

Abstract
The ATP/ADP ratio reflects mitochondrial function and has been reported to be influenced by the size of the Huntington disease gene (HD) repeat. Impaired mitochondrial function has long been implicated in the pathogenesis of Parkinson’s disease (PD) and therefore, we evaluated the relationship of the HD CAG repeat size to PD onset age in a large sample of familial PD cases. PD affected siblings (n=495) with known onset ages from 248 families, were genotyped for the HD CAG repeat. Genotyping failed in 11 cases leaving 484 for analysis, including 35 LRRK2 carriers. All cases had HD CAG repeats (range 15 to 34) below the clinical range for HD, although 5.2 percent of the sample (n=25) had repeats in the intermediate range (the intermediate range lower limit=27; upper limit=35 repeats), suggesting that the prevalence of intermediate allele carriers in the general population is significant. No relation between the HD CAG repeat size and the age at onset for PD was found in this sample of familial PD.
Keywords: Parkinson’s disease, Huntington’s disease, CAG repeat, onset age, genetics, mitochondria
Introduction
PD is a neurodegenerative disorder associated with the loss of neurons of several neurotransmitter systems at many levels of the central and peripheral nervous systems. Its principal symptoms, resting tremor, rigidity, bradykinesia and postural instability, arise from loss of dopaminergic neurons of the substantia nigra. The estimated prevalence is approximately 0.3%, with an average onset age of 60 years.1, 2 Although the majority of cases of PD are of unknown etiology, some inherited forms have been identified.3 Five genes have been cloned that identify monogenic forms of PD. These encode alpha-synuclein (PARK1) 4, parkin (PARK2) 5, PTEN-induced putative kinase 1 (PINK1) (PARK6)6, DJ-1 (PARK7)7, and leucine-rich repeat kinase 2 (LRRK2) (PARK8) 8. Other loci, including PARK3, have been identified, but genes at those sites have not yet been cloned.9 PARK110, PARK211, PARK612, and PARK713 lead to an earlier onset of PD while the onset age for PARK814 is similar to that seen for idiopathic PD.
Accumulating research has implicated mitochondrial dysfunction, possibly triggered by nuclear genetic mutations, in PD pathogenesis.15, 16 We sought to investigate mechanisms influencing mitochondrial function to evaluate their role in PD. Since the size of the triplet repeat CAG in exon 1 of the HD gene influences the ATP/ADP ratio,17 a measure mitochondrial function, the length of this triplet repeat may provide a biomarker of mitochondrial function that influences age at onset of PD. Impaired mitochondrial function has long been implicated in the pathogenesis of Huntington’s disease (HD [MIM 143100]),18, 19, 20, 21 an autosomal dominant neurodegenerative disorder caused by an expanded CAG repeat in exon 1. Among affected individuals, the repeat size is inversely related to the age of onset of the disease,22 although other genetic modifiers may also influence HD onset.23 Notably, the relationship of ATP/ADP ratio to HD repeat size was not confined to the expanded range (>35 repeats), but extends across the normal variation in HD repeats (8 to 35 repeats). Seong et al. 17 proposed that the HD CAG repeat may fortuitously provide a mechanism to evaluate the role of mitochondria in other diseases, such as PD, with potential mitochondrial dysfunction. We have therefore evaluated the relationship of HD CAG repeat size to PD onset age in a large sample of familial PD cases.
Methods
Participants
The GenePD study is an ongoing study of familial PD which recruits families with at least two cases of PD meeting diagnostic criteria.24 Participants were enrolled through thirty academic centers or clinics (19 in the United States, three in Canada, two in Australia, two in Germany, two in Italy, one in the United Kingdom, and one in Denmark).25 The protocol was reviewed and approved by each center’s institutional review board. All participants were advised of the risks of the study and provided written informed consent. Participants met the diagnostic criteria for PD of the United Kingdom PD Society Brain Bank Criteria.26 Eligible participants had at least one first degree relative meeting diagnostic criteria for PD. These familial cases may represent several different modes of transmission (e.g. dominant or recessive forms) as well as possible common environmental risk factors for PD. PD age of onset was described as the youngest age when tremor, rigidity, or bradykinesia was noted by the patient or family. No participants had been diagnosed with HD, and none had a familial history of HD. Participants were screened for known PD causing genes. No cases carrying a-synuclein, DJ-1, or PINK1 mutations were found in this sample. Participants carrying parkin mutations were identified in this sample but only the homozygous parkin carriers were excluded from this study.
