

Abstract
Antimicrobial peptides such as cathelicidins can modulate inflammation by interfering with TLR function. Small fragment hyaluronan (HA) is released following injury, and is an endogenous ligand for TLR4 as well as CD44. In this study, we examined the interactions of cathelicidin with HA. Cathelicidin inhibited HA-induced MIP-2 release from mouse bone marrow derived macrophages in a CD44 dependent manner but did not inhibit MALP2-induced MIP-2 release. This inhibitory activity was more potent than that of a peptide inhibitor of HA binding (Pep-1) and independent of Gi protein coupled or EGF-R signaling, both targets of cathelicidin inhibited HA-induced MIP-2 release. In assay of cell binding to HA, cathelicidins also significantly inhibited this process, suggesting that this antimicrobial peptide can interfere in other membrane binding events mediated by HA. The significance of this inhibition was demonstrated in a skin inflammation model induced by repeated application of 2,4-dinitrofluorobenzene. This induced an increase in HA at the site of application and was partially CD44 dependent. Camp-/- mice lacking cathelcidin demonstrated a large increase in ear swelling, cell infiltration, and MIP-2 expression compared with wild type mice. These results suggest that cathelicidin has anti-inflammatory activity in skin that may be mediated in part by inhibition of HA-mediated processes.
Antimicrobial peptides (AMPs)3 are the first line of host defense against pathogens (1, 2). Among the several dozen distinct AMPs known to exist in mammals, the cathelicidins have been extensively studied and shown to be critical to resisting infection in animal models. These AMPs are expressed in bone marrow derived and epithelial cells, and show antimicrobial action against bacteria, viruses, and fungi (3, 4). Low expression of cathelicidins results in an increased susceptibility to infections in mice and humans (5-7). Furthermore, the expression of cathelicidins is induced by inflammation or wounding, and their abnormal or excess expression can lead to disease (8-10). Progress in understanding the mechanism of action of AMPs in host defense has revealed that these peptides act by both inhibiting the growth of microbes and by alerting surrounding cells of the threat of infection. For example, cathelicidins have multifunctional effects on host cells, including the capacity to promote IL-8 release, leukocyte chemotaxis, and wound repair (8, 11, 12). Recent studies in human systems have shown that high expression of abnormally processed cathelicidin promotes rosacea (9) and that increased cathelicidin in the peptide form LL-37 can complex with self DNA to induce an inflammatory response that may be significant in the pathogenesis of psoriasis (13).
In atopic dermatitis (AD), unlike in psoriasis or rosacea, the expression of cathelicidin is less than expected at sites of inflammation. The decrease in expected expression is explained in part by the excess of Th2 cytokines in AD, such as IL-4 and IL-13, which down-regulate the expression of cathelicidin (7, 14, 15). A consequence of decreased cathelicidin and other AMPs in atopics is that this increases the susceptibility of the skin to infection. Cathelicidins also inhibit inflammatory cytokine release mediated by TLR4 in dendritic cells (DC), and are associated with the suppression of contact hypersensitivity (16). These observations suggest that low expression of cathelicidin may contribute to the disease manifestations of AD in two ways: their decreased expression leads to an increase in microbial susceptibility and an increase in inflammatory mediators released by DC.
The trigger for inflammation in AD is unclear but is known to follow the symptom of itch, and thought to result from trauma due to the scratch response. Small fragment hyaluronan (HA) was recently found to be released from skin following minor trauma such as would occur from scratching, and is an endogenous ligand for TLR4 as well as CD44 (17, 18). Small fragment HA is generated during inflammation or injury (19-21), and has the opposite function to high m.w. HA, which normally exists abundantly in the skin to form an essential part of the extra-cellular matrix and prevents cell activation by coating the cell surface (19). CD44, which is the most common receptor of HA, is involved in HA-induced cytokine release by forming a TLR4-CD44 complex (22).
Because both cathelicidin and HA modulate TLR4 function, in this study we sought to clarify the involvement of these molecules in development of AD using a chronic dermatitis model in mice induced by repeated hapten application, which produces AD-like skin inflammation with Th2 responses (23). We show that cathelicidin inhibits HA function by blocking the HA-TLR4-CD44 interaction, and show that the presence of cathelicidin influences the development of dermatitis in this model. These observations provide new insight into the mechanisms responsible for the development of skin inflammation in AD.
Materials and Methods
Reagents and kits
LPS (Escherichia coli, 026:B6) were obtained from Sigma-Aldrich. Macrophage-activating lipopeptide of 2-kDa molecular mass (MALP-2) was obtained from Alexis Biochemicals. Murine macrophage-CSF (M-CSF) was obtained from R&D Systems. Pertussis toxin (PTX) and the epidermal growth factor receptor (EGFR) inhibitor AG1478 were obtained from Sigma-Aldrich and Calbiochem (EMD Biosciences), respectively. 2,4-dinitrofluorobenzene (DNFB) was obtained from Sigma-Aldrich. Mouse MIP-2 and human IL-8 ELISA kits were obtained from R&D Systems. Mouse IL-6 and Mouse IgE ELISA kits were obtained from BD Biosciences. The mouse TNF ELISA kit was obtained from eBioscience.
Hyaluronan
Hyaluronan was prepared by the method previously described (22). Human umbilical cord HA was purchased from Sigma-Aldrich and contains at wide range of sizes up to 500 kDa based on HPLC analysis. Small 1 ml HA batches were boiled for 1 h to inactivate any enzyme or protein contamination. Samples were then run on endotoxin-removal columns (Associates of Cape Cod) twice to remove any possibility of endotoxin contamination. HA preparations were free of DNA contamination, as preparations showed no peaks at 260 and 280 nm using a spectrometer.
Peptides
LL-37, mouse cathelicidin-related antimicrobial peptide (mCRAMP), KR-20, EK-20, and biotin-labeled-Pep1 were commercially synthesized and purified by HPLC to obtain >95% purity as previously described (11, 24).
Mice
Cathelicidin deficient male mice (Camp-/-) were generated in our laboratory as previously described (6) and backcrossed into the BALB/c background for seven generations. Cd44-/- (Cd44tm1Hbg) and Cd44+/+ control mice on a B6/129 background were purchased from The Jackson Laboratory. All animal experiments were approved by the Veterans Administration of San Diego Committee on Animal Use.
