective aminothiazole aurora kinase inhibitors. ACS Chem Biol. 2008;3:180–192. doi: 10.1021/cb700200w. [DOI] [PubMed] [Google Scholar]
- 26.Niiya F, Xie X, Lee KS, Inoue H, Miki T. Inhibition of cyclin-dependent kinase 1 induces cytokinesis without chromosome segregation in an ECT2 and MgcRacGAP-dependent manner. J Biol Chem. 2005;280:36502–36509. doi: 10.1074/jbc.M508007200. [DOI] [PubMed] [Google Scholar]