

Abstract
Recent studies have suggested that transmitter release facilitation at synapses is largely mediated by presynaptic Ca2+ current facilitation, but the exact contribution of Ca2+ current facilitation has not been determined quantitatively. Here, we determine the contribution of Ca2+ current facilitation, and of an increase in the residual free Ca2+ concentration ([Ca2+]i) in the nerve terminal, to paired-pulse facilitation of transmitter release at the calyx of Held. Under conditions of low release probability imposed by brief presynaptic voltage-clamp steps, transmitter release facilitation at short interstimulus intervals (4 ms) was 227 ± 31% of control, Ca2+ current facilitation was 113 ± 4% of control, and the peak residual [Ca2+]i was 252 ± 18 nm over baseline. By inferring the ‘local’[Ca2+]i transients that drive transmitter release during these voltage-clamp stimuli with the help of a kinetic release model, we estimate that Ca2+ current facilitation contributes to ∼40% to paired-pulse facilitation of transmitter release. The remaining component of facilitation strongly depends on the build-up, and on the decay of the residual free [Ca2+]i, but cannot be explained by linear summation of the residual free [Ca2+]i, and the back-calculated ‘local’[Ca2+]i signal, which only accounts for ∼10% of the total release facilitation. Further voltage-clamp experiments designed to compensate for Ca2+ current facilitation demonstrated that about half of the observed transmitter release facilitation remains in the absence of Ca2+ current facilitation. Our results indicate that paired-pulse facilitation of transmitter release at the calyx of Held is driven by at least two distinct mechanisms: Ca2+ current facilitation, and a mechanism independent of Ca2+ current facilitation that closely tracks the time course of residual free [Ca2+]i.
Synaptic short-term facilitation is a Ca2+-dependent elevation of transmitter release probability during repetitive stimulation of synapses that influences the information transfer between neurons (Abbott & Regehr, 2004). It is well established that facilitation is caused by residual Ca2+ that remains in the nerve terminal from the Ca2+ influx during a previous action potential (AP) (Katz & Miledi, 1968; Zucker & Regehr, 2002). There is also general agreement that simple linear summation of the residual free[Ca2+]i transient and the ‘local’[Ca2+]i at the sites of vesicle fusion cannot explain facilitation, because the residual free [Ca2+]i (amplitude of ∼0.5 μm) is too small to significantly increase the ‘local’[Ca2+]i signal, which has an estimated peak amplitude of 10–25 μm (Bollmann et al. 2000; Schneggenburger & Neher, 2000). Therefore, one class of models, called the ‘facilitation-site’ model (Atluri & Regehr, 1996; Tang et al. 2000; Matveev et al. 2002) propose an additional, high-affinity Ca2+ binding site besides the intermediate-to-low-affinity Ca2+ sensor that drives vesicle fusion. Other models propose that residual Ca2+ remains bound to the Ca2+ sensor for vesicle fusion (‘bound Ca2+ model’; Yamada & Zucker, 1992; Bertram et al. 1996), or else, that non-linearities in the summation of the residual, and the ‘local’ Ca2+ signal due to Ca2+ buffer saturation cause facilitation (‘Ca2+ buffer saturation hypothesis’; Neher, 1998; Blatow et al. 2003; Felmy et al. 2003; Matveev et al. 2004). These three models attempt to explain facilitation without assuming an increased presynaptic Ca2+ influx, because early work at the squid giant synapse demonstrated that facilitation occurs in the absence of changes in Ca2+ influx (Charlton et al. 1982; see also Felmy et al. 2003).