Cell culture and medium
Bone marrow-derived macrophages (BMDMs) were isolated from the tibia and femur of euthanized mice. Cells were seeded on a cell culture plate in RPMI 1640 medium (Invitrogen) supplemented with l-glutamine, 10% FCS, penicillin/streptomycin (100 U/ml and 50 μg/ml, respectively), 0.05 mM 2-ME, and 5 ng/ml M-CSF for 24 h. Nonadherent cells were collected and plated on 96-well plates at a concentration of 1 × 105 cells/well in the above medium. The medium was changed every other day. Experiments on BMDMs were performed on day 7. The human monocyte cell line, THP-1, was purchased from American Type Culture Collection (TIB-202). Cells were grown in RPMI 1640 medium supplemented with l-glutamine, 10% FCS, penicillin/streptomycin (100 U/ml and 50 μg/ml, respectively), and 0.05 mM 2-ME. Normal human epidermal keratinocytes (NHEK; Cascade Biologics) were grown in EpiLife medium (Cascade Biologics) supplemented with 0.06 mM CaCl2, 1% EpiLife defined growth supplement, and penicillin/streptomycin (100 U/ml and 50 μg/ml, respectively). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
Chronic dermatitis model
Chronic dermatitis was induced by the method previously described (23). Age-matched mice were painted with DNFB or vehicle (acetone/olive oil = 3/1) once weekly for 5 wk. Twenty-five microliters of 0.15% DNFB in vehicle or vehicle were painted on each side of both ears. Ear thickness was measured using a micrometer (Mitutoyo) before and 24 h after painting. Serum and tissue samples were obtained 24 h after the fifth painting. Mouse ears were fixed with 10% neutral formalin (Sigma-Aldrich), and embedded in paraffin by standard procedures. Sections were stained with H&E. Number of infiltrating polymorphonuclear leukocytes (PMNs) were counted and averaged in three high-power fields (magnification, ×400).
Measurement of cyokine release from BMDMs
BMDMs were washed once with culture medium without M-CSF before the assay, and stimulated with HA (5 μg/ml) or MALP-2 (100 ng/ml) for 24 h in culture medium without M-CSF at 37°C. For the inhibition assay, cells were pretreated with peptides, Ab or inhibitor for 1 h at 37°C, and then stimulated. After incubation, culture supernatants were centrifuged to obtain a cell-free supernatant. Cytokines in culture supernatants were measured by ELISA according to the manufacturer’s protocols.
Measurement of mRNA expression in tissues
The expression of mRNA was analyzed by real-time PCR using TaqMan Gene Expression Assays (Applied Biosystems). Total RNA was extracted by TRIzol reagent (Invitrogen) according to the manufacturer’s protocols. Tissue samples of ear or cervical lymph node were placed in a tube with 1 ml of TRIzol and 2 mm Zirconia beads (BioSpec Products) and were beaten twice at full speed in a minibead beater apparatus (BioSpec Products) for 1 min. The cDNA was synthesized from 1 μg of extracted RNA by the iScript cDNA Synthesis Kit (Bio-Rad Laboratories) according to the manufacturer’s protocols. GAPDH gene was used to normalize for each sample.
Measurement of MIP-2 in the ear
A mouse ear was placed in a tube with 1 ml of RIPA buffer (50 mM HEPES, 150 mM NaCl, 0.1% SDS, 0.25% deoxycholic acid, and 0.5% Nonidet P-40 (pH 7.4)) containing protease inhibitors (Roche Applied Science), and then beaten with 2 mm Zirconia beads in a minibead beater apparatus as described above, for 45 s on full speed. Beating was performed 5 times with a 5 min interval on ice between each beating to avoid heating. Tubes were centrifuged for 15 min, 12,000 × g at 4°C. The supernatant was transferred to a new tube and used as the ear extract in later experiments. MIP-2 in the ear extract was measured by ELISA as described above. Protein concentration of the ear extract was measured using a BCA protein assay kit (Bio-Rad Laboratories). The values from ELISA were corrected for protein concentration in each sample.
HA staining
HA staining was performed by previously described methods (24). Mouse ears were embedded in OCT compound (Sakura Finetek), frozen at -80°C and sectioned (9 μm thick). Biotin-labeled Pep-1 (25 μg/ml) containing 1% BSA was incubated on slides overnight at 37°C. In some experiments, slides were pretreated with Streptomyces hyaluronidase (Sigma-Aldrich) at 10 U/ml in 100 mM sodium acetate buffer (pH 6.0) for 5 h at 37°C. The slides were developed using 1 h incubation with FITC-conjugated streptavidin (Jackson ImmunoResearch Laboratories) diluted 1/100 in PBS containing 1% BSA and mounted with ProLong Gold with DAPI (Invitrogen). Skin sections were evaluated under an Olympus BX51 fluorescence microscope equipped with an Olympus DP71 digital camera system and DP manager software (Olympus Corporation).
Measurement of HA in the ear
HA in the ear was measured by dot blotting using a biotin-labeled hyaluronic acid binding protein (Associates of Cape Cod) as described previously (22). The ear extract described above was dotted onto a nitrocellulose membrane using dot blot apparatus. The ear extract (2.5 μl) was diluted in 200 μl of TBS; 200 μl of this was placed in one well of the dot blot apparatus and the rest was diluted either five or twenty-five times. The HA described above was used to make a standard curve. The membrane was incubated with HRP-conjugated streptavidin (R&D Systems) and developed with Western Lightning Chemiluminescence (PerkinElmer). The intensity of dot blots was analyzed using Image-J (National Institutes of Health). The values of HA concentration were corrected for protein concentration in each sample.
Measurement of IL-8 release from NHEK
IL-8 release from NHEKs was performed by previously described methods (11). NHEKs were cultured to 70-80% confluence and treated with or without 25 μg/ml HA for 1 h, followed by stimulation with 3 or 10 μM LL-37 for 24 h. The culture supernatants were centrifuged to obtain a cell-free supernatant. IL-8 in culture supernatant was measured by ELISA according to the manufacturer’s protocols.
HA-mediated cell adhesion assay
An HA-mediated cell adhesion assay was performed by previously described methods with some modification (25, 26). HA solutions (10 or 100 μg/ml, 100 μl/well, 0.1 M sodium carbonate (pH 9.5)) were added to 96-well EIA plates (Corning Life Sciences) and incubated overnight at 4°C. The HA-coated wells were washed three times with PBS and blocked with 100 μl of DMEM (Lonza) containing 1% FCS for 1 h at 37°C. THP-1 cells were stimulated with 1 μg/ml LPS (Sigma-Aldrich) overnight, washed three times with DMEM containing 10% FCS, and resuspended in DMEM containing 1% FCS at a concentration of 1 × 106 cells/ml. Cells were incubated with 50 μg/ml BSA or 10 μM LL-37 for 1 h, then 100 μl cell suspension was plated on HA-coated wells, and incubated for 2 h. In some experiments, HA-coated wells were preincubated with 50 μg/ml BSA or 10 μM LL-37 in DMEM containing 1% FCS for 1 h to adjust the final concentration. Alternatively, the cells and HA-coated plates were incubated with 100 μM or 300 μM Pep-1 as described above. After incubation, plates were washed four to six times with warm DMEM containing 1% FCS by pipetting to remove nonadherent cells. The adherent cells were fixed with 10% formalin, stained with 0.1% crystal violet, and eluted with 0.2% Triton X-100. The absorbance at 540 nm in the eluent was measured by a plate reader. Data shown are the mean ± SD of the percentages of total input cells (#, p < 0.05; ##, p < 0.01; n = 3-5 per experiment).
