

Abstract
Biotin carboxylase catalyzes the ATP-dependent carboxylation of biotin, and is one component of the multienzyme complex acetyl-CoA carboxylase that catalyzes the first committed step in fatty acid synthesis in all organisms. Biotin carboxylase from Escherichia coli, whose crystal structures with and without ATP bound have been determined, has served as a model system for this component of the acetyl-CoA carboxylase complex. The two crystal structures revealed a large conformational change of one domain relative to the other domains when ATP is bound. Unfortunately, the crystal structure with ATP bound was obtained with an inactive site-directed mutant of the enzyme. As a consequence the structure with ATP bound lacked key structural information such as for the Mg2+ ions and contained altered conformations of key active site residues. Therefore, nanosecond molecular dynamics studies of the wild-type biotin carboxylase were undertaken to supplant and amend the results of the crystal structures. Specifically, the protein-metal interactionsof the two catalytically critical Mg2+ ions bound in the active site are presented along with a reevaluation of the conformations of active site residues bound to ATP. In addition, the regions of the polypeptide chain that serve as hinges for the large conformational change were identified. The results of the hinge analysis complemented a covariance analysis that identified the individual structural elements of biotin carboxylase that change their conformation in response to ATP binding.
Acetyl-CoA carboxylase (ACC) catalyzes the first committed step in the biosynthesis of long chain fatty acids. ACC is a biotin dependent enzyme found in all bacteria, plants, and animals. Since fatty acids are used for membrane biogenesis and energy storage, ACC is a target for antibiotics (1,2), herbicides (3) and anti-obesity agents (4-6).
ACC produces malonyl-CoA from acetyl-CoA, ATP and bicarbonate, which serves as the source of CO2 for all biotin-dependent carboxylases (7). The reaction mechanism proceeds via two half-reactions (Scheme 1), (1) the first half-reaction is catalyzed by biotin carboxylase (BC), and (2) the second half reaction is catalyzed by carboxyltransferase (CT). In vivo, the vitamin biotin is covalently attached to a protein called the biotin carboxyl carrier protein (designated as enzyme-biotin in Scheme 1). In mammals, these proteins comprise different domains in a single polypeptide chain (8). In contrast, in gram-negative and gram-positive bacteria biotin carboxylase, carboxyltransferase and the biotin carboxyl carrier protein are separate proteins (9). ACC from the gram-negative bacterium E. coli has been used as a model system for mechanistic investigations because the purified BC and CT components retain their activities, and utilize free biotin as a substrate thereby simplifying kinetic analysis (10). Consequently, biotin carboxylase from E. coli has been extensively studied by x-ray crystallography (11,12) and site-directed mutagenesis (13-16).
Scheme 1.

Structural and biochemical studies have provided valuable insights into the mechanism of acetyl-CoA carboxylases; however a number of questions remain to be resolved. Most significant is that the crystal complexed with the cofactor ATP was obtained using an inactive mutant (E288K) of BC (12) because biotin carboxylase has a low ATPase activity in the absence of biotin (17). The activity of biotin carboxylase requires two equivalents of Mg2+ ion. The crystal structure of the BC mutant with ATP does not contain any Mg2+ ions (12). This has severely limited the ability to rationalize the detailed catalytic mechanism based on the structural information. Furthermore, the biochemical studies of biotin carboxylase have primarily focused on mutagenesis of active site residues (13-16), while it is increasingly evident that residues far from the active site are also important to catalysis (18). It is of great interest to carry out dynamics simulations of the wild-type enzymes along with active-site metal ions to provide structural information to aid the design and interpretation of new experiments (19-24). In this paper, we report on results from molecular dynamics simulations of residues in and surrounding the active site, and domain movements in order to understand their roles on the catalytic mechanism of wild-type biotin carboxylase.
Computational Details
Molecular Dynamics Calculations
All calculations were carried out using the CHARMM program (c32a2) (25,26) employing the CHARMM27 force-field (27) for the enzyme, ATP and magnesium ions. The coordinates for the ATP-enzyme complex (PDB code: 1DV2) were used to build the initial configuration. The active site mutant E288K was converted back to a glutamic acid residue by placing a carboxylate group on the Cγ carbon and removing the remaining atoms. All histidines in the crystal structure were analyzed and their protonation states were evaluated based on neighboring atoms. Stochastic boundary conditions were used throughout, dividing the system into three regions: the reaction zone, the buffer zone (Langevin zone) and the reservoir zone (28). The reaction zone consists of residues within a radius of 24 Å of the center (the β–γ-bridging oxygen (O3B) in ATP), and is treated using Newtonian dynamics. Residues within a 24-30 Å sphere (the buffer zone) act as a stochastic heat bath, which are treated by Langevin equations of motion by imposing a friction force and a random force on non-hydrogenic atoms in this region. The friction coefficients were set to 200 ps−1 for all protein atoms and 62 ps−1 for the water atoms. Furthermore, a harmonic restraining function was imposed on the buffer atoms to allow the system to maintain its structural integrity. At the outer edge of the buffer zone, the values of the harmonic force constants used were 1.22 kcal mol−1 Å−2 for main-chain oxygen atoms, 1.30 kcal mol−1 Å−2 for all other main-chain atoms, and 0.73 kcal mol−1 Å−2 for all atoms of side-chain and water molecules. These harmonic force constants are gradually scaled to zero at the reaction region boundary. The remaining atoms, in the reservoir region, were held fixed in the simulations.
The enzyme-ATP complex was solvated by a pre-equilibrated 30 Å sphere of water molecules. A spherical boundary potential was used to prevent water molecules from escaping into “vacuum” and to maintain the correct average distribution (29). Water molecules within 2.5 Å of any non-hydrogenic atom were removed. The solvent addition was repeated three more times while rotating the water sphere along a randomly chosen axis. Thereafter, a 100-step minimization (ABNR) of all water molecules was performed to remove close contacts (25). We placed two magnesium ions, replacing two nearby water molecules, to coordinate with the α, γ– and the β, γ–phosphate oxygens of ATP, respectively. The choice of the positions for the two magnesium ions was based on structures from other enzymes in the ATP-grasp family. Both in the structure of glutathione synthetase complexed with ATP (30), and in glycinamide ribonucleotide transformylase with AMPPNP (31), the two magnesium ions are coordinated to the α, γ– and β, γ–phosphate groups, respectively, along with two glutamic acid residues. Additionally, in the crystal structure of a heavy-atom derivative of BC, a Lu3+ ion is also coordinated with the two glutamatic acid residues E276 and E288 (11).
