

Abstract
A novel, dual-pathway ring-expansion of alkynylcyclopropanols is described. On treatment with a ruthenium catalyst, these compounds undergo highly selective enlargement to either (Z)-alkylidene cyclobutanones or β-substituted cyclopentenones. The unique ability to access the least selective double bond isomers of alkylidene cyclobutanones and the dramatic shift of reactivity observed further illustrate the particular intricacies of ruthenium catalysis when compared to other alkynophilic transition metals.
The fascinating chemistry of small-ring compounds stems almost invariably from the unique reactivity modes allowed by the intrinsic ring strain in these systems.1 In particular, ring-expansion reactions have been abundantly used in organic synthesis to fashion functionalized molecules in an efficient and expeditious manner, and the appearance of various transition metal-catalyzed ring expansion processes has only enriched this landscape. 2-3
There is a considerable body of work on the transition metal-catalyzed ring expansion of vinyl and allenyl cycloalkanols,4 which provide useful tools for the construction of various cyclic ketones. This contrasts with the scarcity of reports of transition metal-promoted skeletal rearrangements of alkynylcycloalkanols.5
Our recent interest in tapping the vast potential of alkynes as selective mediators in metal-catalyzed bond-forming reactions led us to speculate whether ruthenium catalysis would provide an interesting addition to the current arsenal of ring-expansion processes.6 The remote analogy between the isomerization of a propargyl alcohol 1 to an unsaturated carbonyl 1 (termed the redox isomerization reaction7, Scheme 1) and the skeletal rearrangement of a tertiary, cyclopropyl carbinol 4 further spurred our interest. Herein we report that ruthenium catalysis is unique in the activation of alkynyl cyclopropanols 4 as it mediates a highly selective, dual ring-expansion to either four- or five-membered cyclic ketones.
Scheme 1.

Redox isomerization and proposal for a ruthenium-catalyzed ring-expansion of alkynylcyclopropanols
Gratifyingly, our initial forays were successful. Treatment of the TMS-substituted alkynylcyclopropanol 4a with catalytic amounts of ruthenium complex 2 smoothly triggered ring-expansion to alkylidene cyclobutanone 6a in essentially quantitative yield. Interestingly, the least stable (Z)-isomer was formed with nearly 6:1 stereoselectivity (Table 1, entry 1). With our curiosity piqued by these observations, the little precedent found for the expansion of silyl-substituted alkynyl cyclopropanols5c prompted us to examine more in detail this class of substrates. Our results are collected in Table 1.
Table 1.
Ruthenium-catalyzed ring-expansion of silyl-substituted alkynylcyclopropanols
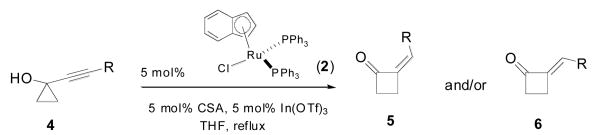 | ||||
|---|---|---|---|---|
| Entry | R | Z/E (5/6) ratioa | Time(h) | Yieldb |
| 1 | TMS 4a | 5.7:1 | 2 | 98% |
| 2 | BDMS 4b | 6.0:1 | 4 | 94% |
| 3 | SiMe2Ph 4c | 6.0:1 | 2 | 96% |
| 4 | TES 4d | 10.0:1 | 2 | 97% |
| 5 | TBS 4e | 11.4:1 | 2 | 98% |
| 6 | TIPS 4f | > 20:1 | 2 | 87%c |
Geometry was assigned by analogy to the Z and E isomers 5a/6a: see Supporting Information for details.
Total yield of two isomers determined by 1H-NMR with mesitylene as internal standard.
Isolated yield. BDMS = benzyl(dimethyl)silyl.
As can be seen, the trend for the preferential formation of (Z)-silylalkylidene cyclobutanone products upon exposure to our conditions appears to be quite general. Strikingly enough, as the steric bulk of the silyl substituent increases, so does the Z:E ratio. The corolary of this premise is that the highly congested TIPS-substituted alkynylcyclopropanol 4f (Table 1, entry 6) leads exclusively (as far as NMR-detection is concerned) to the (Z)-cyclobutanone 5f, a most counter-intuitive result!
Realizing that the electronic properties of silyl moieties might be playing a prominent role in this outcome, we then decided to examine electron-withdrawing substituents. The results of these experiments are compiled in Table 2.
Table 2.
Ruthenium-catalyzed ring-expansion of electron-deficient alkynylcyclopropanols
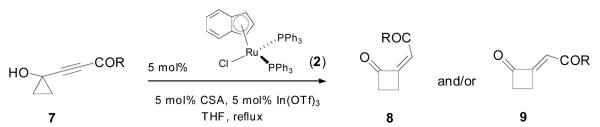 | ||||
|---|---|---|---|---|
| Entry | R | Z/E(8/9) ratioa | Time(h) | Yieldb |
| 1 | Cy 7a | > 20:1 | 12 | 88% |
| 2 | OEt 7b | > 20:1 | 8 | 68% |
| 3 | OBn 7c | > 20:1 | 6 | 81% |
| 4 | O(p-O2NC6H4) 7d | > 20:1 | 12 | 85% |
Olefin geometry was assigned based on 1H-NMR chemical shift (see Supporting Information for details).