Recently, use-dependent changes in Ca2+ entry into nerve terminals have received renewed attention as a possible mechanism for facilitation. One mechanism that might underlie an increase in presynaptic Ca2+ influx is presynaptic action potential (AP) broadening, but its contribution to facilitation is probably restricted to later APs of a train (Jackson et al. 1991; Geiger & Jonas, 2000). Independent from any change in the presynaptic AP waveform, facilitation of presynaptic Ca2+ currents can lead to increased Ca2+ entry during a second of a pair of AP-like stimuli. This mechanism of Ca2+ current facilitation has been discovered in measurements of Ca2+ currents at the large calyx of Held nerve terminals (Cuttle et al. 1998; Borst & Sakmann, 1998b). At these terminals, Ca2+ current facilitation is thought to be mediated by the Ca2+-binding protein neuronal Ca2+ sensor protein-1 (NCS-1) (Tsujimoto et al. 2002), is specific for the P/Q-type channels, as shown in studies of Ca2+ channel α1A-subunit knock-out mice (Inchauspe et al. 2004; Ishikawa et al. 2005), and is caused by a Ca2+-dependent acceleration of the activation kinetics of Ca2+ channels. Work on recombinantly expressed Ca2+ channels has shown that the IQ site in P/Q-type Ca2+ channels, which mediates interaction with calmodulin and other Ca2+-binding proteins (Lee et al. 2000; DeMaria et al. 2001), is responsible for Ca2+ current facilitation. Thus, details about the molecular mechanism of Ca2+ current facilitation are now available, but the impact of Ca2+ current facilitation on transmitter release facilitation is still a matter of debate.
It has been recently suggested that transmitter release facilitation at the calyx of Held synapse might be entirely mediated by Ca2+ current facilitation (Inchauspe et al. 2007; see also Xu et al. 2007). Also, by investigating the role of Ca2+ current facilitation and inactivation in short-term plasticity in superior cervical ganglion neurons, Mochida et al. (2008) recently proposed that bi-directional modulation of Ca2+ currents is the major cause of short-term facilitation and depression (Mochida et al. 2008). On the other hand, robust facilitation of transmitter release was observed in experiments at the calyx of Held when only minimal Ca2+ current facilitation was apparent (Felmy et al. 2003). However, a quantitative estimate of the impact of Ca2+ current facilitation on facilitation of transmitter release is missing.
Here, we quantified the contribution of Ca2+ current facilitation to paired-pulse facilitation of transmitter release, by making paired pre- and postsynaptic voltage-clamp recordings at the calyx of Held synapse. This allowed us to measure the presynaptic Ca2+ current facilitation under voltage clamp, and to image the presynaptic residual [Ca2+]i signal in the nerve terminal, while monitoring paired-pulse facilitation of transmitter release in a simultaneous postsynaptic recording. In combination with kinetic transmitter release simulations that are based on the known intracellular Ca2+ sensitivity of transmitter release at the calyx of Held (Bollmann et al. 2000; Schneggenburger & Neher, 2000; Felmy et al. 2003), our data suggest that Ca2+ current facilitation accounts for ∼40% of the observed transmitter release facilitation. The remaining release facilitation, which is strongly modulated in its time course by the decay of the residual free [Ca2+]i, might be mediated by binding of Ca2+ to a high-affinity facilitation site, and/or by Ca2+ buffer saturation.
Methods
Electrophysiology and slice preparation
Simultaneous pre- and postsynaptic whole-cell recordings were made from visually identified calyx of Held synapses in transverse brainstem slices of 8- to 10-day-old Wistar rats, as previously described (Felmy et al. 2003). The animals were quickly killed by decapitation without prior anaesthesia, in a procedure approved by the cantonal veterinary office of the Canton of Vaud (Switzerland; authorization 1864). The extracellular solution contained (in mm) 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 25 glucose, 0.4 ascorbic acid, 3 myo-inositol and 2 sodium pyruvate, ∼320 mosmol l−1, pH 7.4 when bubbled with 95% O2 and 5% CO2. The extracellular [Ca2+] (at a constant [Mg2+] of 1 mm) was lowered to 0.6 mm in some experiments (Figs 1 and 2). For paired voltage-clamp recordings (Figs 2–8), we added 10 mm tetraethylammonium chloride (TEA-Cl), 0.5 μm tetrodotoxin, 50 μm d-2-amino-5-phophonovaleric acid, and 100 μm cyclothiazide to the extracellular solution. In one set of experiments (Fig. 7), 3 μmω-conotoxin GVIa (Bachem, Heidelberg, Germany), and 0.5 μm SNX-482 (Peptides International, Louisville, KY, USA) were applied to the bath, together with the drugs mentioned above. Bovine serum albumin (0.1 mg ml−1; Sigma) was added to the extracellular solution in this experiment (Fig. 7) to prevent absorption of the peptides to the tubing surface of the perfusion system. The pipette solution used for postsynaptic voltage-clamp recordings contained (in mm): 135 caesium gluconate, 20 TEA-Cl, 10 Hepes, 5 sodium phosphocreatine, 4 MgATP, 0.3 Na2GTP and 5 EGTA. The same solution, without EGTA, was used for presynaptic voltage-clamp recordings, to which either (i) 100 μm fura-6F, (ii) 75 μm EGTA and 100 μm fura-6F, or (iii) 50 μm fura-2 were added. For presynaptic current-clamp recordings (Fig. 1), the pipette solution contained (in mm) 145 potassium gluconate, 10 Hepes, 2 ATP-Mg, 0.3 Na2GTP, 20 KCl and 75 μm EGTA. All experiments were conducted at room temperature (21–24°C).