Having constructed the initial structure, we briefly optimized the Mg2+ positions with 50 steps of ABNR minimization, resulting in a configuration with the ions coordinated to the α, γ– and β, γ–phosphates of ATP, respectively. Then, we energy-minimized the Mg2+-ATP complex by another 50 steps, keeping the rest of the system fixed. This yielded a conformation in which E288 is bridged with both Mg2+ ions and E276 coordinates with one of the Mg2+ ions using both carboxylate oxygen atoms. We next performed yet another 50 steps of minimization for all atoms within a sphere of 5 Å from the center followed by further minimization of a sphere of 10 Å. All water molecules were thereafter relaxed in a 50-step minimization followed by a 10 ps MD simulation (T=50-100 K) to relax all unfavorable contacts. For all solvent water molecules, the three-point-charge model TIP3P (32) was used. The whole enzyme except ATP was then further minimized (50 steps ABNR). The addition of water molecules was repeated four times by randomly rotating this water sphere about a randomly chosen axis. The water molecules were minimized (50 steps ABNR) and then a 10 ps MD simulation was again performed. All atoms in the 30 Å sphere were thereafter minimized (50 steps ABNR). The system was slowly heated from 50-298 K in five cycles over totally 50 ps and equilibrated at 298 K for another 250 ps. Structural analysis was made on 11500 structures extracted from another 2.3 ns simulation. The SHAKE algorithm (33) was used on bonds to hydrogen atoms allowing us to use a time-step of 2 fs in the leapfrog integration scheme. Non-bonded interactions were truncated using a switch function between 12 and 13 Å and the dielectric constant was set to unity.
Covariance Analysis
The degree of correlated movements between different subunit in biotin carboxylase was analyzed by calculating the covariance matrix and correlation coefficients for the α-carbons within a sphere of 15 Å from the origin. A positive correlation coefficient indicates a correlated motion, while a negative correlation coefficient indicates an anti-correlated movement. A threshold of 0.25 for the correlation coefficient has been recommended (24) and is used in the present study to extract correlated movements in the analysis. Each configuration from the trajectory file was aligned to the α-carbons in a reference structure to remove the overall rotation. To test for convergence, plots of the covariance matrix were established both for the full MD-simulation and for the second half of the simulation.
Results and Discussion
(A) Coordinating Interactions of Active Site Metal Ions
The only previous structural information on biotin carboxylase with ATP bound is from the crystal structure of the E288K mutant enzyme (12). However, this structure does not contain the magnesium ions, essential for catalysis. The present molecular dynamics simulation is aimed at providing an analysis of the coordination spheres of the Mg2+ ions in the WT of biotin carboxylase. Figure 1 depicts a snapshot of the metal binding pocket in the dynamics trajectory, in which each of the two Mg2+-ions is seen to form an octahedral complex. Mg2 is bridged between the α- and γ–phosphoryl group of ATP and coordinated to E288, E276 and a water molecule (Table 1). E288 has a bidentate bridge to both Mg1 and Mg2 and its carboxyl group is positioned perpendicular to the plane of the E276 carboxyl group (Figure 1). Mg1 coordinates one β- and one γ-phosphoryl oxygen of ATP and to E288 via a bidentate coordination consisting of one weak and one strong coordination (Table 1 and Figure 1). The hexacoordination is completed by two water molecules. Because E288 plays a key role in bridging both metal ions, this structural organization shows that the E288K mutant in the crystal structure lacks the ability to bind the catalytically essential magnesium cations. In the mutant, the cationic Lys residue interacts with the pyrophosphate groups preventing binding to the magnesium ions (34). The coordination pattern shown in Figure 1 and the role of E288 that forms a metal ion bridge are consistent with crystal structures of other members of the ATP-grasp family, including the corresponding residues E841 in carbamoyl phosphate synthetase (35), E279 in PurT-encoded glycinamide ribonucleotide transformylase (36), and E144 in human glutathione synthetase (37).
Figure 1.
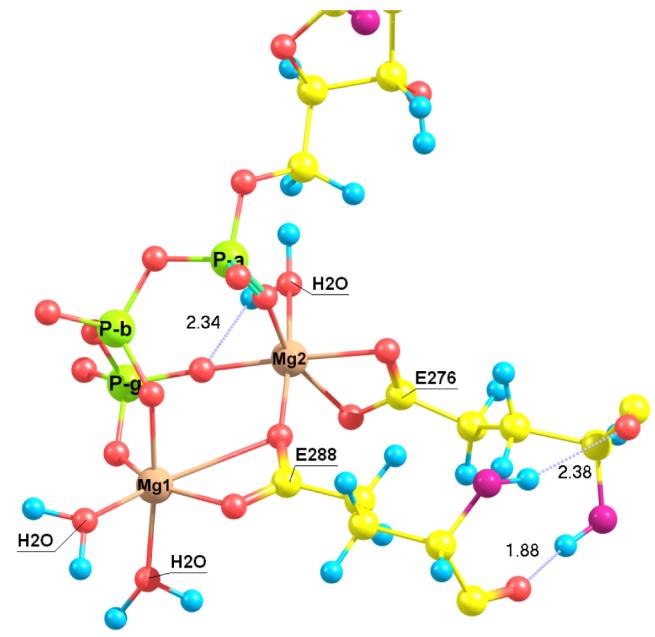
A snapshot of the structure of the magnesium ion binding pocket for the E. Coli wild-type biotin carboxylase from molecular dynamics simulations
Table 1.
Average magnesium-oxygen distances (in Å) in wild-type biotin carboxylase from molecular dynamics simulations.
| Magnesium | Oxygen | Distance |
|---|---|---|
| Mg1 | β (ATP) | 1.858 |
| γ (ATP) | 1.804 | |
| OE2 (E288) | 1.857 | |
| OE1 (E288) | 2.273 | |
| H2O | 1.955 | |
| H2O | 2.018 | |
| Mg2 | α (ATP) | 1.844 |
| γ (ATP) | 1.803 | |
| OE1 (E276) | 1.882 | |
| OE2 (E276) | 2.053 | |
| OE1 (E288) | 1.942 | |
| H2O | 2.067 |
(B) ATP Binding: Comparison of the Crystal Structure with Molecular Dynamics Simulations
In contrast to the magnesium atoms, the nucleotide binding site in BC was initially obtained directly from the E288K mutant structure (12). However, the MD simulations of the wild-type structure with ATP bound revealed several changes in comparison with the mutant crystal structure.