Statistical analysis
A paired Student’s t test for statistical analyses and a value of p < 0.05 was considered significant.
Results
Cathelicidins inhibit HA-induced cytokine release from BMDMs
HA induces abundant MIP-2 release from BMDMs in a TLR4- and CD44-dependent manner (22). To determine whether the presence of cathelicidin peptides could modify HA-induced MIP-2 release from BMDMs, cells were stimulated with HA for 24 h, and MIP-2 levels in culture supernatants were measured. HA induced MIP-2 release from BMDMs in a dose-dependent and CD44-dependent manner (Fig. 1A). In the presence of cathelicidin peptides, this response was inhibited (Fig. 1B). This inhibition was seen with the mouse cathelicidin mCRAMP, which dose-dependently inhibited the HA-induced MIP-2 release from BMDMs (46, 65, and 91% inhibition at 1, 3, and 10 μM mCRAMP, respectively), and the human cathelicidin LL-37, which inhibited HA-induced MIP-2 release at lower concentrations compared with mCRAMP (96 and 98% inhibition at 3 and 10 μM LL-37, respectively). Incubation of BMDMs with mCRAMP or LL-37 alone at 10 μM did not affect MIP-2 release. Furthermore, alternatively processed peptide forms of human cathelicidin, EK-20 and KR-20, which have similar or increased antibacterial potency compared with LL-37 (11), also inhibited HA-induced MIP-2 release (43 and 38% inhibition at 10 μM, respectively), but the inhibitory activity of these LL-37-derived peptides was less than LL-37 (Fig. 1B). Pep-1, a specific inhibitor of HA (26), also dose-dependently inhibited HA-induced MIP-2 release from BMDMs, but the inhibitory activity of Pep-1 (58% inhibition at 300 μM) was at least 100-fold weaker than that of mCRAMP (76% inhibition at 3 μM) (Fig. 1C).
FIGURE 1.

Cathelicidins inhibit HA-induced cytokine release from BMDMs. A, BMDMs from either Cd44+/+ or Cd44-/- mice were stimulated with HA (**, p < 0.01 vs Cd44+/+ mice). B-F, BMDMs from BALB/c mice were stimulated with 5 μg/ml HA (B, C, D, and F) or 100 ng/ml MALP-2 (E). F, BMDMs were pretreated with 100 ng/ml PTX or 2 μM AG1478. Mean ± SD (**, p < 0.01 vs nonstimulant; #, p < 0.05; ##, p < 0.01 vs stimulant alone, n = 5). CR, mCRAMP; LL, LL-37; EK, EK-20; KR, KR-20; Pep, Pep-1.
HA induces the release of several cytokines from macrophages (22) in addition to the CXCL chemokine MIP-2. Analysis of HA induced IL-6 and TNF-α release from BMDMs showed that cathelicidin peptides also inhibited their release (Fig. 1D). Meanwhile, no cytotoxicity was observed in an MTT assay, performed by the incubation of BMDMs with mCRAMP or LL-37 alone for 24 h at 10 μM (data not shown).
To examine the selectivity of inhibition of MIP-2 release by cathelicidins, we tested the effect of cathelicidins against TLR2-induced MIP-2 release. Stimulation with the TLR2 ligand MALP-2 (100 ng/ml) induced similar MIP-2 release as HA stimulation of BMDMs. mCRAMP and LL-37 did not inhibit MALP-2-induced MIP-2 release at 10 μM (Fig. 1E). These results indicate that cathelicidins selectively inhibit HA-induced MIP-2 release.
Previous studies have shown that cell activation and signaling by cathelicidins were Gi protein or EGFR dependent (8, 11, 12). To examine whether these signaling systems contributed to the mechanism of inhibitory activity of cathelicidins against HA-induced cytokine release, BMDMs were preincubated with the Gi protein inhibitor, PTX, or the EGFR tyrosine kinase inhibitor AG1478 for 1 h, before adding mCRAMP. Interestingly, preincubation with PTX or AG1478 alone enhanced the HA-induced MIP-2 release from BMDMs. However, preincubation with PTX or AG1478 did not block the inhibitory activity of mCRAMP (Fig. 1F). These results suggest that the mechanism of inhibitory activity of cathelicidins against HA-induced MIP-2 release is independent of Gi protein or EGFR activation.
HA does not affect cathelicidin-induced IL-8 release in keratinocytes
Previous studies have shown that some cathelicidin peptides, including LL-37, can induce IL-8 release from NHEKs (11). Because cathelicidins are strongly positively charged peptides (27), while HA is slightly negative charged, we next sought to examine the possibility that cathelicidins directly bind to HA to inhibit HA function. As expected, LL-37 induced IL-8 release from NHEKs, but pretreatment of NHEKs with 25 μg/ml HA (5 times the amount required for BMDM stimulation) did not block LL-37-induced IL-8 release (Fig. 2). Furthermore, HA alone did not affect IL-8 release from NHEKs. These results suggest that the mechanism of inhibitory activity of cathelicidin against HA-induced cytokine release is not due to direct binding of cathelicidins to HA, and that the HA-induced cytokine release is cell specific.
FIGURE 2.
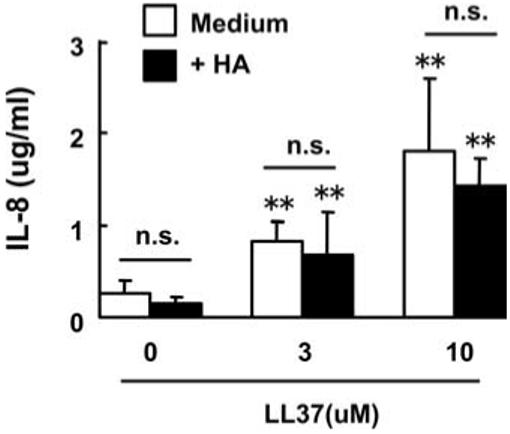
HA does not block cathelicidin-induced IL-8 release in keratinocytes. NHEKs were pretreated with 25 μg/ml HA for 1 h, followed by stimulation with 3 μM or 10 μM LL-37 for 24 h. IL-8 in culture supernatants was measured by ELISA. Mean ± S.D. (**, p < 0.01 vs non LL-37-stimulated cells, n = 4). n.s., not significant.