Figure 1. Presynaptic action potentials during paired-pulse facilitation.

A, presynaptic action potentials (APs) in a calyx of Held (top) and EPSCs (bottom) in response to afferent fibre stimulation. The extracellular Ca2+ concentration ([Ca2+]o) was decreased from 2 mm (black dashed traces) to 0.6 mm (black traces) at a constant [Mg2+]o of 1 mm, revealing paired-pulse facilitation of EPSCs. Note that the presynaptic AP did not change upon lowering [Ca2+]o (compare black and grey traces, top). B, average AP half-widths (n = 10 cells) in response to the first stimulus recorded at 2 mm and 0.6 mm[Ca2+]o, respectively. C, overlay of the first AP (black trace) and the second AP (dotted trace) of an example sweep recorded at 0.6 mm[Ca2+]o. Same cell as in A. D, half-widths (left) and amplitudes (right) of the first and second AP recorded at low [Ca2+]o, when the EPSCs facilitated. Average data from n = 10 cells. Note the slightly longer half-width (left), and the slightly smaller amplitude of the second AP (right, P < 0.001).
Figure 2. Paired-pulse facilitation studied with pairs of identical presynaptic AP-like voltage-clamp waveforms.

A, example recording in which a terminal was stimulated with two identical AP voltage-clamp waveforms applied at an interval of 10 ms (top). The extracellular [Ca2+] was 0.6 mm, and the presynaptic pipette solution contained 75 μm EGTA. Inserting plateaus of different lengths at the peak of the AP waveform (0.2 ms increments) evoked presynaptic Ca2+ currents (middle) and EPSCs (bottom) of increasing amplitudes. B, relationship between the EPSC amplitude and the presynaptic Ca2+ current charge (QCa) during the first stimulus. Fitting a line to the logarithmized data set yielded a Ca2+ current cooperativity of 2.7. C, facilitation of the EPSC amplitudes as a function of the first EPSC. Note the robust facilitation (mean 191%) observed over a wide range of first EPSC amplitudes. Data in A–C are from the same cell. D, mean EPSC–Ca2+ current cooperativity (left), EPSC facilitation (middle), and Ca2+ current facilitation (right) recorded in n = 4 cell pairs.
Figure 8. Direct evidence for transmitter release facilitation that is independent of Ca2+ current facilitation.

A, a terminal filled with 75 μm EGTA was stimulated with two identical voltage-clamp steps to +28 mV (top) at an interstimulus interval of 10 ms. The resulting presynaptic whole-cell Ca2+ currents (ICa, middle, integral indicated in grey), and EPSCs (bottom) facilitated to 106%, and 232%, respectively. B, decreasing the length of the second depolarization by 0.02 ms with respect to the first step (top) produced an almost identical Ca2+ current charge during the second step (99% of control). Nevertheless, the EPSC still facilitated (151% of control, bottom). C, relationship between EPSC facilitation and Ca2+ current facilitation after varying the length of the second depolarization. The data obtained with identical stimulus durations are shown as filled black symbols, and the data which fell in a range of 98–100% of relative Ca2+ current charge during the second step are shown by filled grey symbols. Note that the EPSCs facilitated to ∼140%, although there was no net Ca2+ current facilitation (grey symbols). The dashed grey line is a simple prediction of the expected effect of Ca2+ current facilitation alone, obtained by taking the 3.5th power of the relative Ca2+ charge. Note that the data points show a clear y-offset above this prediction at both positive, and negative Ca2+ current modulation. The grey line is a polynomial fit (3 terms). The data in A–C are from the same cell. D, average EPSC facilitation and Ca2+ current facilitation measured in n = 5 cell pairs. The two bars outlined in black (left) show the data obtained with the same stimulus duration for the first and second pulse. The data shown by the bars outlined in grey (right) were obtained with slightly shorter second depolarizations, resulting in Ca2+ current charges of 98–102% of control (average, 100 ± 0.4%, filled grey bar). Note that despite the absence of Ca2+ current facilitation, the EPSCs still facilitated to 171 ± 11% of control (open grey bar).
