

Abstract
The first general method for the selection of boronic acid-based aptamers that allow for glycan sub-structure focusing is described. Using fibrinogen as a model, we have selected boronic acid-modified DNA aptamers that have high affinities (low nM Kd) and the ability to recognize changes in the glycosylation site. The method developed should also be applicable to the development of aptamers for other glyco-products, such as glycolipids and glycopeptides.
Glycosylation profoundly affects the function and activities of many proteins.1,2 However, detecting and differentiating variations in glycosylation as an integral part of a glycoprotein is not a trivial matter, mostly due to a lack of good tools. Two most powerful methods exist for developing “binders” for glycoproteins: antibody production and nucleic acid-based aptamer selection.2 However, none of these methods has the intrinsic ability to specifically focus on the glycosylation site, which include both the glycan and the surrounding structures, in epitope selection. We are interested in examining the possibility of directing the selection of aptamers to preferentially go after the glycosylation site of a glycoprotein (the sweet spot). By taking advantage of many published methods on incorporating modified nucleotide into DNA/RNA for aptamer selection,3 we decided to incorporate a boronic acid-modified thymidine-5′-triphosphate (B-TTP, Figure 1) into DNA for aptamer selection. Because of the intrinsic ability for the boronic acid moiety to interact with diols4 and single hydroxyl groups,5 we hypothesized that the incorporation of the boronic acid moiety into DNA would allow the selection to gravitate toward the glycosylation site and therefore for the specific recognition of the glycosylation site. When necessary, counter selection can be used to eliminate unwanted cross-reactivity for binders as described in literatures.3, 6 Herein, we report our work that demonstrates the feasibility by using a model protein, fibrinogen, which was chosen because of its commercial availability in large quantities and its known glycan structures.
Figure 1.
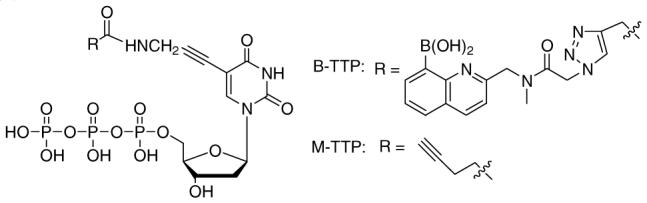
The chemical structures of B-TTP and M-TTP
We used the Systematic Evolution of Ligands by Exponential Enrichment (SELEX) approach for aptamer selection.2,3 Introduction of the boronic acid moiety was accomplished through tethering to the 5-position of TTP (Figure 1) because (1) modification at this position has long been known to have minimal effect on polymerase-catalyzed incorporation;3 (2) 5-position modified TTP has been widely used in aptamer selections to tune their affinity and bestow novel properties,3 and (3) we have demonstrated that the B-TTP can be successfully incorporated into DNA using DNA polymerases, and the synthesized boronic acid-modified DNA (B-DNA) can serve as templates for further amplification.7
For the aptamer selection work, fibrinogen was immobilized to magnetic beads using amidation chemistry. A library of DNA containing 50 randomized positions was used. 32P was used as a radioactive tracer for monitoring the percentage of DNA retained by immobilized fibrinogen. After 13 rounds of selection following procedures described in Scheme 1, nearly 80% of the radioactivity was retained by the beads (Figure 2), indicating a high percentage of specific binders for fibrinogen.
Scheme 1.

SELEX scheme for selecting DNA aptamers with high affinity for fibrinogen
Figure 2.