First, in the crystal structure of ATP with E288K mutant there are no hydrogen bonding interactions between K159 and the nucleotide base, whereas the present MD studies reveal that K159 is directly hydrogen-bonded to the N7 atom of the adenine base (Figure 2A and Table 2, entry b). Moreover, K159 also interacts with the α-phosphoryl group of ATP through a water molecule (entry c and d). The third hydrogen of the K159 side chain forms a salt-bridge with E201 (entry a), which, in turn, hydrogen bonds to the N6-amino group (entry f), either via a direct contact (not shown) or via a bridging water molecule (Figure 2A). Thus, K159 plays a key role in anchoring ATP, which is supported by the observation that the Km value for ATP increases by 90-fold in the mutation of K159 into Gln (14). Analyses of the MD trajectory also revealed that the origin of the K159 structural shift is associated with the conformational flexibility of an eleven-residue loop consisting of residues 159-169, called the T-loop after a similar loop in glycinamide ribonucleotide transformylase (36). The T-loop closes and covers the pyrophosphate groups upon binding (Figure 3) (12). The E288K mutation perturbs the metal binding and affects the T-loop closure, thereby the ability of K159 to form hydrogen bonds with the nucleotide base.
Figure 2.
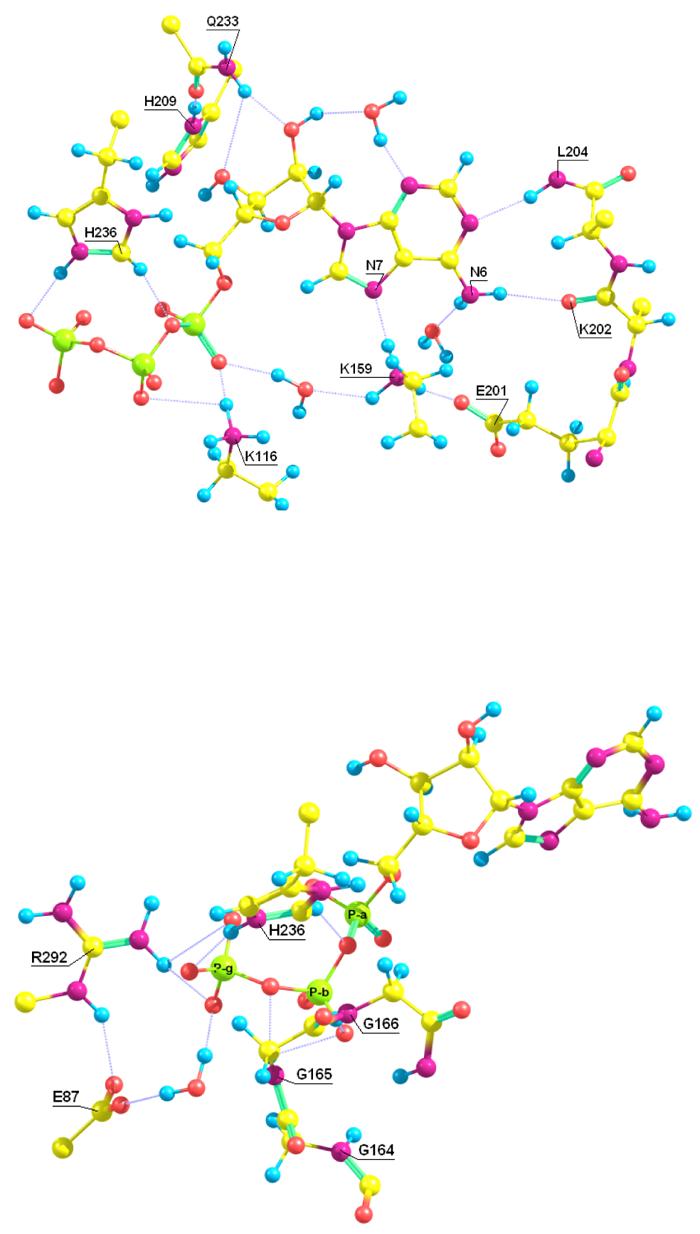
Figure 2A and Figure 2B. The hydrogen bond network to ATP in wild-type biotin carboxylase from MD simulations.
Table 2.
Selected average distances (in Å) in, or near, the ATP binding site from molecular dynamics simulations of wild-type of biotin carboxylase.
| Entry | Interacting residues | Distance |
|---|---|---|
| a | K159-E201 | 1.69 |
| b | K159-N7(ATP) | 1.96 |
| c | K159-H2O | 1.88 |
| d | O1A-H2O | 2.74 |
| e | Y199-E201 | 3.05 |
| f | E201-N6H2(ATP) | 2.60 |
| g | K202-N6H2(ATP) | 2.01 |
| h | L204-N1(ATP) | 2.13 |
| i | Q233-O-ribose(ATP) | 2.25 |
| j | H236-G165 | 3.78 |
| k | N290-E276 | 2.53 |
| l | R292-E276 | 2.88 |
| m | H209-E211 | 1.69 |
| n | G165-O1B(ATP) | 2.34 |
| o | G165-O3B(ATP) | 2.49 |
| p | G166-O1B(ATP) | 1.95 |
| q | G166-O3B(ATP) | 2.76 |
| r | G164-O1B(ATP) | 2.04 |
| s | K116 NH1-O1A(ATP) | 2.79 |
| t | K116 NH1-O2B(ATP) | 2.96 |
| u | K116 NH2-O1A(ATP) | 2.81 |
| v | K116 NH3-O1A(ATP) | 2.37 |
| w | K116 NH3-O2B(ATP) | 3.01 |
| x | H236-O2G(ATP) | 1.71 |
| y | H236-O3B(ATP) | 2.60 |
| z | R292-O1G(ATP) | 2.57 |
Figure 3.
The glycine rich T-loop (red) and its interactions with ATP (black).
The lack of two metal ions in the phosphate binding site also affects the interaction patterns of K116, which is hydrogen bonded only to the α-phosphoryl oxygen of ATP in the crystal structure of the mutant enzyme. In the molecular dynamics simulations, K116 is bridged between both the α– and β-phosphoryl groups of ATP (Figure 2A and Table 2). Mutation of K116 into Gln affects ATP binding, decreasing the enzyme affinity for ATP by 50-fold (14).
Second, although H236 interacts with the γ-and β-phosphate groups (Table 2, entry x and y) both in the crystal structure and throughout the dynamic trajectory, the imidazole ring of H236 rotates about 30° relative to the orientaion of the crystal structure. This makes the imidazole ring to face the carbonyl oxygen of the T-loop residue G165 (entry j in Table 2), as shown in Figure 2B. This spatial arrangement favors electrostatic interactions between the two residues, which are positioned as outposts in two loops. These interactions help stabilize the loops in position to embrace the phosphate portion of ATP, which may prevent hydrolysis of the unstable carboxyphosphate intermediate by bulk water. A similar loop arrangement has been observed in the crystal structure of carbamoyl phosphate synthetase (35) and in glutathione synthetase (38).