Figure 7. Ca2+ current facilitation and EPSC facilitation under block of N-type and R-type Ca2+ channels.

A, example of presynaptic Ca2+ currents (ICa, top) and EPSCs (bottom) in response to two identical voltage-clamp steps to +28 mV applied at an interstimulus interval of 10 ms under control conditions (75 μm EGTA was present in the presynaptic pipette solution). B, presynaptic Ca2+ currents and EPSCs in the same cell pair after applying ω-conotoxin (3 μm) and SNX-482 (0.5 μm) to the bath solution. C, average relative values of Ca2+ current charge (QCa, left), and EPSC amplitude (right) in the presence of ω-conotoxin and SNX (n = 3 cell pairs). D, time course of paired-pulse EPSC facilitation (mean ±s.e.m.; top), and Ca2+ current facilitation (bottom) in the presence of ω-conotoxin and SNX (same cell as in A and B). Data were fitted with single exponential functions, with time constants of 24 and 70 ms for EPSC facilitation and Ca2+ current facilitation, respectively (grey lines). E, average Ca2+ current facilitation (left), and EPSC facilitation (right) recorded in the presence of ω-conotoxin and SNX in n = 3 cell pairs. Note that both Ca2+ current facilitation and EPSC facilitation were similar to control conditions without ω-conotoxin and SNX (see Fig. 3D).
Current-clamp and voltage-clamp recordings were performed using EPC-9/2 or EPC-10/2 double patch-clamp amplifiers (HEKA Elektronik, Lambrecht, Germany). Series resistances (Rs) ranged between 3 and 8 MΩ (compensation up to 90%) for postsynaptic recordings, and 15−30 MΩ (55% compensation) for presynaptic recordings. Presynaptic current-clamp recordings were made under Cfast cancellation and Rs compensation (55%) of the EPC-10 amplifier. Postsynaptic EPSC traces were corrected for the remaining Rs error off-line. During the simultaneous pre- and postsynaptic recordings shown in Figs 3–7, we stimulated the terminals by short (0.6–1.2 ms), identical presynaptic step depolarizations from −70 mV to +28 mV. The length of the pulses was adjusted in each cell pair such that the resulting EPSC amplitudes were 1–2 nA (Figs 3 and 4). In the experiment shown in Fig. 8, the duration of the second pulse was shortened by up to 1 ms with respect to the first one (see Results). In the experiments of Fig. 2, release was stimulated by two identical AP voltage-clamp command waveforms. The command waveform was generated by averaging APs recorded in n = 9 calyces, and the clamp voltage before and after the AP was set to −70 mV. The resulting AP voltage-clamp command was from −70 mV to +49 mV, and had a 20–80% rise time of 0.21 ms and a half-width of 0.44 ms. We inserted plateaus of different lengths (0.2 ms increments) at the peak of the AP voltage-clamp commands to increase the Ca2+ current amplitude (Borst & Sakmann, 1998a). For these experiments (Fig. 2), we attempted to cut calyceal axons by introducing a mediolateral angle during the slice preparation, with the aim of improving the presynaptic voltage-clamp conditions (Borst & Sakmann, 1998a).
Figure 3. Measuring Ca2+ current facilitation and presynaptic residual [Ca2+]i during transmitter release facilitation.

