

Abstract
Calcium/calmodulin-dependent protein kinase II (CaMKII) is a pivotal signaling molecule in both the brain and the heart. In this issue of Cell, Erickson et al. (2008) demonstrate a mechanism for CaMKII activation by reactive oxygen species that provides a direct link between kinase activation and cardiac dysfunction.
The regulation of calcium/calmodulin-dependent protein kinase II (CaMKII) is of intense interest to neuroscientists due to its role in neuronal plasticity. However, CaMKII also has important functions in other tissues such as the heart. In this issue of Cell, Erickson et al. (2008) add a new piece to the puzzle of CaMKII regulation with their demonstration that reactive oxygen species (ROS) involve activation of CaMKII and cardiac pathology. These findings may have important implications for our understanding of disease mechanisms in both the heart and nervous system.
In the brain, the coupling of cell excitation and Ca2+ influx to the calmodulin(Ca2+/CaM) triggered activation of CaMKII is a critical signaling mechanism. The cardinal feature of CaMKII, which is believed to underlie its role in neuronal plasticity, is its ability to remain active independent of Ca2+/CaM binding after its initial activation (for review, see Griffith, 2004). During CaMKII activation, binding of Ca2+/CaM to the regulatory domain of CaMKII disrupts the interaction between the autoinhibitory sequences and the N-terminal catalytic domain to allow for substrate phosphorylation. Activation also catalyzes autophosphorylation within the autoinhibitory domain at threonine 287 (T287 for β, γ, δ isozymes; T286 for αCaMKII). It is this threonine phosphorylation that gives CaMKII its ability to remain active even after dissociation of Ca2+/CaM, as the modification prevents the reassociation of the CaMKII autoinhibitory domain even in the absence of Ca2+/CaM. This property of the kinase allows it to remain active for a prolonged period of time after the Ca2+ concentration in the cytoplasm has returned to basal levels, thereby allowing neurons to retain a “memory” of the excitation. The CaM-binding domain of the kinase can also undergo autophosphorylation at T306 and T307. Phosphorylation at these sites blocks binding of CaM and provides a mechanism for inhibiting kinase activation (Lu et al., 2003).
Neurons are of course not the only excitable cells in the body—heart cells are also excitable. In the heart, CaMKII is known to be critically involved in coupling excitation and contraction of muscle through regulation of voltage-gated Na+, K+, and Ca2+ channels as well as a variety of Ca2+ handling proteins (Maier and Bers, 2007). Constitutively active CaMKII is elevated in a number of pathological conditions of the heart and has a pivotal role in mediating cardiac remodeling induced by hyperactivation of β-adrenergic receptors (Zhang et al., 2005). Intriguingly, CaMKII is also activated by the pro-oxidant cellular environment produced in many other forms of cardiac dysfunction. Shear stress, volume overload, and reperfusion after anoxia all induce the production of ROS through activation of NADPH oxidase. In addition, a diverse collection of agents, including angiotensin II, induce translocation of the NADPH oxidase subunit p47 to the cell membrane. This change in p47 localization results in the production of ROS and the activation of kinases and transcriptional responses that can lead to apoptosis or cardiac hypertrophy.
Erickson and colleagues set out to determine how ROS activate CaMKII by first examining the effects of a powerful ROS, hydrogen peroxide (H2O2), on purified δCaMKII. They found that in the presence of Ca2+/CaM, H2O2 treatment caused CaMKII to become constitutively active. Importantly, this activation was entirely independent of phosphorylation at T287, given that it occurred even when the threonine was mutated to alanine (T287A). Reasoning that this activation had to involve the well-characterized regulatory domain of CaMKII, the authors used protein chemistry to look for oxidative modifications of cysteine and methionine residues. They found that oxidation of either methionine 281 or methionine 282 (M281 or M282) could activate the kinase, whereas oxidation of cysteines or other methionines in this domain had no effect. These findings suggest that binding of Ca2+/CaM induces a conformational change that opens up the regulatory domain of the kinase and makes these methionines available for modification. The requirement for initial Ca2+/CaM binding is also a feature of T287 phosphorylation, suggesting that the regulatory domain is normally protected by intramolecular interactions in the inactive state (Figure 1).
Figure 1. Modifying the δCaMKII Regulatory Domain.
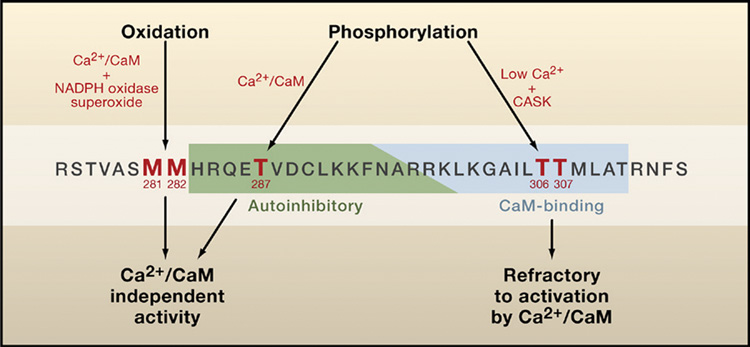
The CaMKII regulatory domain is defined as amino acids 282–311 (numbering for δCaMKII; see Hudmon and Schulman, 2002 for review), with an overlapping autoinhibitory domain (green) and a CaM-binding domain (blue). Binding of Ca2+/CaM activates CaMKII by disrupting interactions between the autoinhibitory domain and the catalytic substrate-binding site and ATP-binding site. Activation of CaMKII results in autophosphorylation of threonine 287 (T287), which keeps the enzyme active even if Ca2+/CaM dissociates. Phosphorylation within the CaM-binding domain at T306 or T307 can occur when CaM dissociates from CaMKII phosphorylated on T287 (Patton et al., 1990) or when unphosphorylated CaMKII is in a complex with CASK (at low Ca2+ concentrations), a MAGUK scaffolding protein (Lu et al., 2003). This phosphorylation blocks subsequent Ca2+/CaM binding and activation of CaMKII. Erickson et al. (2008) show that M281/M282 can also be modified by oxidation to generate Ca2+/CaM-independent activity, suggesting that our definition of the regulatory domain of CaMKII should be expanded to include these residues.