Third, a major structural change in the dynamics simulations is the formation of a hydrogen bond between R292 and the γ-phosphoryl oxygen of ATP. Initially, R292 is located about 5 Å away in the crystal structure, whereas it moves within 2.6 Å during the MD simulation (Figure 2B and Table 2, entry z). For comparison, the corresponding residue in carbamoyl phosphate synthetase (R845) is also within hydrogen bonding distance to the γ-phosphate of AMPPNP (35), suggesting that this residue plays a role to stabilize the negative charge of ATP. R292 is strictly conserved in biotin utilizing enzymes in the ATP-grasp family (39), however, the Km value for bicarbonate, biotin, and ATP only changes slightly in R292A mutants (13).
R292 also forms an ion pair with E87 and the latter residue is shifted along with R292 during the dynamics simulations, bringing them close to the γ-phosphoryl group through an intervening water bridge (Figure 2B). In addition, E87 forms a hydrogen bond with one of the water molecules coordinated to Mg1. A similar structure feature involving the corresponding residue E84 has been found in the PurT-encoded glycinamide ribonucleotide transformylase (36). Since BC and GTPases (40) both involve a nucleophilic attack at a γ-phosphate, it is worth mentioning that GTPases utilize an arginine finger to help a glutamine residue align the nucleophilic (water) for attack (41). Thus, the present simulations suggest an analogous network of interactions exists in biotin carboxylase, where R292 mediates E87 to bind bicarbonate at the Mg1 center. The residue E87 is expected to be a key catalytic residue since a similar glutamic acid residue has been identified at this position in several members of the ATP-grasp superfamily, including: glutathione synthetase (E94, ref. 30), D:Ala-D:Ala ligase (E68, ref. 42), PurT-encoded glycinamide ribonucleotide transformylase (E84, ref. 31), carboxyaminoimidazole ribonucleotide synthetase (E51, ref. 43), and pyruvate carboxylase (E65, ref. 44). Conformation of the significance of the E87 to the catalytic mechanism will have to await site-directed mutageneis studies.
Fourth, the conserved residue H209 interacts with the side chain amide oxygen of Q233 (Figure 2A). In turn, Q233 forms two bridged hydrogen bonds with the ribosyl hydroxyl groups (Table 2). The intervention of a Gln residue in these interactions is different from that found in other ATP-grasp enzymes, where the corresponding H209 residue is directly hydrogen bonded to the ribose hydroxyl groups. Thus, H209 adopts the role similar to H781 in carbamoyl phosphate synthetase (35) which interacts with the ribose hydrogen bonding residue E761, or H223 in PurT-encoded glycinamide ribonucleotide transformylase (31), which interacts with E203.
(C) Analyses of the Conformational Change
The defining structural feature of the ATP-grasp superfamily of enzymes is a significant quarternary conformational change accompanying nucleotide binding (12,30,35). In the case of biotin carboxylase, a rotation of ca. 45° of the B-domain relative to the A and C domains is observed after ATP is bound (12). Analyses of the protein dynamic fluctuations have been used to help identify structural segments that undergo the conformational changes associated with ATP binding in biotin carboxylase. In the present study, two approaches are utilized. First, statistical and informatic approaches were used to identify segments of the polypeptide chain that serve as hinges for domain movement. In the second approach, we examined regions that show correlated and anti-correlated movements using the covariance analysis method of Ichiye and Karplus (45).
The conformational change associated with ATP binding in biotin carboxylase is best described as a hinge motion (46) where the B-domain moves relative to the other domains of the protein (47). Comparison of the ATP-bound and the apo enzyme suggests that the hinge in biotin carboxylase is located in random loops of the polypeptide chain that link the B-domain with the A and C domains. The goal is to identify the specific residues that serve as the hinge. The program HingeMaster by Gerstein et al. (48) was used to search for hinges that move the B-domain relative to the rest of the enzyme. The procedure is to iteratively dissect the polypeptide chain into two fragments to analyze the intra-molecular potential of the two fragments. The joints that result in low energies are considered to be part of a hinge (a more detailed description of this program can be found in ref 48). The results of this analysis for BC are shown in Figure 4 where the energy is plotted against residue number. Three hinge pairs (i.e. the end points for each hinge) were identified; H1: E93-Q94 and M123-K124, H2: C130-V131 and Y203-L204, H3: K238-V239 and V328-H329.
Figure 4.
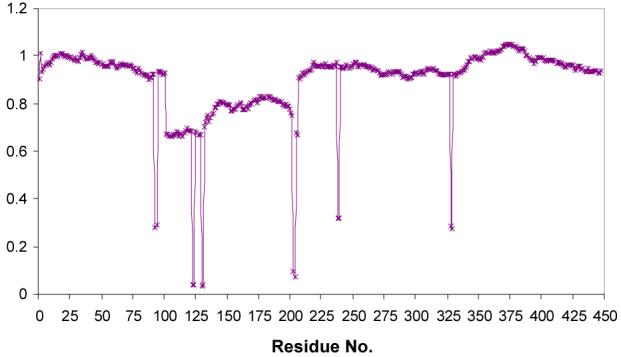
Histogram of potential hinges for domain motions in biotin carboxylase from the analysis using the program HingeMaster (48). A minimum in the plot represents an end point of a hinge-region. The y-axis represents a normalized score.
In the second analysis, we examine correlated motions in biotin carboxylase. Characteristically, if two structural elements move in the same direction the correlation is positive, whereas if the motion is towards each other the correlation is negative. The degree of correlated movement can be quantified where a positive correlation coefficient has a maximum value of 1 (same phase and period), while completely negative correlated movements have correlation coefficients with a maximum value of −1 (45). Figure 5 shows the correlation matrix between residue pairs of biotin carboxylase. Not surprisingly, many of the structural elements of biotin carboxylase showing correlated motion are also related to the hinges that allow the conformational change to occur. Thus, the subsequent discussion will integrate the results of both types of analyses.
Figure 5.
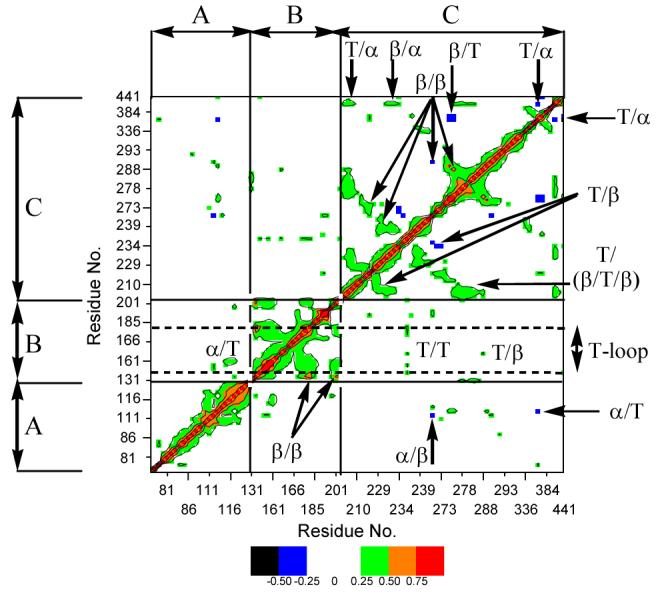
average covariant matrix of pair residues in E. Coli biotin carboxylase. The A-, B-, and C-domains are indicated along with α-helices, β-sheets and turns (T).