A, presynaptic whole-cell Ca2+ currents (ICa, integral highlighted in grey), EPSCs (middle) and transmitter release rates obtained from EPSC deconvolution analysis (bottom, see Methods), elicited by rectangular voltage-clamp steps applied at two different interstimulus intervals (Δt). Fura-6F (100 μm) and EGTA (75 μm) were present in the presynaptic pipette solution. Note the slight facilitation of the Ca2+ currents at the intervals of 10 ms (Aa, arrowhead), and the slight depression of Ca2+ currents at the longest interval (Ab), respectively. B, paired-pulse facilitation of the peak transmitter release rate (mean ±s.e.m.; top), and of the presynaptic Ca2+ current charges (bottom) as a function of the interstimulus interval. The data were fitted with single exponential functions, with time constants of 24 and 70 ms for transmitter release facilitation, and Ca2+ current facilitation, respectively (grey lines). C, presynaptic residual Ca2+ signal measured with 100 μm fura-6F in response to single voltage-clamp steps to +28 mV (average of n = 24 sweeps). The decay of the signal was fitted with a double-exponential function (grey fit line), that was back-extrapolated to the time of the stimulus (arrowhead). The open black data symbols indicate the [Ca2+]i values that were used to model the contribution of residual free [Ca2+]i to facilitation (see Figs 5 and 6). The grey line before the stimulus onset corresponds to the temporal average of ‘baseline’[Ca2+]i. Data in A–C are from the same cell. D, average Ca2+ current facilitation (‘QCa’, left), and release-rate facilitation (right) of n = 4 cell pairs recorded with 100 μm fura-6F and 75 μm EGTA.
Figure 4. Probing the effect of presynaptically added Ca2+ buffers on Ca2+ current facilitation, transmitter release facilitation, and residual [Ca2+]i.

A, average time courses of paired-pulse facilitation of transmitter release recorded in the presynaptic presence of 100 μm fura-6F (n = 4; black), 100 μm fura-6F and 75 μm EGTA (n = 4; blue), and 50 μm fura-2 (n = 4; red). B, average time course of Ca2+ current facilitation (same colour code as in A). C, average residual [Ca2+]i signals in response to single depolarizations measured under the three different presynaptic Ca2+-buffering conditions. The [Ca2+]i traces were offset on the y-axis such that the baseline [Ca2+]i values before stimulus onset overlapped. Note that the time course of transmitter release facilitation (A) and the decay of [Ca2+]i (C) are more sensitive to adding EGTA than the time course of Ca2+ current facilitation (B). D, average control EPSC amplitudes (top), and control Ca2+ charges (bottom) under the different Ca2+-buffering conditions. In the presence of fura-2, the Ca2+ charge was significantly larger as compared with fura-6F alone (P = 0.04), but the EPSC amplitudes were not significantly different between the various Ca2+-buffering conditions, indicating a slight suppression of transmitter release by 50 μm fura-2. The average data in A–D are from the same data set (n = 4 cells for each Ca2+-buffering condition).
Presynaptic Ca2+ currents (Figs 2, 3, 5, 7 and 8) are shown after leak-current correction and capacitive-current correction by using a ‘p/5’ protocol with leak pulses from −80 to −58 mV. For afferent fibre stimulation (Fig. 1), presynaptic axons were stimulated with a bipolar electrode that was custom-made from platinum–iridium wire (diameter: 0.125 mm), and placed close to the midline of the brainstem slice (Borst et al. 1995; Meyer et al. 2001).
Figure 5. Modelling the contributions of Ca2+ current facilitation and residual Ca2+ to paired-pulse facilitation.

Aa, presynaptic whole-cell Ca2+ currents (ICa) recorded at an interstimulus interval of 10 ms. The Ca2+ current integral is highlighted in red. Ab, the inverted Ca2+ current waveforms (black traces), and the waveforms of local [Ca2+]i signals inferred by the ‘five-site model’ (blue, red and dashed green traces). The blue trace represents the ‘local’[Ca2+]i waveform that predicted the transmitter release rate during the first pulse (see Ac). The red and green traces are simulated local [Ca2+]i waveforms for the second pulse, which take into account Ca2+ current facilitation alone (‘Q’; dashed red trace; hardly visible), or Ca2+ current facilitation plus a linear summation of residual free [Ca2+]i and the local [Ca2+]i signal (‘Q+R’; red trace), or both mechanisms plus an assumed supra-linearity in the summation of the [Ca2+]i signals (‘Q+R+S’; dashed green trace). Ac, measured transmitter release rates (black trace) and model fits, under the same assumptions and with the same colour code as shown in Ab. Note that Ca2+ current facilitation together with a direct action of residual free [Ca2+]i (red trace) explains only 42% of the transmitter release facilitation in this example, as visualized by the red arrow. B, peak transmitter release rates as predicted by driving the five-site model (black trace; parameters as in Felmy et al. 2003), and the allosteric model (dashed black trace; parameters as in Lou et al. 2005) with local [Ca2+]i waveforms as shown in Ab, over a wide range of [Ca2+]i amplitudes. The red lines with circles and squares indicate slopes in double-log coordinates at two ranges of [Ca2+]i. Note that a near-maximal slope of the five-site model is only reached at exceedingly low [Ca2+]i (slope, 4.8 at 1–1.5 μm[Ca2+]i). The grey line shows the prediction of the five-site model with the same parameter set, but using square [Ca2+]i elevations. The rightward shift of the predicted transmitter release rates driven by [Ca2+]itransients is caused by the briefness of the rise and fall of the local [Ca2+]i signal.