Cathelicidins inhibit HA-mediated cell adhesion of THP-1 cells
CD44 is best known as a receptor for HA (28), and has been shown in Fig. 1A and in prior reports (22) to be necessary for maximum cytokine release from HA-stimulated BMDMs. Hence, we next examined whether cathelicidins inhibit the CD44-HA interaction in the human monocyte THP-1 cell line, which expresses CD44 abundantly following LPS stimulation and binds to HA in a CD44-dependent manner (25). LPS-stimulated THP-1 cells were shown to adhere to a solid plastic matrix in an HA dependent manner, and LL-37 or mCRAMP significantly blocked this binding at a concentration of 10 μM (Fig. 3, A and B). Pep-1 also inhibited THP-1 cell binding at a concentration of 300 μM, but no inhibition was observed at 100 μM (Fig. 3B). To compare the potency of inhibition, both THP-1 cells and HA-coated wells were preincubated with cathelicidins or Pep-1. However, preincubation of HA-coated wells with cathelicidins was not necessary to demonstrate inhibitory activity and simultaneous addition yielded similar results (data not shown).
FIGURE 3.
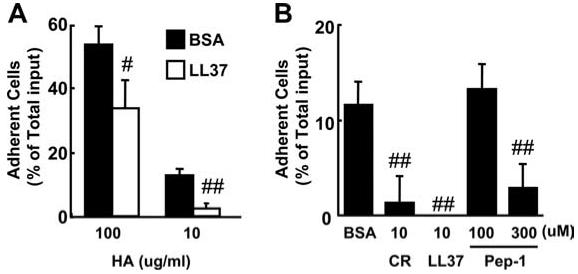
Cathelicidin inhibits HA-mediated cell adhesion of THP-1 cells. THP-1 cells were incubated with 50 μg/ml BSA (control) or peptides as indicated on HA-coated wells. A, 10 μM LL-37; B, 10 μg/ml HA. Mean ± SD of the percentages of total input cells (#, p < 0.05; ##, p < 0.01 vs BSA-treated cells, n = 3(A), n = 5(B)). CR, mCRAMP.
Cathelicidin deficiency increases chronic allergic inflammation in mice
Patients with AD have diminished induction of cathelicidin in lesional skin compared with expression in lesional skin of other inflammatory skin disorders (7, 15). Furthermore, Pep-1, a HA specific inhibitor, has been shown to inhibit the development of DNFB-induced chronic dermatitis in mice (26). In the above experiments, we observed that cathelicidins had more potent anti-inflammatory activity than Pep-1 in the inhibition of HA function. These results encouraged us to examine the involvement of a cathelicidin-HA system for the development of AD in an in vivo model using mice deficient in cathelicidin (Camp-/-).
The function of cathelicidin and HA was examined in a chronic dermatitis model induced by repeated DNFB application. This dermatitis model produces AD-like skin inflammation with high serum IgE levels in an IL-4 dependent manner (23). Camp-/- mice and wild-type (WT) mice were painted with either DNFB or vehicle on both ears once a week for a total of five applications over 29 days. The application of DNFB to mice induced a scratch response to the ear, and repeated tissue trauma. To confirm that this skin trauma from hapten exposure and scratching resulted in the expected increase in HA expression, HA abundance was evaluated by immunhistochemistry and by quantitative dot-blot analysis. HA expression increased in both the epidermis and dermis of DNFB treated mice compared with vehicle treated mice (Fig. 4A). A particularly strong HA signal was detected in the intercellular space of the dermis. In accordance with the histological observations, quantitative analysis of HA by dot blotting showed a significant increase of HA in the ear of DNFB treated mice as compared with vehicle treated mice. There were no differences observed between Camp-/-mice and WT mice in the DNFB or vehicle treated groups (Fig. 4B).
FIGURE 4.
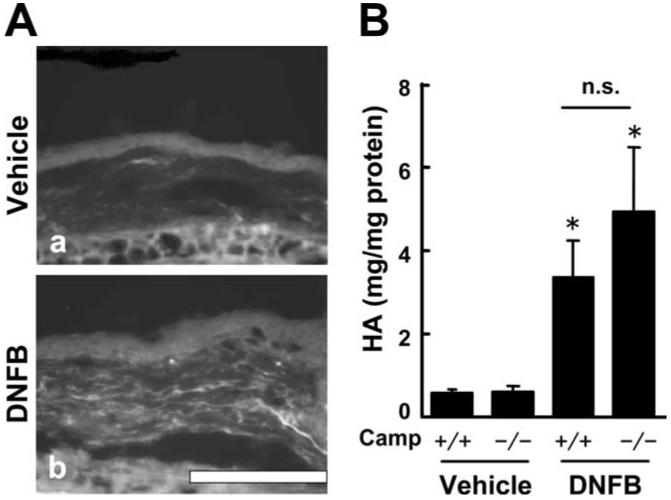
HA expression increases in chronic allergic dermatitis skin. A, The ear sections from BALB/c mice 24 h after the fifth application of vehicle (a) or DNFB (b) were stained with Pep-1 (white). Scale bar, 100 μm. B, HA concentration in the ear was measured by dot blotting. Mean ± SE (*, p < 0.05; **, p < 0.01 vs vehicle-treated mice, n = 6). n.s., not significant.
To quantify the inflammatory response of mice in this AD-like skin inflammation model, ear thickness was measured 24 h after each painting and cytokine responses quantified in local tissue and draining lymph nodes. Camp-/- mice treated with DNFB showed a significantly larger increase in ear thickness compared with WT mice (Fig. 5A). There was a 2.2-fold increase in ear thickness in Camp-/- mice 24 h after the fifth painting of DNFB when compared with ear thickness of WT mice. There was no significant increase in ear thickness and no significant difference in ear thickness between the two groups of mice painted with vehicle alone. Histological analysis revealed epidermal hypertrophy and an increase in PMNs in the ears of all DNFB-treated mice but this was more severe in Camp-/- mice compared with WT mice (Fig. 5B). The number of PMNs in DNFB-treated Camp-/- mice were 3.2-fold larger than in WT mice (Fig. 5C). The ratio of eosinophils to infiltrated PMN cells was 68% in both Camp-/- and WT mice. No apparent differences in cell numbers were observed in each vehicle treated mouse group. IL-4 expression in the ear and cervical lymph node (Fig. 5D) and serum IgE levels (Fig. 5E) were elevated in DNFB-treated mice. However, no differences were observed between Camp-/- mice and WT mice in the DNFB or vehicle treated groups. In contrast, MIP-2 expression in ears from DNFB treated Camp-/- mice was significantly higher than that from WT mice when measured by ELISA and real-time PCR (Fig. 5F).
FIGURE 5.