Our initial study included 495 siblings with confirmed PD diagnosis and known onset ages from 248 families. Genotyping of the HD gene failed in 11 of these cases leaving a final sample of 484 siblings for analysis (Table 1). Thirty-five participants from 18 families were found to be LRRK2 carriers and these were studied separately as “LRRK2 cases”. The remaining 449 non-LRRK2 carriers are termed “Familial PD” cases.
Table 1.
Summary statistics including average age of onset and sex distribution for LRRK2 and other Familial PD cases.
| Number of cases | Number of Families | Percent male | Average age of onset | |
|---|---|---|---|---|
| Total | 484 | 248 | 57.0 | 62.2 |
| Familial PD | 449 | 230 | 57.9 | 62.4 |
| LRRK2 PD | 35 | 18 | 45.7 | 60.7 |
Both LRRK2 and Familial PD cases have similar onset ages, although the proportion of male cases is greater for the Familial PD sample.
Genotyping
Each participant provided a blood sample for the purpose of DNA genotyping. DNA extraction occurred on site for samples collected at Washington University in St. Louis, Germany, Australia, and Milan. For all other locations, blood samples were shipped to the Center for Human Genetic Research at the Massachusetts General Hospital, where DNA was extracted.27 All genotyping was performed by the ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com/). CAG repeat sizes were determined by polymerase chain reaction of the number of CAG trinucleotide repeats responsible for the HD gene, using a modified protocol that eliminated an adjacent proline (CCG) repeat.28, 29
Statistical analysis
The mean, median, standard deviation and range of CAG repeats were calculated for the entire sample and separately for the LRRK2 cases and the other familial PD cases. Association to PD onset age was examined by (1) studying the larger CAG repeat carried by each participant, (2) by dichotomizing at the median CAG length of larger repeat and (3) studying the sum of both repeats. The average age of onset of PD was compared by larger repeat size and by high repeat size versus low repeat size determined by having at least one repeat greater than 19 versus having only repeats sizes of 19 or less. Generalized estimating equation (GEE) models were used to test the association between repeat size and PD onset age to account for repeated observations within a family. Models were tested predicting PD onset age by larger repeat size, high repeat size versus low repeat size and by the sum of repeat sizes.
Results
The median HD CAG repeat size among the 484 study participants was 17 with a standard deviation of 3.15 and a range of 9 to 34. The maximum CAG repeat size (larger of the two alleles) ranged from 15 to 34 and had a median of 19. The distribution of CAG repeats was similar between the LRRK2 and other familial PD cases (Table 2).
Table 2.
Distribution of CAG repeats in LRRK2 and other Familial PD.
| Number of alleles | Mean repeat size | Median repeat size | St. Dev | Range | |
|---|---|---|---|---|---|
| Total Sample | |||||
| All repeats | 968 | 18.47 | 17 | 3.11 | 9–34 |
| Larger Repeat | 484 | 19.92 | 19 | 3.41 | 15–34 |
| Familial PD | |||||
| All repeats | 898 | 18.51 | 17 | 3.15 | 9–34 |
| Larger Repeat | 449 | 19.96 | 19 | 3.47 | 15–34 |
| LRRK2 PD | |||||
| All repeats | 70 | 18.04 | 17 | 2.57 | 10–28 |
| Larger Repeat | 35 | 19.43 | 19 | 2.59 | 17–28 |
CAG repeat size information is presented as either “All repeats” representing both alleles from each of the 484 study participants, or as “Larger Repeat”, representing only the larger of the two repeats for each of the 484 study participant.
Alleles with repeats above 26 units are considered “intermediate” and may be prone to expansion into the clinical range of 40 or more repeats with paternal transmission.30 In this sample, there were twenty-five participants (5.2%) with repeats above 26 CAG units (eight with 27 CAG repeats, three with 28, one with 29, five with 30, three with 32, three with 33, and two with 34).
Of the 449 familial PD cases not carrying a LRRK2 mutation, 196 (43.7%) had a HD repeat size greater than 19 while 253 (56.3%) participants had repeats of 19 or less. Mean PD onset age was 62.3 and 62.4, respectively (Table 2). Among the 35 participants with a LRRK2 mutation, 14 (40%) had HD repeats greater than 19, and the remaining 21 (60%) participants had repeats smaller than or equal to 19. Mean PD onset age was 61.6 and 60.0, respectively. The mean PD onset age for participants with HD repeat sizes greater than 19 was not statistically different from participants with repeat sizes less than 19, regardless of LRRK2 status (p=0.92 for Familial PD and p=0.60 for LRRK2 PD).