Paired-pulse facilitation was studied by applying pulse pairs at various interstimulus intervals (4, 10, 25, 50, 100 and 400 ms) in a pseudo-randomized sequence. Each pulse pair was separated by 15 or 20 s.
Ca2+ imaging
Fura-6F (100 μm), or fura-2 (50 μm, both from Molecular Probes, Eugene, OR, USA) were added to the caesium gluconate-based presynaptic pipette solution (see above) to image [Ca2+]i. The Ca2+-imaging system (TILL-Photonics, Gräfelfing, Germany) used a monochromator to excite the fura derivates at 355 nm (a slightly lower wavelength than the isosbestic point of the dyes), and at 380 nm. A 12-bit CCD camera was used to detect the emitted light. Sampling rate was ∼140 Hz (5 ms exposure time plus 2 ms wait time between each image), pixel binning was 8 × 15, and the off-line analysis was identical to the one previously described (Müller et al. 2007). The fluorescence ratios were converted to the intracellular free Ca2+ concentration ([Ca2+]i) using the equation of Grynkiewicz et al. (1985).
The calibration constants were first determined by an in vitro calibration, followed by an in-cell calibration of Rmin (fluorescence ratio at lowest Ca2+ load), as previously described (Felmy et al. 2003; Müller et al. 2007). The apparent Kd values of each dye were 14.7 ± 0.6 μm for fura-6F (n = 6 calibrations) and 0.2 μm for fura-2 (n = 1 calibration).
Data analysis
Transmitter-release rates were obtained by deconvolution of EPSCs under correction of glutamate spill-over current as described in detail by Neher & Sakaba (2001). Facilitation of transmitter release at a given interstimulus interval is expressed as (%):
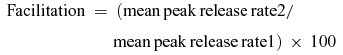 |
(Figs 3–6). Alternatively, facilitation was analysed as (see Figs 2, 7 and 8) (%):
Figure 6. The time course of paired-pulse facilitation predicted by Ca2+ current facilitation and residual [Ca2+]i under different presynaptic Ca2+-buffering conditions.

A, top: average transmitter release facilitation measured in the presynaptic presence of 100 μm fura-6F (black squares; same data as Fig. 4A, black symbols). Also shown are the predictions of transmitter release facilitation based on (i) Ca2+ current facilitation alone (‘Q’; open red symbol), (ii) based on Ca2+ current facilitation plus linear summation of residual [Ca2+]i and ‘local’[Ca2+]i (‘Q+R’; red circles), and (iii) based on both of the latter mechanisms plus supra-linear summation of the [Ca2+]i signals (‘Q+R+S’; open green symbols). Exponential fits (lines) to the data and to the predictions revealed that the predicted facilitation time courses decayed with a similar time constant (τ) as the measured facilitation (τ∼70 ms). Bottom: relative peak amplitudes of the derived ‘local’[Ca2+]i signals for transmitter release that underlie the simulations shown in the top panel. B, average transmitter release facilitation measured with 100 μm fura-6F and 75 μm EGTA in the presynaptic patch pipette (black squares; same data as Fig. 4A, blue symbols). Symbols and lines have the same meaning as in A. Note that facilitation predicted by Ca2+ current facilitation alone (red squares), and by Ca2+ current facilitation and a linear summation of residual free [Ca2+]i and local [Ca2+]i (red circles) had a smaller maximal amplitude, and a slower decay than the observed facilitation.
Applying these two analysis methods to the same data sets yielded similar amounts of facilitation.