Examination of the B-domain closure in the crystal structures suggests that a hinge to allow for the closing motion of the B-domain lies in the two stretches of loops that led to and from the B-domain. The hinge pair H2 is located in this section of the protein (Figure 6). The hinge pair end point C130-V131 is in the stretch of polypeptide leading from the A-domain to the B-domain, while the other end point of the pair, Y203-L204, is in the section going from the B-domain to the C-domain. Since the side-chain of L204 makes a hydrophobic contact with the side-chain of V131 the two sides of the hinge are connected. Y203 makes an edge-face aromatic interaction with the adenosine ring in ATP (Figure 7), while the backbone amide hydrogen of L204 hydrogen bonds with N1 in ATP (Figure 2A and Table 2). Within the ATP-grasp family, these two positions are always occupied by two bulky hydrophobic residues (36).
Figure 6.
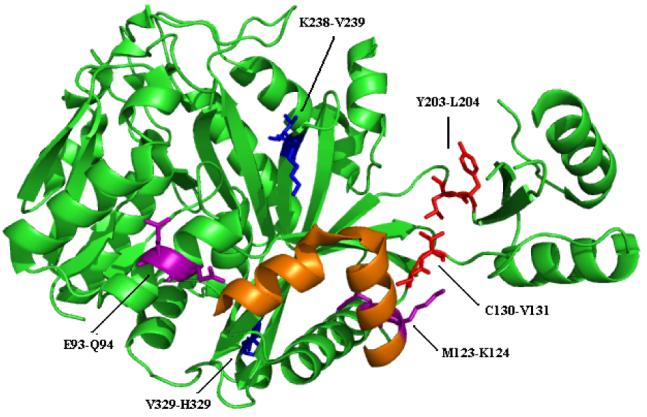
Ribbon drawing of the monomer of biotin carboxylase without any ligands showing the residues identified as hinges for the conformational change. Hinge pair H1: E93-Q94 and M123-K124 is colored purple; hinge pair H2: C130-V131 and Y203-L204 is red; and hinge pair H3: K238-V239 and V328-H329 is blue. Residues 107-126 that form helices oriented orthogonal to one another are colored in orange.
Figure 7.
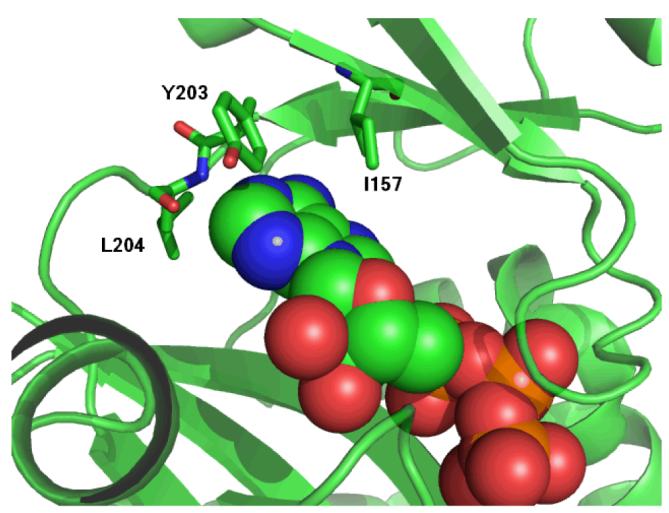
Interaction of I157, Y203 and L204 with the adenine portion of ATP.
The correlation analysis revealed that movement of several residues and structural elements of biotin carboxylase are coupled to hinge pair H2. For instance, the movement of I157 and E201 has a strong correlation with Y203. I157 is part (along with Y203 and L204) of the hydrophobic pocket in which the adenine portion of ATP is buried (Figure 7). Moreover, E201 is part of a β-strand consisting of Y199-Y203 that exhibits positive correlated movement with the T-loop-including K159 with which it forms a salt bridge (Figure 2A).
Positive correlated motions are observed between E201 and D115-V117, which are part of two α-helices encompassing residues 107-126. These two helices are oriented orthogonal to one another and bend towards the active site when ATP is present, which is manifested as positive correlated movements between the two helices. Movement of these two helices towards ATP is consistent with the observation that K116 interacts with the α- and β-phosphoryl groups of ATP (Figure 2A and Table 2). Moreover, K116 which lies at the bend between the helices, forms backbone hydrogen bonds with the T-loop residues S161 and G162. These interactions help keep the A- and B-domains of biotin carboxylase connected when ATP is bound. Similar interactions were also detected in an MD study of human glutathione synthetase (53). The hinge analysis indicates that hinge pair H1: E93-Q94 and M123-K124 (Figure 6) controls the movement of these two α-helices towards ATP. It is interesting to note that the thiol of C130 from hinge pair H2 is in close proximity to K124 in hinge pair H1 suggesting that the movements controlled by these two hinges are likely to be connected. In addition, the sulfur in M123 interacts with the phenyl rings of F275 (which rotates about 60° when ATP binds) and F286, which are part of the β-strands that ATP rest on, implying M123 aids in controlling the movement of the β-strands. Thus, the hinge and correlation analyses illustrate that ATP binding results in coordinated movement of the hinge residues and several different structural elements of biotin carboxylase.
A few other distinct patterns of positive correlated motion are found in biotin carboxylase (Figure 8). Most of the residues within the T-loop and a B-domain helix defined by residues 185-192 show strong positive correlated movement. In addition, the β-strands upon which ATP rests (especially residues E276 and E288) in the C-domain exhibit strong positive correlated motion. The α-helix encompassing residues 437 to 441 with the neighboring turn defined by residues 204 to 207 demonstrate strong positive correlated movement. The common feature of all of the positive correlated motions is the structural elements move in concert in the same direction towards the ATP binding site.
Figure 8.