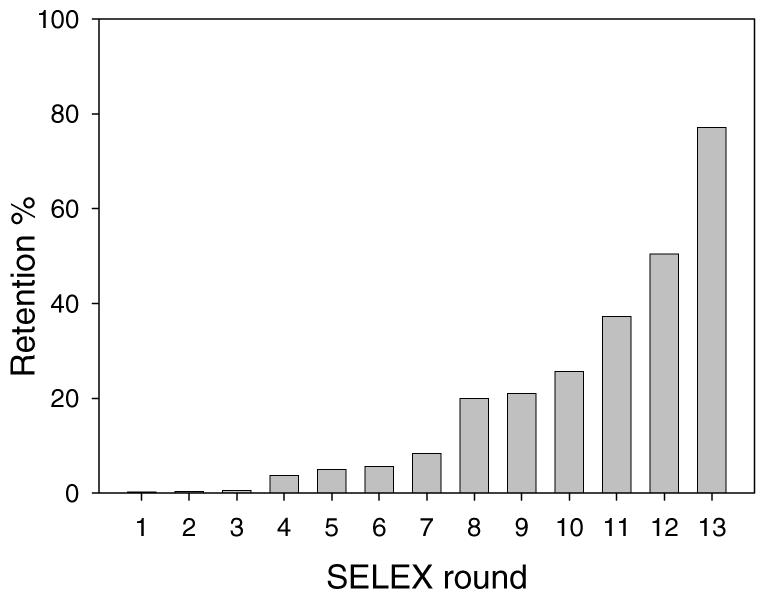
Retention of radioactive DNA on fibrinogen-immobilized beads during 13 rounds of selection
We recognize that many aptamers without boronic acid incorporation might bind to fibrinogen. There may also be other aptamers that contain boronic acid, but do not use the boronic acid moiety for binding. However, without the “pull” of the boronic acid moiety toward the glycans, such aptamers would likely bind randomly to various parts of fibrinogen without a predilection for the glycosylation site, which would be undesirable. Therefore, we needed to “select out” this pool that had no intrinsic preference for the glycosylation sites. For this reason, after the 13th round, we amplified the library using all natural dNTPs (without B-TTP) and collected only those that remained unbound to the immobilized fibrinogen. In this way, aptamers that could bind fibrinogen tightly without the involvement of boronic acid interactions were eliminated, thus removing those without an intrinsic preference for carbohydrates.
The enriched DNA library was cloned into E. coli. Twenty out of the several hundred colonies appeared were randomly selected for sequence analysis (Figure S1). Among the 20 colonies, three sequences, 85A-C, were randomly selected for further analysis. Then the dissociation constants (Kd) of these aptamers were measured using an equilibrium filtration method with 32P-label aptamers (Table 1, Figure 3, and Figures S2-19).6 All boronic acid-labeled aptamers, referred to as B-TTP aptamers, bound fibrinogen with Kd in the low nM range. In contrast, the DNA pool after the 13th round of selection showed an average Kd of about 5 μM.
Table 1.
Calculated dissociation constants (nM) of aptamers with fibrinogen and glycan-modified fibrinogen*
| Aptamer | Fibrinogen | Deglycosylated Fibrinogen | Periodated fibrinogen | |||
|---|---|---|---|---|---|---|
| B-TTP | TTP | B-TTP | TTP | B-TTP | TTP | |
| 85A | 6.2±1.4 | 102±39.7 | 390±145 | 60.2±18.0 | 70.7±18.4 | 67.4±25.0 |
| 85B | 6.4±0.81 | 63.8±16. 9 |
86.2±16.4 | 140±15.9 | 118±17.0 | 130.2±18. 7 |
| 85C | 17.1±2.1 | 122±43.7 | 371±61.9 | 256±28.9 | 202±28.1 | 321±39.7 |
All studies were done in triplicate.
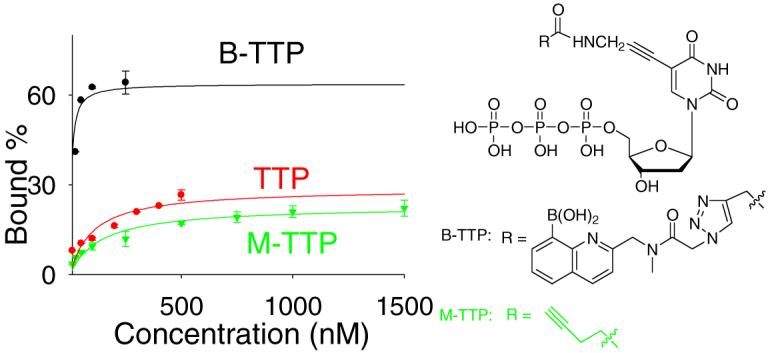
Figure 3.

Binding curves of B-TTP-, TTP- and M-TTP-derived 85A with fibrinogen (left), deglycosylated fibrinogen (middle) and periodated fibrinogen (right)
In order to examine whether binding indeed involves the glycosylation site and involves boronic acid interactions with the glycan, we designed a series of 11 types of experiments to probe this issue. These experiments include binding studies using fibrinogen (1) after glycan removal using literature procedures,8 (2) after periodate treatment, which should modify the glycan portion and thus decrease binding affinity, and (3) after sialidase treatment, which should remove the terminal sialic acid moiety. We also examined the roles that the boronic acid moiety plays in binding by conducting the binding studies using (1) TTP-aptamer, which do not have the boronic acid moiety and should have reduced affinity, (2) H2O2-treated B-TTP aptamer, which should have the boronic acid moiety removed and decreased binding, (3) DNA synthesized using H2O2-treated B-TTP, which should have decreased binding, and (4) DNA synthesized using M-TTP (Figure 1), which has a thymidine-5-position side chain, but not a boronic acid. As additional controls, we also performed the aptamer binding studies (1) in the presence of an added boronic acid, (2) in the presence of added fructose, and (3) in the presence of the glycan cleaved from fibrinogen. All three experiments should show competitive inhibition if glycan-boronic acid interactions were involved in binding. We also studied whether the aptamers identified compete against each other for binding.