To assess Ca2+ current facilitation, we first integrated the Ca2+ current traces to obtain Ca2+ charge values (QCa). Integration started at the zero intercept following the positive Ca2+ current artifact. As we mostly observed a second, slowly decaying component to the Ca2+ tail current that accounted for ∼5–10% of the total Ca2+ current amplitude (see also Borst & Sakmann, 1998a), integration stopped at 3 ×τ of an exponential fitted to the fast Ca2+ current decay. Otherwise integration stopped at the end of the tail current (zero intercept). Ca2+ current facilitation was expressed as (%):
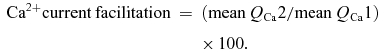 |
The spatially averaged presynaptic [Ca2+]i transients in response to single voltage-clamp steps (Figs 3C and 4C) are averages of 18–38 sweeps (fura-6F), or 6–10 sweeps (fura-2), which were separated by at least 20 s. The decay of the average [Ca2+]i transients were fitted with either double-exponential, or mono-exponential functions (Müller et al. 2007). Since we noticed that the peak of the [Ca2+]i transient did not always coincide with the first data point following the stimulus onset, we back-extrapolated the exponential fits of the decay of [Ca2+]i to the time corresponding to the stimulus onset (Fig. 3C). The amplitude of the average [Ca2+]i transient of a given terminal was calculated as the difference between the back-extrapolated [Ca2+]i amplitude, and the average of 20 points before stimulus onset (Fig. 3C).
All analysis routines and simulations were custom-written in IGOR (Wavemetrics Inc., Lake Oswego, OR, USA). Results are reported as mean ±s.e.m., and mean ±s.d. in bar graphs. Statistical significance was tested using Student's t test and accepted at P < 0.05.
Simulations of transmitter release
The ‘local’[Ca2+]i waveform compatible with the measured release rates and the previously obtained Ca2+ sensitivity of transmitter release was inferred similarly as described before (Schneggenburger & Neher, 2000; Felmy et al. 2003; Lou et al. 2005). In short, we used a template [Ca2+]i waveform that was composed of two half-Gaussian functions which were attached to each other at their peaks. This ‘local’[Ca2+]i waveform (Fig. 5Ab, blue trace) was used to drive the models of cooperative calcium binding and vesicle fusion (the ‘five-site model’ and the ‘allosteric model’; Schneggenburger & Neher, 2000; Lou et al. 2005) with the (control) parameters published in Felmy et al. 2003 in the case of the 5-site model. The rise of the local Ca2+ waveform was constrained to the rise of the Ca2+ tail currents measured in the corresponding cells (e.g. 20–80% rise time of 0.087 ms in the cell shown in Fig. 5A). We then ran the release model with local [Ca2+]i waveforms of different amplitude, delay and decay rate, until the simulated transmitter release rate described the measured one (Fig. 5Ac left, blue trace).
Results
Action potential broadening does not contribute to facilitation of release
It is generally accepted that paired-pulse facilitation of transmitter release is not caused by changes in the presynaptic action potential (AP) waveform (see Zucker & Regehr, 2002 for a review), but this assumption has not been tested in CNS synapses. In a first series of experiments (Fig. 1), we therefore investigated whether paired-pulse facilitation of EPSCs is accompanied by changes in the presynaptic AP waveform. Simultaneous presynaptic current-clamp recordings, and postsynaptic voltage-clamp recordings were made at the calyx of Held, and afferent fibres were stimulated by an extracellular stimulation electrode with pairs of stimuli (interval, 10 ms). At the start of each experiment, the Ca2+ concentration ([Ca2+]o) of the bath solution was 2 mm, and the EPSCs showed paired-pulse depression, as expected (Borst et al. 1995; Schneggenburger et al. 1999). Reducing [Ca2+]o to 0.6 mm at a constant [Mg2+]o of 1 mm led to a decrease in the first EPSC amplitude, and converted paired-pulse depression into paired-pulse facilitation (Fig. 1A, black traces, bottom).
We verified whether reducing the [Ca2+]o leads to a change in the AP half-width, but no significant change was observed (Fig. 1B), with half-widths of 0.44 ± 0.02 ms and 0.45 ± 0.02 ms at 2 mm and 0.6 mm[Ca2+]o, respectively (P = 0.37, n = 10 cells). The amplitude of the presynaptic AP was similar at the two [Ca2+]o, although we sometimes observed a slight (∼6%) decrease in AP amplitude during prolonged recording (not shown). Overall, lowering the [Ca2+]o does not have a significant effect on the presynaptic AP waveform.