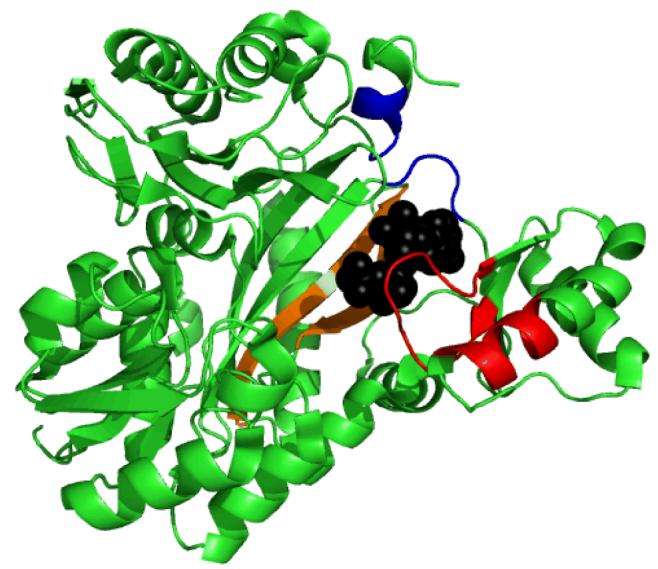
The T-loop and α helix defined by residues 185-192 are shown in red. ATP is represented in a black space-filling model and rests on β-strands (including residues E276 and E288) colored orange. The α-helix encompassing residues 437 to 441 and the neighboring turn defined by residues 204 to 207 are colored blue.
The common theme of stabilizing ATP binding is also evident in segments that exhibit negative correlated movements (i.e. structural elements that move towards each other instead of in the same direction). Strong negative correlations are only observed involving the A- and C-domains (Figure 9). For instance, the loop composed of residues D382 and S383 shows strong negative correlated motions with the 437-441 helix mentioned above and residues F275-F277, both of which are in the C-domain, as well as residues L112 and M113 from the A-domain (Figure 9). Moreover, even though P80 from the A-domain is 15 Å from E276 in the C-domain these residues exhibit a strong negative correlation (Figure 9). Notice that D382 and S383 and P80 are vertically aligned on one side of biotin carboxylase and move towards (i.e. negative correlated movement) the 437-441 helix, L112 and M113 and F275-F277 that are vertically aligned on the other side. Moderate negatively correlated movements are also observed between Y269 and residues L112 and M113 and N290. In addition, a stretch of residues including Y269 (Y269-G273), which define a turn and the beginning of a β-strand, exhibits negative correlated motion with H236. Thus, both the positive and negative correlated movements can all be summed up as structural elements of biotin carboxylase move either in the same direction or towards each other just so long as they move towards the bound ATP molecule.
Figure 9.
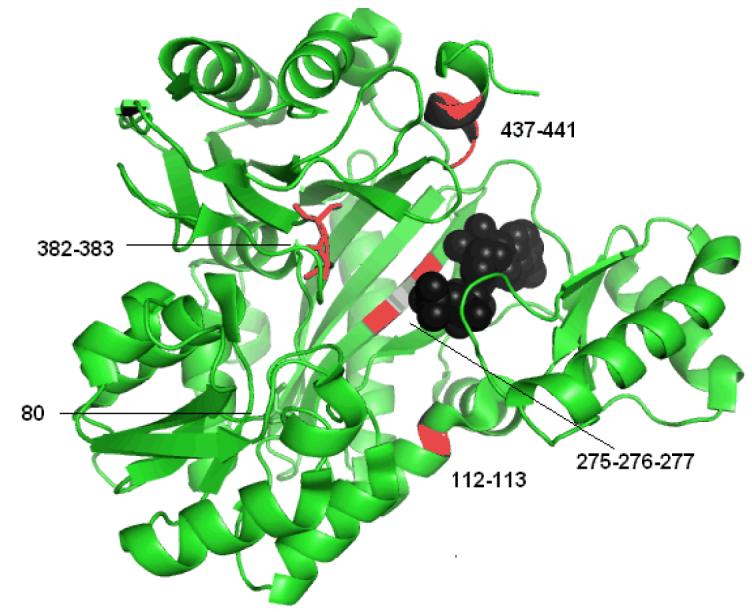
Structural elements that undergo negative correlated motions. D382 and S383 (orange) and P80 (cyan) are shown in stick format. The 437-441 helix, L112 and M113 and F275-F277 are colored in red, while E276 is in blue. ATP is represented in a black space-filling model.
The last hinge pair H3, K238-V239 and V328-H329, is located in the C-domain (Figure 6). Unlike the hinge pairs H1 and H2, it is not obvious what role the hinge H3 plays in controlling conformational changes. However, the observation that mutation of K238Q results in an inactive enzyme for biotin carboxylation along with an 85-fold increase in Km for ATP suggests that K238 (and by inference hinge pair H3) is essential for catalysis. In addition, the V328-H329 endpoint of hinge H3 also helps to explain the kinetics of heterodimers of biotin carboxylase. Heterodimeric forms of BC where one subunit is the wild-type enzyme and the other subunit is mutant with 100-fold reduced activity showed activity only slightly greater than the homodimer of the mutant (34). If the subunits acted independently the heterodimer would have about 50% of the wild-type activity, however, the mutation exhibited a dominant negative effect on the wild-type subunit. Mathematical modeling studies suggested the subunits alternate their catalytic cycles so that as one subunit is undergoing catalysis the other is releasing products (54). This type of model would require residues at the dimer interface to be coupled to catalysis. The identification that one half of the hinge pair H3, V328-H329, is involved in the dynamics of BC and is located at the interface between the two monomers in the homodimer of BC suggests it plays a role in subunit-subunit communication (Figure 10). H329 forms a hydrogen bond with R331, which has a face-to-face side-chain interaction with the corresponding R331 in the other monomer. Such Arg-Arg interactions have been shown to play a major role at protein interfaces (55, 56).
Figure 10.
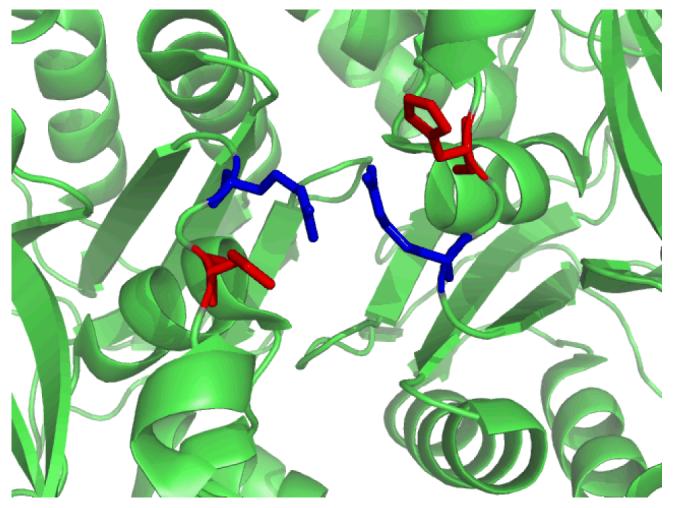
Hydrogen bonding interactions between H329 (red) and R331 (blue) across the biotin carboxylase dimer.