Overall, all results suggest that the aptamers identified indeed bind to fibrinogen through recognition of the glycosylation site with the involvement of the boronic acid moiety. Specifically, the Kd values of TTP aptamers were 10-20 fold higher than that of B-TTP aptamers (Table 1), which suggests an important role in binding for the boronic acid moiety. For example, for 85A the Kd of the B-TTP aptamer for fibrinogen was 6 nM, while the Kd of the TTP aptamer was about 100 nM. When M-TTP was incorporated instead of B-TTP, aptamer 85A showed a Kd value (138 nM) that is over 20 fold higher than that of the B-TTP aptamer. Similarly, aptamer treated with H2O2 showed a much higher Kd value (173 nM) than the B-TTP aptamer (Figure S15). Control experiments showed that the same H2O2 treatment of the TTP aptamer (no boronic acid incorporation) resulted in no significant changes in its Kd (139 nM), indicating that the DNA was stable under the hydrogen peroxide treatment conditions (Figure S15). We have also used H2O2-treated B-TTP, which had the boronic acid replaced by a hydroxyl group, in the synthesis of modified DNA aptamer. The resulting aptamer (85A) showed a Kd of about 300 nM. The fact that the Kd of the aptamer synthesized from H2O2-treated B-TTP is higher than that of post-synthesis H2O2 treated B-TTP aptamer (173 nM) suggests the possibility of incomplete boronic acid cleavage when intact DNA was treated with H2O2.
When fibrinogen with modified glycan or complete removal of glycan was used in binding, significantly reduced binding affinity was observed (Table 1), indicating an important role the glycan plays in binding. For example, with deglycosylated fibrinogen, the B-TTP aptamer 85A showed a 60-fold lower affinity when compared to binding with unmodified fibrinogen, with a Kd of 390 nM. With periodate-treated fibrinogen, the B-TTP aptamer 85A showed an approximately 12-fold lower affinity (Kd 70 nM) when compared to binding with unmodified fibrinogen. After treatment of fibrinogen with sialidase, B-TTP aptamer 85A showed about 26-fold lower affinity (Kd 165 nM) than toward the original fibrinogen (Figure S16). Also very important is the fact that the aptamer also binds to fibrinogen even after glycan modification. This indicates that the aptamer also recognizes the protein portion of fibrinogen, which is very much desirable, as aptamers that bind only to the sugar portion can be prepared in a more direct fashion and would not be as useful for the detection of intact glycoproteins.
The binding of B-TTP aptamer was also inhibited by the addition of fructose and boronic acid in a concentration dependent manner, further indicating the important roles that boronic acid-glycan interactions play in binding. For example, At 50 mM of fructose, the apparent Kd of aptamer 85A for fibrinogen was about 500 nM (Figure S17), which was close to 100 fold higher than in the absence of fructose (6 nM). Similarly, 1 mM of 4-isoquinolinylboronic resulted in an increase of the apparent Kd from about 6 to 130 nM (Figure S18). In the presence of the glycan cleaved from fibrinogen, the apparent Kd of 85A increased by 12 fold to about 80 nM (Figure S19). We have also found that all aptamers compete with each other for binding, indicating that they all bind to the same general site. Any single experiment above would not allow us to draw unequivocal conclusions. However, combined together, they all indicate that the aptamer binding was indeed through recognition of the glycosylation sites through boronic acid-mediated actions.
In order to further demonstrate the crucial role that the boronic acid moiety plays in the aptamer selection process, a control selection by using natural TTP (instead of B-TTP) was conducted. After the 12th round of SELEX, there was about 35% of radioactivity retention by the immobilized fibrinogen. After 16 rounds, only 40% of the radioactivity was retained by fibrinogen immobilized on magnetic beads (Figure S20). Selection was stopped after 16th round because it seemed to have reached a plateau.