Examining the larger CAG repeat size as a continuous variable also showed no effect of repeat size on PD onset age for either the familial PD (GEE effect estimate= 0.06 and p-value =0.73) or LRRK2 PD (GEE effect estimate= −0.36 and p-value =0.55). Figure 1 illustrates the absence of effect of the larger HD CAG repeat size on the onset age of PD among the familial PD sample.
Figure 1.
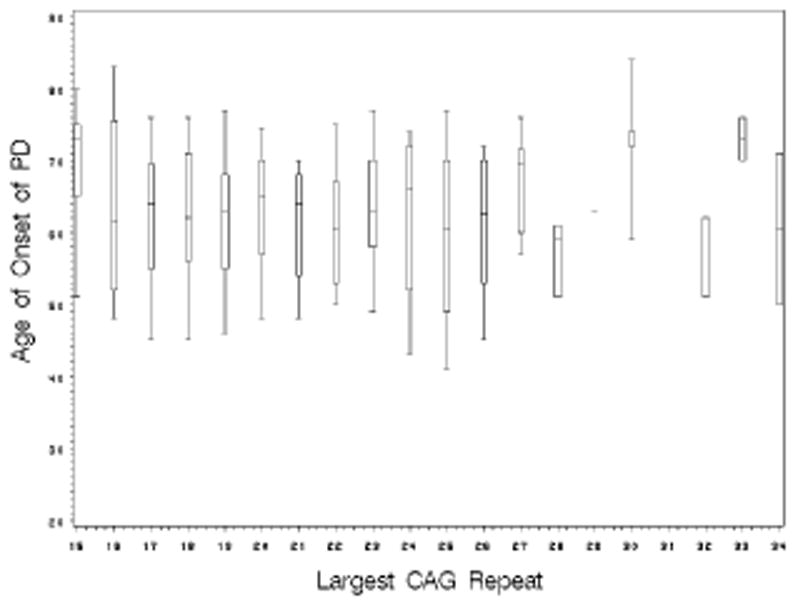
The box plot shows the median and 25th and 75th percentiles with whiskers extending to the 5th and 95th percentiles of the onset age of PD for each CAG repeat length.
Association between PD onset age and both repeats was also studied by examining the sum of both repeats and no significant difference was seen for either the Familial PD sample (GEE effect estimate= 0.02 and p-value =0.86) or the LRRK2 sample (GEE effect estimate= 0.06 and p-value =0.90).
Discussion
In this analysis of familial PD cases recruited by the GenePD study, the CAG repeat size in the HD gene is not associated with the age at onset of PD. All cases had HD CAG repeats (range 15 to 34) below the clinical range for HD, although 5.2 percent of the sample had repeats in the intermediate range (27 to 35 repeats), suggesting that the prevalence of intermediate allele carriers in the general population is significant.
While several working hypotheses propose a link between huntingtin and mitochondrial dysfunction31, 32 others have suggested that the pathogenesis of HD does not involve a defect in energy metabolism. 33 Seong et al. 26 observed that persons with HD may show reduced mitochondrial ATP/ADP ratios in damaged neurons. Dysfunction within the mitochondrial electron transport chain occurs in victims of both PD and HD, suggesting a shared dysfunctional pathway.15, 24 It was hypothesized that the various alleles of the HD gene, which have been shown to influence the onset age of HD,21 might also influence the onset age of PD. Although PD and HD may be linked by potential defects in mitochondrial function and CAG repeat length may correlate with mitochondrial function, we did not find a relationship between CAG repeat size and age of PD onset. This suggests that PD and HD are probably not related and that huntingtin is not implicated in PD. These findings suggest that the process(es) that yield altered energetics and loss of neurons in PD are distinct from the mechanism by which the HD CAG repeat regulates mitochondrial function.
Table 3.
Comparison of PD cases with CAG repeats ≤ 19 versus cases with CAG repeats > 19.
| N | Mean age (± S.D.) | Age range | |
|---|---|---|---|
| Familial PD | |||
| Larger repeat >19 | 196 | 62.3 ± 10.6 | 25 – 84 |
| Larger repeat ≤19 | 253 | 62.4 ± 11.3 | 27 – 88 |
| LRRK2 PD | |||
| Larger repeat >19 | 14 | 61.6 ± 8.2 | 50 – 75 |
| Larger repeat ≤19 | 21 | 60.0 ± 10.7 | 43 – 77 |
The Familial PD sample and the LRRK2 PD sample show no significant difference in onset age between the high repeat size and low repeat size categories.