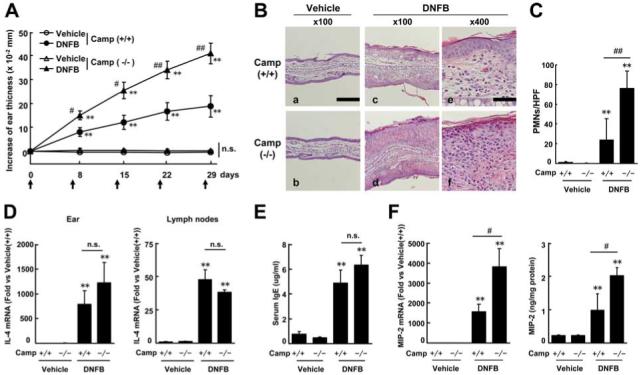
Cathelicidin deficiency increases chronic allergic inflammation in mice. A, Increase in ear thickness from day 0. Arrows indicate the day of application. B, H&E staining; Scale bars, 200 μm (A-D) and 50 μm (E and F). C, Number of infiltrating PMNs in the ear; HPFs, high-power fields. D, IL-4 expression. E, Serum IgE levels. F, MIP-2 expression in the ear. Mean ± SE (*, p < 0.05; **, p < 0.01 vs vehicle-treated mice; #, p < 0.05; ##, p < 0.01 vs Camp+/+ mice, n = 6-7). n.s., not significant.
To confirm the involvement of HA in this model, we next examined Cd44-/- mice because CD44 is a receptor that recognizes HA. Consistent with the previous reports of Th2 responses in B6/129 strains of mice (29, 30), we observed Th2 responses in Cd44+/+ mice (B6/129 mice), such as infiltration of eosinophils, elevation of serum IgE levels, and elevation of IL-4 in skin by the repeated DNFB application (data not shown). Cd44-/- mice painted with DNFB had a significantly smaller increase in ear thickness than WT mice (Fig. 6A). The increase of ear thickness in Cd44-/- mice was approximately half of that in WT mice 24 h after the fifth painting. Ear thickness was not significantly increased and was not significantly different in Cd44-/- and WT mice painted with vehicle alone. The number of infiltrated PMN cells in DNFB treated Cd44-/- mice was 46% of that in WT mice (Fig. 6B). The ratio of eosinophils to infiltrated PMNs cells was 37 and 49% in WT mice and Cd44-/- mice, respectively. No apparent differences were observed in each vehicle-treated mouse group. MIP-2 expression in ears from DNFB-treated Cd44-/- mice were significantly lower than that in WT mice (Fig. 6C).
FIGURE 6.
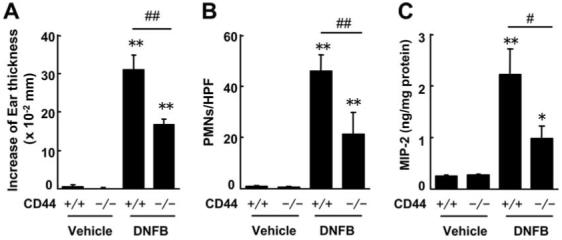
CD44 deficiency diminishes chronic allergic inflammation in mice. A, Increase in ear thickness 24 h after the fifth application (at 29 days). B, Number of infiltrated PMNs in the ear; HPFs, high-power fields. C, MIP-2 expression in the ear. Mean ± SE (*, p < 0.05; **, p < 0.01 vs vehicle-treated mice; #, p < 0.05; ##, p < 0.01 vs Cd44+/+ mice, n = 5-6). n.s., not significant.
Discussion
HA is a glycosaminoglycan polymer, exists abundantly in the skin as a major component of the extracellular matrix, and is thought to prevent cell activation by coating cell surfaces (19). Recent studies have shown that small fragment HA is generated by inflammation or injury, and that this induces cytokine release from macrophages (19, 20). These findings have led to the hypothesis that the release of HA fragments after physical or chemical trauma serves as an endogenous signal of inflammation in both the lung (17), and skin (22). In this study, we demonstrated that cathelicidins inhibit HA function and produce anti-inflammatory activity in vitro and in vivo. Cathelicidins inhibited HA-induced cytokine release in macrophages and HA-mediated cell binding, and cathelicidin deficiency exacerbated the development of chronic allergic dermatitis.
The functions of cathelicidins and other AMPs in the host immune response are complex and dependent on the cell type and assay systems used to examine their activity (11, 31). Cathelicidins inhibit LPS-TLR4 signaling and show anti-inflammatory activity in vitro and in vivo. Cathelicidins inhibit LPS-induced DC activation, and inhibit LPS-induced cytokine release from macrophages and DC in vitro (16, 31, 32). Cathelicidins also protect against LPS-induced sepsis (27), and cathelicidin deficiency exacerbates allergic contact dermatitis in vivo (16). The suggested mechanism of inhibitory activity of cathelicidins against LPS is the direct binding to LPS or the blockage of TLR4 action in the cell membrane (16, 27, 31, 33). In the current study, we show that cathelicidins block the function of HA, known as an endogenous CD44 and TLR4 ligand. Both mCRAMP and human LL-37 showed inhibitory activity against HA function. The inhibitory activity of cathelicidins against HA was selective and not due to nonspecific toxic effects because cathelicidins did not affect TLR2 ligand MALP2-induced cytokine release, and because no significant cytotoxic effect was observed by MTT assay. Furthermore, the inhibitory activity of cathelicidins was sequence specific and independent of antimicrobial activity because the inhibitory activity of the LL-37-derived peptides, EK-20 and KR-20, which have same antibacterial potency (11), was apparently weaker than that of original LL-37. The anti-LPS activity of cathelicidin is less likely to explain the observed observations because the presence of LPS is not dependent on the development of inflammation in vivo and the in vitro assays were done under conditions with undetectable levels of LPS in the HA preparations.
Previous studies have shown that some cellular activation events attributable to cathelicidins are Gi protein or EGFR dependent (8, 11). In our experiments, the inhibitory activity of mCRAMP against HA was independent of Gi protein or EGFR activation, because pretreatment with PTX (Gi coupled receptor inhibitor) or AG1478 (EGFR inhibitor) failed to block the inhibitory activity of mCRAMP. Furthermore, we investigated the possibility that cathelicidins bind directly to HA and neutralize HA function. Precedence for glycosaminoglycan binding to cathelicidin has been reported for heparin, which can bind and neutralize antibacterial activity of LL-37 (34). However, preincubation of LL-37 with an excess amount of HA did not affect LL-37-induced IL-8 release in keratinocytes. In addition, preincubation of LL-37 with HA-coated plates was not necessary for the inhibitory activity of LL-37 against HA-mediated THP-1 cell binding. These results suggest that even if cathelicidins and HA bind to each other, the impact of binding is negligible for their function. Furthermore, the inhibitory activity of Pep-1 supports this explanation. Pep-1 can bind selectively to HA and inhibit HA function (26), but the maximal inhibitory activity of Pep-1 against HA-induced MIP-2 release was ∼50% at 300 μM. Compared with Pep-1, cathelicidins completely block HA-induced MIP-2 release. This suggests that the mechanism of inhibitory activity of cathelicidins is different from that of Pep-1 (i.e., not direct binding to HA).