Yields refer to pure, isolated products.
In contrast to the silyl-substituted substrates, in this case the conversion was slower, which could be ascribed to the lower electron-density at the alkyne (vide infra). Nonetheless, good yields of alkylidene cyclobutanones 8 were obtained and this regardless of the electron-withdrawing substituent being a ketone (entry 1) or ester (entries 2-4) group. It should be noted that the nature of the ester group (aliphatic, benzylic or nitroaromatic) also does not affect the outcome of the reaction. Importantly, and in analogy with the case of silyl-substituted alkynylcyclopropanols (cf. Table 1), a single isomer was obtained in all cases, which was assigned the (Z)-configuration. It is important to note that the stereochemical outcome for these reactions is the precise opposite of what was reported using gold-catalysis, suggesting that different mechanistic pathways may be operative in each case.5
Having witnessed the ability of our catalytic system to efficiently convert silyl- and acceptor-substituted alkynylcyclopropanols to stereodefined alkylidene cyclobutanones, we were eager to probe the stereoselectivity of the analogous process employing electron-“neutral” alkyl substituents at the alkyne.
To our surprise, when we exposed the hexyl-substituted alkynylcyclopropanol 10a to our reaction conditions (equation 1), a new product was formed which was not the anticipated cyclobutanone 11a. We quickly realized that the unexpected β-substituted cyclopentenone 12a had been generated instead!
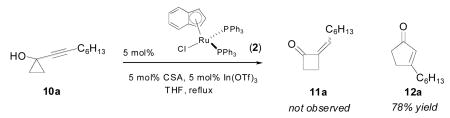 |
(1) |
Impressed by this complete shift in reactivity, we set out to examine the generality of this observation and briefly examined the alkyl-substituted substrates depicted in Table 3.
Table 3.
Ruthenium-catalyzed ring-expansion of alkyl-substituted alkynylcyclopropanols to cyclopentenones
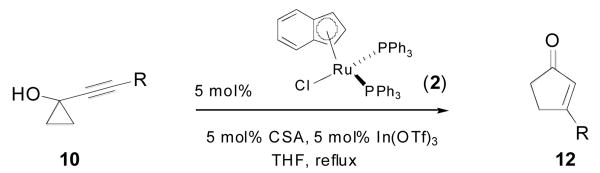 | ||||
|---|---|---|---|---|
| Entry | R | Product | Time(h) | Yielda |
| 1 | n-C6H13 10a | 12a | 4 | 78% |
| 2 | Bn 10b | 12b | 6 | 81% |
| 3 | Cy 10c | 12c | 4 | 88% |
| 4 | (CH2)3OBn 10d | 12d | 2 | 76% |
| 5 | (CH2)4OBn 10e | 12e | 2 | 68% |
| 6 | (CH2)3Cl 10f | 12f | 2 | 74% |
Yields refer to pure, isolated products.
Interestingly enough, substrates comprising benzyl (entry 2), cycloalkyl (entry 3), or remote alkoxy (entries 4-5) and halide (entry 6) substituents all underwent completely selective ring-enlargement to the corresponding cyclopentenones. In all of these cases cyclopentenones 12 were obtained exclusively, with only trace amounts of the analogous cyclobutanones detectable by 1H-NMR analysis of the crude mixtures. To the best of our knowledge, only one example of a metal-catalyzed direct cyclopropanol-cyclopentenone rearrangement was reported prior to our findings.5a,b
Our working mechanistic hypothesis to accomodate these results is presented in Scheme 2.7 We believe that, in the case of both silyl and electron-withdrawing substituents, the electronic properties of the system are presumably exacerbated upon coordination to the metal catalyst. Thus, the ability of silicon to stabilize a developing β-positive charge (Scheme 2, R = SiR3) and the propensity of ynones and propiolate derivatives to undergo Michael addition (Scheme 2, R = COR) probably favor a rapid, substrate-controlled 1,2-alkyl shift. It is worthy of note that the observed (Z)-selectivity in these cyclopropanol/cyclobutanone rearrangements, suggests that internal chelation of the putative vinylmetal intermediate by the cyclobutanone carbonyl is not operative.
Scheme 2.

Mechanistic proposal for the dual ring-expansions
On the other hand, the electron-“neutral” substrates studied (Table 3) should be more prone to metal insertion into a carbon-carbon bond of the cyclopropane moiety (Scheme 2, R = alkyl). Such a process would provide ruthenacyclohexenone 13, from which reductive elimination accounts for the observed products. The fact that only trace amounts of the analogous cyclobutanones are obtained implies that a net 1,2-alkyl shift is much less favoured in these systems.