Acknowledgments
This study was supported by PHS grants R01 NS36711-09 “Genetic Linkage Study in PD” and P50 NS16367-27 “Huntington disease Center without walls”, the Huntington’s Disease Society of America, the Huntington Society of Canada, the Hereditary Disease Foundation, the Massachusetts Huntington’s Disease Society of America, the Coalition for the Cure of HDSA, the Bumpus Foundation, and the Jerry MacDonald HD research fund. DNA samples contributed by the Parkinson Institute - Istituti Clinici di Perfezionamento, Milan, Italy, were from the “Human genetic bank of patients affected by PD and parkinsonisms”, supported by Italian Telethon grant n. GTB07001 and by the “Fondazione Grigioni per il Morbo di Parkinson”.
Footnotes
Disclosure: The authors report no conflicts of interest.
References
- 1.de Lau LM, Giesbergen PC, de Rijk MC, Hofman A, Koudstaal PJ, Breteler MM. Incidence of parkinsonism and Parkinson disease in a general population: the Rotterdam Study. Neurology. 2004;63(7):1240–4. doi: 10.1212/01.wnl.0000140706.52798.be. [DOI] [PubMed] [Google Scholar]
- 2.Morgante L, Rocca WA, Di Rosa AE, De Domenico P, Grigoletto F, Meneghini F, Reggio A, Savettieri G, Castiglione MG, Patti F, et al. Prevalence of Parkinson’s disease and other types of parkinsonism: a door-to-door survey in three Sicilian municipalities. The Sicilian Neuro-Epidemiologic Study (SNES) Group. Neurology. 1992 Oct;42(10):1901–7. doi: 10.1212/wnl.42.10.1901. [DOI] [PubMed] [Google Scholar]
- 3.Klein C, Schlossmacher MG. The genetics of Parkinson disease: implications for neurological care. Nature Clinical Practice Neurology. 2006;2(3):136–46. doi: 10.1038/ncpneuro0126. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 5.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 6.Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. 2001;68(4):895–900. doi: 10.1086/319522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Duijn CM, Dekker MCJ, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJLM, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, van Swieten JC, Oostra BA, Heutink P. PARK7, A novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet. 2001;69:629–34. doi: 10.1086/322996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 9.Sharma M, Mueller JC, Zimprich A, Lichtner P, Hofer A, Leitner P, Maass S, Berg D, Dürr A, Bonifati V, De Michele G, Oostra B, Brice A, Wood NW, Muller-Myhsok B, Gasser T European Consortium on Genetic Susceptibility in Parkinson’s Disease (GSPD) The sepiapterin reductase gene region reveals association in the PARK3 locus: analysis of familial and sporadic Parkinson’s disease in European populations. J Med Genet. 2006;43(7):557–62. doi: 10.1136/jmg.2005.039149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spira PJ, Sharpe DM, Halliday G, Cavanaugh J, Nicholson GA. Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann Neurol. 2001;49:313–9. [PubMed] [Google Scholar]
- 11.Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denèfle P, Wood NW, Agid Y, Brice A The European Consortium on Genetic Susceptibility in Parkinson’s Disease, The French Parkinson’s Disease Genetics Study Group. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342(21):1560–7. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 12.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 13.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MCJ, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256–9. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 14.Goldwurm S, Di Fonzo A, Simons EJ, Rohé CF, Zini M, Canesi M, Tesei S, Zecchinelli A, Antonini A, Mariani C, Meucci N, Sacilotto G, Sironi F, Salani G, Ferreira J, Chien HF, Fabrizio E, Vanacore N, Dalla Libera A, Stocchi F, Diroma C, Lamberti P, Sampaio C, Meco G, Barbosa E, Bertoli-Avella AM, Breedveld GJ, Oostra BA, Pezzoli G, Bonifati V. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J Med Genetics. 2005;42(11):e65. doi: 10.1136/jmg.2005.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1(8649):1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 16.Abou-Sleiman PM, Muqit MMK, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nature Reviews Neuroscience. 2006;7(3):207–19. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 17.Seong IS, Ivanova E, Lee J, Choo YS, Fossale E, Anderson M, Gusella JF, Laramie JM, Myers RM, Lesort M, MacDonald ME. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Gen. 2005;14(19):2871–80. doi: 10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- 18.Brennan WA, Jr, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington’s disease brain. Journal of Neurochemistry. 1985;44(6):1948–50. doi: 10.1111/j.1471-4159.1985.tb07192.x. [DOI] [PubMed] [Google Scholar]