Having identified a new mode of residue modification in the regulatory domain, the authors then set out to explore the physiological role of methionine oxidation in CaMKII. Erickson et al. compared the effects of two agents that are known to be involved in cardiac tissue remodeling: the hormone angiotensin II, which increases the generation of ROS, and the β-adrenergic agonist isoproterenol, which increases intracellular Ca2+ and activates CaMKII. Using an oxidation-specific antibody, they showed that oxidation of M281/282 occurs in vivo in the hearts of angiotensin II-treated mice but not in the hearts of mice treated with isoproterenol. This suggested that the modification is dependent on the presence of ROS. Consistent with this, CaMKII oxidation was blocked by mutation of p47, the NADPH oxidase subunit responsible for increased ROS following angiotensin II treatment. Finally, mutation of MsrA, the enzyme that regenerates methionine from its oxidized form, enhanced oxidation of CaMKII. Though both angiotensin II and isoproterenol stimulate phosphorylation of δCaMKII at T287, angiotensin II-stimulated phosphorylation was blocked by mutation of p47. This suggested that ROS also have effects, possibly mediated by regulation of phosphatases, on the steady-state levels of phosphorylated T287. Both oxidation and phosphorylation of the regulatory domain were blocked in mice expressing AC3I, a peptide inhibitor of CaMKII, supporting the role of initial kinase activation in the generation of these distinct constitutively active species.
The authors go on to establish the clinical relevance of CaMKII activation by oxidation in a very elegant set of experiments. In cultured cardiac myocytes, they showed that following downregulation of endogenous δCaMKII with a short hairpin RNA (shRNA), expression of shRNA-resistant wild-type δCaMKII supported both angiotensin II- and isoproterenol-stimulated apoptosis. In contrast, expression of a mutant kinase in which M281 and M282 are replaced by valines (M281/282V) only supported β-agonist isoproterenol-triggered cell death. The authors coupled these exciting results with additional experiments showing that mice with surgically induced myocardial infarction had increased CaMKII M281/282 oxidation and that MsrA mutant mice have worse outcomes in terms of cardiac function and mortality. These findings together build a strong case for oxidative modification of CaMKII as a significant contributor to ROS-mediated pathology.
This study, together with previous work from this group (Zhang et al., 2005), places CaMKII activation at the heart of the pathological mechanisms of both β-adrenergic and ROS-mediated cardiac damage. Both pathways are activated in the context of elevated Ca2+ levels in the beating heart and can cause autophosphorylation-dependent activation of CaMKII. This study demonstrates that signaling molecules that generate ROS have an additional mechanism for the activation of CaMKII via oxidation. Given that the localization of CaMKII is critical to its specificity (Schulman, 2004), it will be interesting to see if oxidation-modified CaMKII and phosphorylated CaMKII are differentially localized within the myocyte or whether they act on different sets of effectors. In both cases, inhibiting CaMKII in the heart may be an appealing therapeutic approach for preventing sudden death following a heart attack. Moreover, ROS-dependent pathology is important in many neurological conditions including Alzheimer's disease (Infanger et al., 2006) and the vascular dysfunction that follows subarachnoid hemorrhage (Zemke et al., 2007). The ability of CaMKII to be directly stimulated by the generation of ROS may connect this versatile enzyme to a variety of other diseases.
REFERENCES
- Erickson JR, Joiner MA, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, et al. Cell, this issue. 2008 doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith LC. J. Neurosci. 2004;24:8394–8398. doi: 10.1523/JNEUROSCI.3604-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Annu. Rev. Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- Infanger DW, Sharma RV, Davisson RL. Antioxid. Redox Signal. 2006;8:1583–1596. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- Lu CS, Hodge JJ, Mehren J, Sun XX, Griffith LC. Neuron. 2003;40:1185–1197. doi: 10.1016/s0896-6273(03)00786-4. [DOI] [PubMed] [Google Scholar]
- Maier LS, Bers DM. Cardiovasc. Res. 2007;73:631–640. doi: 10.1016/j.cardiores.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Patton BL, Miller SG, Kennedy MB. J. Biol. Chem. 1990;265:11204–11212. [PubMed] [Google Scholar]
- Schulman H. J. Neurosci. 2004;24:8399–8403. doi: 10.1523/JNEUROSCI.3606-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemke D, Farooq MU, Mohammed Yahia A, Majid A. Vasc. Med. 2007;12:243–249. doi: 10.1177/1358863X07081316. [DOI] [PubMed] [Google Scholar]
- Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr., Thiel W. Nat. Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]