The mechanism through which cathelicidin blocks HA-dependent activation and skin inflammation is not clear but previous studies have shown that cathelicidins can alter cell membrane structure and fluidity in a manner that inhibits TLR signaling (11, 16). The same mechanism might be responsible for the effects of cathelicidins in BMDMs. Cathelicidins might block the formation of the TLR4-CD44 complex needed for HA-induced cytokine release (22). This mechanism may also explain the inhibition of HA-mediated THP-1 cell binding because, for HA-mediated cell binding to occur, oligomerization of CD44 on the cell membrane is required (35). Blockage of CD44 oligomerization inhibits binding of HA to CD44, and inhibits CD44-HA-induced uPA expression (36). Precedence for this exists as cathelicidins block the aggregation of ICAM-1 on cell membrane in DCs (16).
CD44-HA signaling has previously been shown to have several effects on inflammation, with the nature of this effect depending on the inflammatory process and the type of HA present. For example, in a lung injury model, CD44 deficiency exacerbates inflammation associated with a lack of clearance of HA (37) and administration of Pep-1 diminished inflammation (17). The size of HA is critical for this function. HA in the 200-500 kDa range induces inflammatory cytokines, but smaller HA fragments less than 100 kDa have been reported to show different effects on host cells (38-43). The HA prepared for the experiments described herein contained a wide range of sizes up to 500 kDa based on HPLC analysis, and has overall been reported to have proinflammatory activity (21, 22). Different responses to HA can also be mediated by cell or tissue specificity due to expression of different forms of glycosylated CD44 (44), or involvement of other receptors LYVE-1 (45) and TSG-6 (46, 47). Therefore, the HA response is complex and varied depending on HA size and cell specific receptor patterns. Further work is needed to clarify the mechanism of inhibition of HA by cathelicidin, but our findings clearly show that these peptides have potent capacity to block proinflammatory mediators released by BMDM in response to HA.
The utility of repeated DNFB application as a model of AD is supported by observations in WT mice that no increase in expression of cathelicidin mRNA was observed in ears by real-time PCR, or protein by immunohistochemical analysis, with the development of dermatitis. Increased expression of cathelicidin was only detected in infiltrated PMNs (data not shown). This is consistent with the diminished induction of cathelicidin in lesional skin of AD, and with data from a recent study that has shown that the expression of cathelicidin in skin is decreased with the development of dermatitis induced by repeated Oxazolone application (48). Furthermore, this model increases IL-4 and IgE levels, but no differences were observed between Camp-/- and WT mice in this response. Thus, our findings suggest that cathelicidin is not able to modulate Th1/Th2 balance in mice and does not exert its influence on inflammation through this mechanism.
There is much evidence to indicate that HA is involved in the development of allergic skin inflammation and thereby linking our observations in BMDM with the findings in Camp-/- mice. Administration of anti-CD44 Ab reduced ear swelling in mice with contact dermatitis (49). CD44 deficiency was associated with diminished skin inflammation and low leukocyte infiltration in an allergic dermatitis model (50). Furthermore, administration of Pep-1 inhibits inflammation in the chronic dermatitis model (26, 51). In this study, we demonstrated that cathelicidins had more potent inhibitory activity than Pep-1 against HA function. We also observed that CD44 deficiency reduced skin inflammation in the chronic allergic dermatitis model, but that the features of Cd44-/- mice with dermatitis were totally different to those of Camp-/- mice (e.g., cell infiltration or MIP-2 expression). We therefore hypothesize that the effects of cathelicidin to suppress HA effects in BMDMs may partly explain the exacerbation of tissue inflammation seen in Camp-/- mice after repeated hapten exposure. Supporting this hypothesis are observations that HA expression and fragmentation are induced by inflammation or injury (20) and our current observations that HA expression was increased in both the epidermis and dermis of DNFB-treated mice. Because scratching is an important factor in development of inflammation in hapteninduced chronic dermatitis models (52), as well as in human AD (53), the increase in HA may be due to destruction of the skin barrier and subsequent accelerated production of HA. Interestingly, in our experiments, no difference in HA expression was observed between Camp-/- and WT mice despite the large differences in ear swelling. This supports our hypothesis that the lack of cathelicidin fails to inhibit HA-induced inflammatory responses rather than suppressing the tissue injury itself.
In this research, no species differences were observed in the inhibition of HA function by cathelicidin, further work using human AD skin samples are needed to clarify the involvement of HA in the development of AD. Furthermore, additional experiments are needed to fully understand the influence of cathelicidin in inflammatory responses because cathelicidin can also exacerbate inflammation in skin in models of rosacea and psoriasis (9, 13). Several mechanisms have been proposed to be responsible for proinflammatory responses to cathelicidins including activation of FPRL-1 on monocytes (12, 54) and complexing with self-DNA in psoriasis to activate TLR9 that stimulate an IFN response in plasmacytoid DCs (13). The target cell type is essential to interpretation of these effects and the in vivo endpoint can guide interpretation. For example, in our experiments HA neither affected LL-37-induced IL-8 release, nor induced cytokine release from keratinocytes. This suggests that the functions of LL-37 and HA are different in different cell types and in different diseases. Supporting this, and in contrast to the proposed model of cathelicidin-dependent TLR9 activation of plasmacytoid DCs in psoriasis, murine cathelcidin does not appear to be necessary for IFN production in the skin as no difference in IFN-γ production was seen Camp-/- mice compared with WT mice after trauma (M. Gilliet, unpublished observations). It is possible that high expression of LL-37 in keratinocytes would exacerbate the development of psoriasis or rosacea, whereas low expression of cathelicidin in AD will exacerbate this inflammatory disorder. Thus, our findings demonstrate that in addition to increased susceptibility to infections, low expression of cathelicidin itself may be a risk factor of the development of dermatitis in AD.
Footnotes
This work was supported by National Institutes of Health Grants NIH R01AR052728, NIH R01 AI052453, and a Veterans Affairs Merit Award (to R.L.G.) and NIH AR41256 (to D.L.).
- AMP
- antimicrobial peptide
- AD
- atopic dermatitis
- WT
- wild type
- DC
- dendritic cell
- HA
- hyaluronan
- M-CSF
- macrophage-CSF
- PTX
- pertussis toxin
- EGFR
- epidermal growth factor receptor
- DNFB
- 2,4-dinitrofluorobenzene
- mCRAMP
- mouse cathelicidin-related antimicrobial peptide
- BMDM
- bone marrow derived macrophage
- NHEK
- normal human epidermal keratinocyte
- PMNs
- polymorphonuclear leukocyte
- MALP-2
- macrophage-activating lipopeptide of 2-kDa molecular mass
Disclosures
The authors have no financial conflict of interest.