Conclusions
Since fatty acid synthesis is essential for membrane biogenesis in bacteria, biotin carboxylase is a target for antibiotics (1,2). Recent evidence suggested that consideration of protein flexibility is becoming increasingly important for drug design (57). Any tight binding inhibitor of BC will likely have to be trapped by the conformational change that occurs with ATP binding. Thus, a detailed understanding of the flexible segments of the enzyme involved in the conformational change will be essential for the design of ligands that bind tightly to biotin carboxylase. Moreover, identification of residues and structural elements of biotin carboxylase important for catalysis will help to minimize development of resistance to inhibitors serving as antibiotics.
In the present study, wild-type E. Coli biotin carboxylase has been reconstructed from the E288K mutant crystal structure. This allowed us to model the magnesium binding site and further analyze the active site interactions and correlated motions relevant to domain hinge movements. Each magnesium ion is coordinated with two phosphoryl groups of ATP, while E288 plays a key role by forming a bridge between the two metal ions. Mg1 is more exposed to the solvent and along with E87 and R292 may play a role in binding the bicarbonate substrate. Sequence and energy analyses identified three pairs of hinges, and covariant correlation analyses of the molecular dynamics trajectory indicate that they are associated with, respectively, the large B-domain motion relative to the A-C domain (C130-V131 and Y203-L204), two α-helices bending upwards toward the ATP binding site (E93-Q94 and M123-K124), and biotin carboxylase dimer interface communications (K238-V239 and V328-H329). These studies provide insight into active site interactions and dynamics of wild-type biotin carboxylase, which will provide the neccessary background for the design and interpretation of further experiments.
Supplementary Material
Acknowledgments
This work is partially supported by the National Institutes of Health grant number GM46376 (JG) and by the The Foundation BLANCEFLOR Boncompagni-Ludovisi, née Bildt for a fellowship (SONL).
Abbreviations
- ACC
acetyl-CoA carboxylase
- BC
biotin carboxylase
- CT
carboxyltransferase
- MD
molecular dynamics
- ABNR
Adopted Basis Newton-Raphson
- WT
wild-type
Footnotes
SONL gratefully acknowledges a post-doctoral stipend from The Foundation BLANCEFLOR Boncompagni-Ludovisi, née Bildt.
Supporting Information Available: Plots of selected interatomic distance along the trajectory. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Campbell JW, Cronan JE., Jr. Ann. Rev. Microbiol. 2001;55:305. doi: 10.1146/annurev.micro.55.1.305. [DOI] [PubMed] [Google Scholar]
- 2.Heath RJ, White SW, Rock CO. Prog. Lipid Res. 2001;40:467. doi: 10.1016/s0163-7827(01)00012-1. [DOI] [PubMed] [Google Scholar]
- 3.Delye C. Weed Science. 2005;53:728. [Google Scholar]
- 4.Waldrop GL, Stephens JM. Curr. Med. Chem.- Imun., Endoc. & Metab. Agents. 2003;3:229. [Google Scholar]
- 5.Harwood HJ. Expert Opinion on Therapeutic Targets. 2005;9:267. doi: 10.1517/14728222.9.2.267. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y, Burn P. Nature Reviews-Drug Discovery. 2004;3:695. doi: 10.1038/nrd1469. [DOI] [PubMed] [Google Scholar]
- 7.Knowles JR. Ann. Rev. Biochem. 1989;58:195. doi: 10.1146/annurev.bi.58.070189.001211. [DOI] [PubMed] [Google Scholar]
- 8.Tanabe T, Wada K, Okazaki T, Numa S. Eur. J. Biochem. 1975;57:15. doi: 10.1111/j.1432-1033.1975.tb02272.x. [DOI] [PubMed] [Google Scholar]
- 9.Cronan JE, Waldrop GL. Prog. Lipid Res. 2002;41:407. doi: 10.1016/s0163-7827(02)00007-3. [DOI] [PubMed] [Google Scholar]
- 10.Guchhait RB, Polakis SE, Dimroth P, Stoll E, Moss J, Lane MD. J. Biol. Chem. 1974;249:6633. [PubMed] [Google Scholar]
- 11.Waldrop GL, Rayment I, Holden HM. Biochemistry. 1994;33:10249. doi: 10.1021/bi00200a004. [DOI] [PubMed] [Google Scholar]
- 12.Thoden JB, Blanchard CZ, Holden HM, Waldrop GL. J. Biol. Chem. 2000;275:16183. doi: 10.1074/jbc.275.21.16183. [DOI] [PubMed] [Google Scholar]
- 13.Blanchard CZ, Lee YM, Frantom PA, Waldrop GL. Biochemistry. 1999;38:3393. doi: 10.1021/bi982660a. [DOI] [PubMed] [Google Scholar]
- 14.Sloane V, Blanchard CZ, Guillot F, Waldrop GL. J. Biol. Chem. 2001;276:24991. doi: 10.1074/jbc.M101472200. [DOI] [PubMed] [Google Scholar]
- 15.Levert KL, Lloyd RB, Waldrop GL. Biochemistry. 2000;39:4122. doi: 10.1021/bi992662a. [DOI] [PubMed] [Google Scholar]
- 16.Sloane V, Waldrop GL. J. Biol. Chem. 2004;279:15772. doi: 10.1074/jbc.M311982200. [DOI] [PubMed] [Google Scholar]
- 17.Climent I, Rubio V. Arch. Biochem. Biophys. 1986;251:465. doi: 10.1016/0003-9861(86)90353-x. [DOI] [PubMed] [Google Scholar]
- 18.Rajagopalan PTR, Lutz S, Benkovic SJ. Biochemistry. 2002;41:12618. doi: 10.1021/bi026369d. [DOI] [PubMed] [Google Scholar]
- 19.Antoniou D, Caratzoulas S, Kalyanaraman C, Mincer JS, Schwartz SD. Eur. J. Biochem. 2002;269:3103. doi: 10.1046/j.1432-1033.2002.03021.x. [DOI] [PubMed] [Google Scholar]
- 20.Gao J. Curr. Opin. Struct. Biol. 2003;13:184. doi: 10.1016/s0959-440x(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 21.Benkovic SJ, Hammes-Schiffer S. Science. 2003;301:1196. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 22.Radkiewicz JL, Brooks CL. J. Am. Chem. Soc. 2000;122:225. [Google Scholar]