This selected pool was then cloned into E. coli and ten colonies were randomly picked for sequencing (Figure S21). Among these 10 sequences, two aptamers were randomly selected for further binding analysis. The dissociation constants (Kd) of these aptamers were measured using an equilibrium filtration method (Figures S22-29). Binding of the TTP aptamers from the control selection work was much weaker (e.g., aptamer 121A: Kd = 450 ± 150 nM; 121B: Kd = 500 ± 150 nM) compared to the aptamers selected using B-TTP (Kd, 3-30 nM, Table 1). Furthermore, the binding affinity of the TTP aptamers from their control selection with deglyco-fibrinogen was very similar to the binding with intact fibrinogen. For example, the binding of 121A with deglycofibrinogen has a Kd of 410 ± 120 nM, which is within experimental error of its binding with intact fibrinogen. Such results indicate that the binding site for aptamers from the control selection was not the glycosylation site. In addition, incorporation of the boronic acid moiety into the aptamer selected using TTP did not result in a significant improvement in binding either. For example, the Kd for 121A binding with fibrinogen after boronic acid incorporation was 300 ± 80 nM, which is within experimental error of the binding without boronic acid incorporation. Such results indicate that random introduction of the boronic acid moiety does not help to improve binding. Overall, the control results support the hypothesis that the incorporation of the boronic acid moiety into the DNA during the selection process confers crucial advantage in guiding the selection toward the glycosylation site.
In conclusion, we have described the first method for the selection of boronic acid-modified aptamers that allow for carbohydrate sub-structure focused selection of aptamers for a glycoprotein. The same approach should also be applicable for the selection of aptamers for the recognition of other glyco-products, such as glycolipids. It is interesting to note that with the aptamers identified, there is no consensus in their sequences. This might be because the glycan is large and the aptamers identified might not all bind to the same specific position. Work is on-going to sequence more aptamers and to conduct detailed structural studies.
Supplementary Material
Acknowledgment
We gratefully acknowledge the financial support from the Georgia Cancer Coalition, Georgia Research Alliance, and the National Institutes of Health (CA123329, CA113917, and GM084933).
Footnotes
Supporting Information Available: Description of the experimental procedure and detailed results. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Fukuda M. In: Molecular Glycobiology. Fukuda M, Hindsgaul O, editors. Oxford University Press; New York: 1994. pp. 1–52. [Google Scholar]
- (2)a).Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]; b) Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]; c) Robertson DL, Joyce GF. Nature. 1990;344:467–468. doi: 10.1038/344467a0. [DOI] [PubMed] [Google Scholar]
- (3)a).Klussmann S. The Aptamer Handbook-Functional Oligonucleotides and Their Applications. Wiley-VCH Werlag GMbH & Co. KGaA; Weinheim: 2006. Famulok M, Hartig JS, Mayer G. Chem. Rev. 2007;107:3715–3743. doi: 10.1021/cr0306743. and references cited therein.
- (4)a).Hall DG, editor. Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2005. Manimala JC, Wiskur SL, Ellington AD, Anslyn EV. J. Am. Chem. Soc. 2004;126:16515. doi: 10.1021/ja0478476.James TD, Shinkai S. Top. Curr. Chem. 2002;218:159–200.Springsteen G, Wang B. Tetrahedron. 2002;58:5291–5300.Wang W, Gao X, Wang B. Curr. Org. Chem. 2002;6:1285–1317.Yan J, Fang H, Wang B. Med. Res. Rev. 2005;25:490–520. doi: 10.1002/med.20038. and references cited therein.
- (5)a).Groll M, Berkers CR, Ploegh HL, Ovaa H. Structure. 2006;14:451–456. doi: 10.1016/j.str.2005.11.019.Yang W, Gao X, Wang B. Med. Res. Rev. 2003;23:346–368. doi: 10.1002/med.10043. and references cited therein.
- (6).Lin N, Yan J, Huang Z, Altier C, Li M, Carrasco N, Suyemoto M, Johnston L, Wang S, Wang Q, Fang H, Caton-Williams J, Wang B. Nucleic Acids Res. 2007;35:1222–9. doi: 10.1093/nar/gkl1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7)a).Jenison RD, Gill SC, Pardi A, Polisky B. Science. 1994;263:1425–1429. doi: 10.1126/science.7510417. [DOI] [PubMed] [Google Scholar]; b) Huang Z, Szostak JW. RNA. 2003;9:1456–63. doi: 10.1261/rna.5990203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Edge ASB, Faltynek CR, Hof L, Reichert LE, Jr., Weber P. Anal. Biochem. 1981;118:131–7. doi: 10.1016/0003-2697(81)90168-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.