References
- 1.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 2.Braff MH, Bardan A, Nizet V, Gallo RL. Cutaneous defense mechanisms by antimicrobial peptides. J. Invest. Dermatol. 2005;125:9–13. doi: 10.1111/j.0022-202X.2004.23587.x. [DOI] [PubMed] [Google Scholar]
- 3.Zanetti M. The role of cathelicidins in the innate host defenses of mammals. Curr. Issues Mol. Biol. 2005;7:179–196. [PubMed] [Google Scholar]
- 4.Howell MD, Jones JF, Kisich KO, Streib JE, Gallo RL, Leung DY. Selective killing of vaccinia virus by LL-37: implications for eczema vaccinatum. J. Immunol. 2004;172:1763–1767. doi: 10.4049/jimmunol.172.3.1763. [DOI] [PubMed] [Google Scholar]
- 5.Chromek M, Slamova Z, Bergman P, Kovacs L, Podracka L, Ehren I, Hokfelt T, Gudmundsson GH, Gallo RL, Agerberth B, Brauner A. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat. Med. 2006;12:636–641. doi: 10.1038/nm1407. [DOI] [PubMed] [Google Scholar]
- 6.Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414:454–457. doi: 10.1038/35106587. [DOI] [PubMed] [Google Scholar]
- 7.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 8.Carretero M, Escamez MJ, Garcia M, Duarte B, Holguin A, Retamosa L, Jorcano JL, Rio MD, Larcher F. In vitro and in vivo wound healing-promoting activities of human cathelicidin LL-37. J. Invest. Dermatol. 2008;128:223–236. doi: 10.1038/sj.jid.5701043. [DOI] [PubMed] [Google Scholar]
- 9.Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, Dorschner RA, Bonnart C, Descargues P, Hovnanian A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat. Med. 2007;13:975–980. doi: 10.1038/nm1616. [DOI] [PubMed] [Google Scholar]
- 10.Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J. Invest. Dermatol. 2001;117:91–97. doi: 10.1046/j.1523-1747.2001.01340.x. [DOI] [PubMed] [Google Scholar]
- 11.Braff MH, Hawkins MA, Di Nardo A, Lopez-Garcia B, Howell MD, Wong C, Lin K, Streib JE, Dorschner R, Leung DY, Gallo RL. Structure-function relationships among human cathelicidin peptides: dissociation of antimicrobial properties from host immunostimulatory activities. J. Immunol. 2005;174:4271–4278. doi: 10.4049/jimmunol.174.7.4271. [DOI] [PubMed] [Google Scholar]
- 12.Kurosaka K, Chen Q, Yarovinsky F, Oppenheim JJ, Yang D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J. Immunol. 2005;174:6257–6265. doi: 10.4049/jimmunol.174.10.6257. [DOI] [PubMed] [Google Scholar]
- 13.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 14.Howell MD, Gallo RL, Boguniewicz M, Jones JF, Wong C, Streib JE, Leung DY. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24:341–348. doi: 10.1016/j.immuni.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 15.Howell MD, Wollenberg A, Gallo RL, Flaig M, Streib JE, Wong C, Pavicic T, Boguniewicz M, Leung DY. Cathelicidin deficiency predisposes to eczema herpeticum. J. Allergy Clin. Immunol. 2006;117:836–841. doi: 10.1016/j.jaci.2005.12.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Nardo A, Braff MH, Taylor KR, Na C, Granstein RD, McInturff JE, Krutzik S, Modlin RL, Gallo RL. Cathelicidin antimicrobial peptides block dendritic cell TLR4 activation and allergic contact sensitization. J. Immunol. 2007;178:1829–1834. doi: 10.4049/jimmunol.178.3.1829. [DOI] [PubMed] [Google Scholar]
- 17.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 18.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stern R, Asari AA, Sugahara KN. Hyaluronan fragments: an information-rich system. Eur. J. Cell Biol. 2006;85:699–715. doi: 10.1016/j.ejcb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu. Rev. Cell Dev. Biol. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- 21.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 22.Taylor KR, Yamasaki K, Radek KA, Di Nardo A, Goodarzi H, Golenbock D, Beutler B, Gallo RL. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. J. Biol. Chem. 2007;282:18265–18275. doi: 10.1074/jbc.M606352200. [DOI] [PubMed] [Google Scholar]
- 23.Nagai H, Ueda Y, Ochi T, Hirano Y, Tanaka H, Inagaki N, Kawada K. Different role of IL-4 in the onset of hapten-induced contact hypersensitivity in BALB/c and C57BL/6 mice. Br. J. Pharmacol. 2000;129:299–306. doi: 10.1038/sj.bjp.0703054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zmolik JM, Mummert ME. Pep-1 as a novel probe for the in situ detection of hyaluronan. J. Histochem. Cytochem. 2005;53:745–751. doi: 10.1369/jhc.4A6491.2005. [DOI] [PubMed] [Google Scholar]
- 25.Gee K, Kozlowski M, Kumar A. Tumor necrosis factor-α induces functionally active hyaluronan-adhesive CD44 by activating sialidase through p38 mitogen-activated protein kinase in lipopolysaccharide-stimulated human monocytic cells. J. Biol. Chem. 2003;278:37275–37287. doi: 10.1074/jbc.M302309200. [DOI] [PubMed] [Google Scholar]
- 26.Mummert ME, Mohamadzadeh M, Mummert DI, Mizumoto N, Takashima A. Development of a peptide inhibitor of hyaluronan-mediated leukocyte trafficking. J. Exp. Med. 2000;192:769–779. doi: 10.1084/jem.192.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mookherjee N, Rehaume LM, Hancock RE. Cathelicidins and functional analogues as antisepsis molecules. Expert Opin. Ther. Targets. 2007;11:993–1004. doi: 10.1517/14728222.11.8.993. [DOI] [PubMed] [Google Scholar]
- 28.Miyake K, Underhill CB, Lesley J, Kincade PW. Hyaluronate can function as a cell adhesion molecule and CD44 participates in hyaluronate recognition. J. Exp. Med. 1990;172:69–75. doi: 10.1084/jem.172.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, et al. Prostaglandin D2 as a mediator of allergic asthma. Science. 2000;287:2013–2017. doi: 10.1126/science.287.5460.2013. [DOI] [PubMed] [Google Scholar]
- 30.Herrick CA, Xu L, McKenzie AN, Tigelaar RE, Bottomly K. IL-13 is necessary, not simply sufficient, for epicutaneously induced Th2 responses to soluble protein antigen. J. Immunol. 2003;170:2488–2495. doi: 10.4049/jimmunol.170.5.2488. [DOI] [PubMed] [Google Scholar]
- 31.Mookherjee N, Brown KL, Bowdish DM, Doria S, Falsafi R, Hokamp K, Roche FM, Mu R, Doho GH, Pistolic J, et al. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J. Immunol. 2006;176:2455–2464. doi: 10.4049/jimmunol.176.4.2455. [DOI] [PubMed] [Google Scholar]