- 23.Luo J, Bruice TC. Proc. Natl. Acad. Sci. USA. 2004;101:13152. doi: 10.1073/pnas.0405502101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schiøtt B. Intl. J. Quant. Chem. 2004;99:61. [Google Scholar]
- 25.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J. Comput Chem. 1983;4:187. [Google Scholar]
- 26.MacKerell AD, Jr., Brooks B, Brooks CL, III, Nilsson L, Roux B, Won Y, Karplus M. Encyclopedia of Computational Chemistry. Vol. 1. John Wiley & Sons; Chichester: 1998. p. 271. [Google Scholar]
- 27.Foloppe N, MacKerell AD., Jr. J. Comp. Chem . 2000;21:86. [Google Scholar]
- 28.Brooks CL, III, Karplus M. J. Mol. Biol. 1989;208:159. doi: 10.1016/0022-2836(89)90093-4. [DOI] [PubMed] [Google Scholar]
- 29.Brooks CL, III, Karplus M. J. Chem. Phys. 1983;79:6312. [Google Scholar]
- 30.Yamaguchi H, Kato H, Hata Y, Nishioka T, Kimura A, Oda J, Katsube Y. J Mol Biol. 1993;229:1083. doi: 10.1006/jmbi.1993.1106. [DOI] [PubMed] [Google Scholar]
- 31.Thoden JB, Firestine S, Nixon A, Benkovic SJ, Holden HM. Biochemistry. 2000;39:8791. doi: 10.1021/bi000926j. [DOI] [PubMed] [Google Scholar]
- 32.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 33.Ryckaert JP, Ciccotti G, Berendsen HJC. J. Comput. Phys. 1977;23:327. [Google Scholar]
- 34.Janiyani K, Bordelon T, Waldrop GL, Cronan JE., Jr. J. Biol. Chem. 2001;276:29864. doi: 10.1074/jbc.M104102200. [DOI] [PubMed] [Google Scholar]
- 35.Thoden JB, Wesenberg G, Raushel FM, Holden HM. Biochemistry. 1999;38:2347. doi: 10.1021/bi982517h. [DOI] [PubMed] [Google Scholar]
- 36.Thoden JB, Firestine SM, Benkovic SJ, Holden HM. J. Biol. Chem. 2002;277:23898. doi: 10.1074/jbc.M202251200. [DOI] [PubMed] [Google Scholar]
- 37.Dinescu A, Cundari TR, Bhansali VS, Luo JL, Anderson ME. J. Biol. Chem. 2004;279:22412. doi: 10.1074/jbc.M401334200. [DOI] [PubMed] [Google Scholar]
- 38.Hara T, Kato H, Katsube Y, Oda J. Biochemistry. 1996;35:11967. doi: 10.1021/bi9605245. [DOI] [PubMed] [Google Scholar]
- 39.Galperin MY, Koonin EV. Protein Sci. 1997;6:2639. doi: 10.1002/pro.5560061218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pasqualato P, Cherfils J. Structure. 2005;13:533. doi: 10.1016/j.str.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 41.Scheffzek K, Ahmadian MR, Wittinghofer A. Trends Biochem. Sci. 1998;23:257. doi: 10.1016/s0968-0004(98)01224-9. [DOI] [PubMed] [Google Scholar]
- 42.Fan C, Moews PC, Walsh CT, Knox JR. Science. 1994;266:439. doi: 10.1126/science.7939684. [DOI] [PubMed] [Google Scholar]
- 43.Thoden JB, Kappock TJ, Stubbe J, Holden HM. Biochemistry. 1999;38:15480. doi: 10.1021/bi991618s. [DOI] [PubMed] [Google Scholar]
- 44.Kondo S, Nakajima Y, Sugio S, Yong-Biao J, Sueda S, Kondo H. Acta Crystallogr., Sect. D. 2004;60:486. doi: 10.1107/S0907444904000423. [DOI] [PubMed] [Google Scholar]
- 45.Ichiye T, Karplus M. Proteins: Struct., Funct., and Gen. 1991;11:205. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 46.Joseph D, Petsko GA, Karplus M. Science. 1990;249:1425. doi: 10.1126/science.2402636. [DOI] [PubMed] [Google Scholar]
- 47.Gerstein M, Lesk AM, Chothia C. Biochemistry. 1994;33:6739. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 48.Flores S, Echols N, Milburn D, Hespenheide B, Keating K, Lu J, Wells S, Yu EZ, Thorpe M, Gerstein M. Nucleic Acids Res. 2006;34:D296. doi: 10.1093/nar/gkj046. The analysis has been performed using HingeMaster, a linear combination of FlexOracle, GNM, StoneHinge, and TLSMD as implemented in the web-based tool found at http://molmovdb.org. In short, identification of a hinge in FlexOracle is done by examination of the solvation energy after separating two domains thought to be joined by a hinge. If the solvation energy of the fragments is favorable, or at least not very unfavorable, then the domains are identified as being separated at a hinge. In addition, the cost to break van der Waals, Coulomb, or hydrogen-bonded interactions is small if the domains are separated at a hinge. The intra-molecular potential energies of the two fragments are evaluated using CHARMM (25). StoneHinge uses the FIRST algorithm to locate hinges (49), GNM is based on a Gaussian Network Model (50, 51), and TLSMD partitions the enzyme structure in a set of translation/libration/screw rigid-body motion of large groups of atoms, for example domains (52). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rader AJ, Hespenheide BM, Kuhn LA, Thorpe MF. Proc. Natl. Acad. Sci. 2002;99:3540. doi: 10.1073/pnas.062492699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bahar I, Atilgan AR, Erman B. Folding and Design. 1997;2:173. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 51.Haliloglu T, Bahar I, Erman B. Phys. Rev. Lett. 1997;79:3090. [Google Scholar]
- 52.Painter J, Merritt EA. Acta Crystallogr., Sect. D. 2006:439. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 53.Dinescu A, Anderson ME, Cundari TR. Biochem. Biophys. Res. Comm. 2007;353:450. doi: 10.1016/j.bbrc.2006.12.050. [DOI] [PubMed] [Google Scholar]
- 54.de Queiroz MS, Waldrop GL. J. Theor. Biol. 2007;246:167. doi: 10.1016/j.jtbi.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 55.Brinda KV, Vishveshwara S. BMC Bioinformatics. 2005;6:296. doi: 10.1186/1471-2105-6-296. See also, http://www.biochem.ucl.ac.uk/bsm/sidechains/Arg/Arg/sindex.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mukherjee A, Bagchi B. Biochemistry. 2006;45:5129. doi: 10.1021/bi0522899. [DOI] [PubMed] [Google Scholar]
- 57.Teague SJ. Nature Reviews-Drug Discovery. 2003;2:527. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.