In summary, we have developed a novel ruthenium-catalyzed ring-expansion of alkynylcyclopropanols. This atom-economical8 reaction appears to proceed by two different pathways. The unique ability of ruthenium to selectively mediate either of the two pathways depending on the electronic properties of the substrate bears testament to the versatile nature of this metal in catalysis. In particular, the ability to access functionalized β-substituted cyclopentenones through a direct two-carbon homologation is very appealing. Moreover, the exclusive obtention of the (Z)-alkylidene cyclobutanone isomers through the cyclopropanol/cyclobutanone expansion manifold is unprecedented and serves to further distinguish ruthenium from other, alkynophilic transition metals.
Supplementary Material
Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Science Foundation and the National Institutes of Health (NIH-13598) for their generous support of our programs. N.M. is grateful to the Fundação para a Ciência e Tecnologia (FCT) for the awarding of a Post-doctoral fellowship. We thank Johnson-Matthey for a generous gift of ruthenium complexes.
References
- 1.See Trost BM. In: Small Ring Compounds in Organic Synthesis. de Meijere A, editor. Springer-Verlag; Berlin: 1986. pp. 3–82.Wong HNC, Lau KL, Tam KF. In: Small Ring Compounds in Organic Synthesis. de Meijere A, editor. Springer-Verlag; Berlin: 1986. pp. 83–157.
- 2.(a) Gutsche CD, Redmore D. Carbocyclic Ring Expansion Reactions. Academic Press; New York: 1968. [Google Scholar]; (b) Hudlicky T, Becker DA, Fan RL, Kozhushkov S. Carbocyclic Three- and Four-membered Ring Compounds. In: de Meijere A, editor. Houben-Wey Methods of Organic Chemistry. El7c. Thieme; Stuttgart: 1997. p. 2538. [Google Scholar]; (c) Krief A. In: Small Ring Compounds in Organic Synthesis II. de Meijere A, editor. Springer-Verlag; Berlin: 1987. pp. 1–76. [Google Scholar]
- 3.(a) Iwasawa N, Narasaka K. Top Curr Chem. 2000:70–88. [Google Scholar]; (b) Yoshida M. Yakugaku Zasshi. 2004;124:425–35. doi: 10.1248/yakushi.124.425. [DOI] [PubMed] [Google Scholar]; (c) Muzart J. Tetrahedron. 2005;61:9423–9463. [Google Scholar]; (d) Muzart J. Tetrahedron. 2008;64:5815–5849. [Google Scholar]
- 4.For leading references, see: Snider BB, Vo NH, Foxman BM. J Org Chem. 1993;58:7228–37.Kim S, Uh K. Tetrahedron Lett. 1996;37:3865–3866.Nemoto H, Miyata J, Yoshida M, Raku N, Fukumoto K. J Org Chem. 1997;62:6450–6451.Trost BM, Yasukata T. J Am Chem Soc. 2001;123:7162–7163. doi: 10.1021/ja010504c.Yoshida M, Sugimoto K, Ihara M. Org Lett. 2004;6:1979–82. doi: 10.1021/ol049438k.Owada Y, Matsuo T, Iwasawa N. Tetrahedron. 1997;53:11069–11086.Nemoto H, Miyata J, Ihara M. Tetrahedron Lett. 1999;40:1933–1936.Yoshida M, Sugimoto K, Ihara M. Tetrahedron. 2002;58:7839–7846.Nagao Y, Ueki A, Asano K, Tanaka S, Sano S, Shiro M. Org Lett. 2002;4:455–7. doi: 10.1021/ol010287k.Trost BM, Xie J. J Am Chem Soc. 2006;128:6044–5. doi: 10.1021/ja0602501.Trost BM, Xie J. J Am Chem Soc. 2008;130:6231–42. doi: 10.1021/ja7111299.
- 5.Cobalt: Iwasawa N. Chem Lett. 1992:473–476.Iwasawa N, Matsuo T, Iwamoto M, Ikeno T. J Am Chem Soc. 1998;120:3903–3914.Gold: Markham JP, Staben ST, Toste FD. J Am Chem Soc. 2005;127:9708–9709. doi: 10.1021/ja052831g.Yeom H, Yoon S, Shin S. Tetrahedron Lett. 2007;48:4817–4820.Sordo LT, Ardura D. Eur J Org Chem. 2008:3004–3013.Palladium: Larock RC, Reddy CK. Org Lett. 2000;2:3325–3327. doi: 10.1021/ol000219i.Larock RC, Reddy CK. J Org Chem. 2002;67:2027–2033. doi: 10.1021/jo010577e.Yoshida M, Komatsuzaki Y, Nemoto H, Ihara M. Org Biomol Chem. 2004;2:3099–107. doi: 10.1039/b410362a.For a related reaction, see: Sugimoto K, Yoshida M, Ihara M. Synlett. 2006:1923–1927.
- 6.Trost BM, Weiss AH. Angew Chem, Int Ed. 2007;46:7664–7666. doi: 10.1002/anie.200702637.and references therein. Trost BM, Ball ZT, Laemmerhold KM. J Am Chem Soc. 2005;127:10028–10038. doi: 10.1021/ja051578h.
- 7.(a) Trost BM, Livingston RC. J Am Chem Soc. 1995;117:9586–9587. [Google Scholar]; (b) Trost BM, Livingston RC. J Am Chem Soc. 2008;130:11970–11978. doi: 10.1021/ja804105m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.