- 19.Brouillet M, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA. 1995;92(15):7105–9. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milakovic T, Johnson GV. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem. 2005;280(35):30773–82. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- 21.Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nature Neuroscience. 2002;5(8):731–6. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 22.Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M, Gray J, Conneally P, Young A, Penney J, Hollingsworth Z, Shoulson I, Lazzarini A, Falek A, Koroshetz W, Sax D, Bird E, Vonsattel J, Bonilla E, Alvir J, Bickham Conde J, Cha JH, Dure L, Gomez F, Ramos M, Sanchez-Ramos J, Snodgrass S, de Young M, Wexler N, Moscowitz C, Penchaszadeh G, MacFarlane H, Anderson M, Jenkins B, Srinidhi J, Barnes G, Gusella J, MacDonald M. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nature Genetics. 1993;4(4):387–92. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- 23.Djoussé L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, Ross C, Margolis R, Rosenblatt A, Durr A, Dode C, Morrison PJ, Novelletto A, Frontali M, Trent RJA, McCusker E, Gómez-Tortosa E, Mayo D, Jones R, Zanko A, Nance M, Abramson R, Suchowersky O, Paulsen J, Harrison M, Yang Q, Cupples LA, Gusella JF, MacDonald ME, Myers RH. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. American Journal Of Medical Genetics. 2003;119a:279–282. doi: 10.1002/ajmg.a.20190. [DOI] [PubMed] [Google Scholar]
- 24.DeStefano Al, Golbe LI, Mark M, et al. Genome-wide scan for Parkinson ’s disease: The GenePD study. Neurology. 2001;57(6):1124–6. doi: 10.1212/wnl.57.6.1124. [DOI] [PubMed] [Google Scholar]
- 25.Maher NE, Golbe LI, Lazzarini AM, et al. Epidemiologic study of 203 sibling pairs with Parkinson ’s disease: the GenePD study. Neurology. 2002;58(1):79–84. doi: 10.1212/wnl.58.1.79. [DOI] [PubMed] [Google Scholar]
- 26.Gibb WRG, Lees AJ. The revelance of the Lewy body to the pathogenesis of idiopathic Parkinson ’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745–52. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson MA, Gusella JF. Use of cyclosporin A in establishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro. 1984;20:856–8. doi: 10.1007/BF02619631. [DOI] [PubMed] [Google Scholar]
- 28.Barron LH, Warner JP, Porteous M, Holloway S, Simpson S, Davidson R, Brock DJ. A study of the Huntington’s disease associated trinucleotide repeat in the Scottish population. J Med Genet. 1993;30(12):1003–7. doi: 10.1136/jmg.30.12.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuhlke C, Riess O, Schroder K, Siedlaczck I, Epplen JT, Engel W, Thies U. Expansion of the (CAG)n repeat causing Huntington’s disease in 352 patients of German origin. Hum Mol Genet. 1993;2(9):1467–9. doi: 10.1093/hmg/2.9.1467. [DOI] [PubMed] [Google Scholar]
- 30.Myers RH. Huntington’s Disease Genetics. NeuroRx. 2004;1:255–262. doi: 10.1602/neurorx.1.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Leavitt BR, van Raamsdonk JM, Dragatsis I, Goldowitz D, MacDonald ME, Hayden MR, Friedlander RM. Huntingtin inhibits caspase-3 activation. The EMBO Journal. 2006;25:5896–906. doi: 10.1038/sj.emboj.7601445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zourlidou A, Gidalevitz T, Kristiansen M, Landles C, Woodman B, Wells DJ, Latchman DS, de Belleroche J, Tabrizi SJ, Morimoto RI, Bates GP. Hsp27 overexpression in the R6/2 mouse model of Huntington’s disease: chronic neurodegeneration does not induce Hsp27 activation. Hum Mol Genet. 2007;16(9):1078–90. doi: 10.1093/hmg/ddm057. [DOI] [PubMed] [Google Scholar]
- 33.Powers WJ, Videen TO, Markham J, McGee-Minnich L, Antenor-Dorsey JV, Hershey T, Perlmutter JS. Selective defect of in vivo glycolysis in early Huntington’s disease striatum. Proc Natl Acad Sci USA. 2007;104(8):2945–9. doi: 10.1073/pnas.0609833104. [DOI] [PMC free article] [PubMed] [Google Scholar]