- 32.Kandler K, Shaykhiev R, Kleemann P, Klescz F, Lohoff M, Vogelmeier C, Bals R. The anti-microbial peptide LL-37 inhibits the activation of dendritic cells by TLR ligands. Int. Immunol. 2006;18:1729–1736. doi: 10.1093/intimm/dxl107. [DOI] [PubMed] [Google Scholar]
- 33.Rosenfeld Y, Papo N, Shai Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides: peptide properties and plausible modes of action. J. Biol. Chem. 2006;281:1636–1643. doi: 10.1074/jbc.M504327200. [DOI] [PubMed] [Google Scholar]
- 34.Baranska-Rybak W, Sonesson A, Nowicki R, Schmidtchen A. Glycosaminoglycans inhibit the antibacterial activity of LL-37 in biological fluids. J. Antimicrob. Chemother. 2006;57:260–265. doi: 10.1093/jac/dki460. [DOI] [PubMed] [Google Scholar]
- 35.Liu D, Sy MS. Phorbol myristate acetate stimulates the dimerization of CD44 involving a cysteine in the transmembrane domain. J. Immunol. 1997;159:2702–2711. [PubMed] [Google Scholar]
- 36.Suzuki M, Kobayashi H, Fujie M, Nishida T, Takigawa M, Kanayama N, Terao T. Kunitz-type protease inhibitor bikunin disrupts phorbol ester-induced oligomerization of CD44 variant isoforms containing epitope v9 and subsequently suppresses expression of urokinase-type plasminogen activator in human chondrosarcoma cells. J. Biol. Chem. 2002;277:8022–8032. doi: 10.1074/jbc.M108545200. [DOI] [PubMed] [Google Scholar]
- 37.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science. 2002;296:155–158. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- 38.McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, Noble PW. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages: the role of HA size and CD44. J. Clin. Invest. 1996;98:2403–2413. doi: 10.1172/JCI119054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKee CM, Lowenstein CJ, Horton MR, Wu J, Bao C, Chin BY, Choi AMK, Noble PW. Hyaluronan fragments induce nitric-oxide synthase in murine macrophages through a nuclear factor κB-dependent mechanism. J. Biol. Chem. 1997;272:8013–8018. doi: 10.1074/jbc.272.12.8013. [DOI] [PubMed] [Google Scholar]
- 40.Horton MR, Shapiro S, Bao C, Lowenstein CJ, Noble PW. Induction and regulation of macrophage metalloelastase by hyaluronan fragments in mouse macrophages. J. Immunol. 1999;162:4171–4176. [PubMed] [Google Scholar]
- 41.Noble PW, Lake FR, Henson PM, Riches DW. Hyaluronate activation of CD44 induces insulin-like growth factor-1 expression by a tumor necrosis factor-α-dependent mechanism in murine macrophages. J. Clin. Invest. 1993;91:2368–2377. doi: 10.1172/JCI116469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodge-Dufour J, Noble PW, Horton MR, Bao C, Wysoka M, Burdick MD, Strieter RM, Trinchieri G, Pure E. Induction of IL-12 and chemokines by hyaluronan requires adhesion-dependent priming of resident but not elicited macrophages. J. Immunol. 1997;159:2492–2500. [PubMed] [Google Scholar]
- 43.Horton MR, McKee CM, Bao C, Liao F, Farber JM, Hodge-DuFour J, Pure E, Oliver BL, Wright TM, Noble PW. Hyaluronan fragments synergize with interferon-γ to induce the C-X-C chemokines mig and interferon-inducible protein-10 in mouse macrophages. J. Biol. Chem. 1998;273:35088–35094. doi: 10.1074/jbc.273.52.35088. [DOI] [PubMed] [Google Scholar]
- 44.Katoh S, Zheng Z, Oritani K, Shimozato T, Kincade PW. Glycosylation of CD44 negatively regulates its recognition of hyaluronan. J. Exp. Med. 1995;182:419–429. doi: 10.1084/jem.182.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banerji S, Ni J, Wang SX, Clasper S, Su J, Tammi R, Jones M, Jackson DG. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J. Cell Biol. 1999;144:789–801. doi: 10.1083/jcb.144.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blundell CD, Almond A, Mahoney DJ, DeAngelis PL, Campbell ID, Day AJ. Towards a structure for a TSG-6.hyaluronan complex by modeling and NMR spectroscopy: insights into other members of the link module superfamily. J. Biol. Chem. 2005;280:18189–18201. doi: 10.1074/jbc.M414343200. [DOI] [PubMed] [Google Scholar]
- 47.Blundell CD, Mahoney DJ, Almond A, DeAngelis PL, Kahmann JD, Teriete P, Pickford AR, Campbell ID, Day AJ. The link module from ovulation- and inflammation-associated protein TSG-6 changes conformation on hyaluronan binding. J. Biol. Chem. 2003;278:49261–49270. doi: 10.1074/jbc.M309623200. [DOI] [PubMed] [Google Scholar]
- 48.Man MQ, Hatano Y, Lee SH, Man M, Chang S, Feingold KR, Leung DY, Holleran W, Uchida Y, Elias PM. Characterization of a hapten-induced, murine model with multiple features of atopic dermatitis: structural, immunologic, and biochemical changes following single versus multiple oxazolone challenges. J. Invest. Dermatol. 2008;128:79–86. doi: 10.1038/sj.jid.5701011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Camp RL, Scheynius A, Johansson C, Pure E. CD44 is necessary for optimal contact allergic responses but is not required for normal leukocyte extravasation. J. Exp. Med. 1993;178:497–507. doi: 10.1084/jem.178.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonda A, Gal I, Szanto S, Sarraj B, Glant TT, Hunyadi J, Mikecz K. CD44, but not l-selectin, is critically involved in leucocyte migration into the skin in a murine model of allergic dermatitis. Exp. Dermatol. 2005;14:700–708. doi: 10.1111/j.0906-6705.2005.00348.x. [DOI] [PubMed] [Google Scholar]
- 51.Mummert DI, Takashima A, Ellinger L, Mummert ME. Involvement of hyaluronan in epidermal Langerhans cell maturation and migration in vivo. J. Dermatol. Sci. 2003;33:91–97. doi: 10.1016/s0923-1811(03)00160-9. [DOI] [PubMed] [Google Scholar]
- 52.Inagaki N, Shiraishi N, Igeta K, Itoh T, Chikumoto T, Nagao M, Kim JF, Nagai H. Inhibition of scratching behavior associated with allergic dermatitis in mice by tacrolimus, but not by dexamethasone. Eur. J. Pharmacol. 2006;546:189–196. doi: 10.1016/j.ejphar.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 53.Bender BG, Ballard R, Canono B, Murphy JR, Leung DY. Disease severity, scratching, and sleep quality in patients with atopic dermatitis. J. Am. Acad. Dermatol. 2008;58:415–420. doi: 10.1016/j.jaad.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 54.De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med. 2000;192:1069–1074. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]