We next compared the first and second AP at low [Ca2+]o (Fig. 1C). The second AP was slightly broader than the first one, with half-widths 0.44 ± 0.02 ms and 0.46 ± 0.02 ms for the first and the second AP, respectively (P < 0.001; n = 10 cell pairs; Fig. 1D, left panel). At the same time, the second AP was slightly smaller (112 ± 3 mV) than the first one (117 ± 4 mV; measured from baseline; n = 10 cells; P < 0.001; Fig. 1D, right panel). Since the observed changes in AP amplitude and half-widths were small, and since they have opposite expected effects on presynaptic Ca2+ influx, we estimate that changes of the presynaptic AP waveform do not contribute substantially to paired-pulse facilitation of transmitter release.
Studying paired-pulse facilitation with pairs of identical AP waveforms
The results in Fig. 1 suggest that paired-pulse facilitation is not caused by a change in the presynaptic AP waveform. If this is correct, then paired-pulse facilitation should also be observed when identical APs are used in a presynaptic voltage-clamp experiment. To test this, we used an AP-waveform command was based on a previously recorded presynaptic APs (Fig. 1; see also; Borst & Sakmann, 1998a; Bischofberger et al. 2002).
We made paired pre- and postsynaptic whole-cell recordings at low [Ca2+]o (0.6 mm). The AP-voltage-clamp command was applied twice at an interval of 10 ms (Fig. 2A). The Ca2+ current in response to the first AP voltage-clamp command in the cell shown in Fig. 2A had an amplitude and a half-width of 1.31 nA and 0.4 ms, respectively, but the relatively small Ca2+ charge associated with these currents (0.5–0.6 pC) evoked only small EPSCs (< 0.5 nA). In order to obtain larger first EPSC amplitudes (up to ∼2 nA), we prolonged the AP waveform at its peak, thereby evoking Ca2+ tail currents and EPSCs of increasing amplitudes (Fig. 2A). This experiment also allowed us to investigate the relationship between presynaptic Ca2+ currents and EPSC amplitudes upon increasing the number of activated Ca2+ channels.
Figure 2B shows the relationship between EPSC amplitude and presynaptic Ca2+ current charge for the experiment of Fig. 2A, displayed in double-logarithmic scales. When we fitted the logarithmized data sets of this relation with a line (Fig. 2B), we found a slope of 2.7 for this cell, and an average value of 2.8 (n = 4 cells; Fig. 2D). Knowing this slope value, which represents the Ca2+ current–release cooperativity, is necessary for estimating the impact of changes in the presynaptic Ca2+ current on the amount of transmitter release. The value obtained here (∼3) at low [Ca2+]o confirms earlier results which reported a cooperativity value of 3–3.5 (Schneggenburger et al. 1999; Xu & Wu, 2005), but it does not agree with a recent estimate of ∼5.5 that was obtained at the calyces of Held of young mice at low [Ca2+]o (Fedchyshyn & Wang, 2005).
We next analysed paired-pulse facilitation of EPSC amplitudes (Fig. 2A and C). As shown in the plot of the paired-pulse ratio as a function of the first EPSC amplitude (Fig. 2C), the average paired-pulse facilitation in the cell shown in Fig. 2 was 191%, for a range of first EPSC amplitudes of 0.5–2 nA, and 191 ± 8% for all cells (n = 4; Fig. 2D; middle panel). Thus, robust paired-pulse facilitation of EPSCs is also seen when pairs of identical AP voltage-clamp commands are used. This demonstrates that paired-pulse facilitation of transmitter release is independent of any small changes in AP waveform that might occur during paired afferent fibre stimuli (Fig. 1).
In the voltage-clamp experiment shown in Fig. 2, we observed that the presynaptic Ca2+ current facilitated to 106 ± 1% of its control charge integral (Fig. 2D, right panel; n = 4 cells), in good agreement with a previous study that used an AP as a voltage-clamp command (Borst & Sakmann, 1998b). We next investigated the contribution of Ca2+ current facilitation, and residual free [Ca2+]i to paired-pulse facilitation of transmitter release.
Measuring Ca2+ current facilitation and residual free Ca2+ during paired-pulse facilitation of transmitter release under different presynaptic Ca2+-buffering conditions
To examine the contribution of Ca2+ current facilitation (Borst & Sakmann, 1998b; Cuttle et al. 1998) to facilitation of transmitter release, we made paired pre- and postsynaptic recordings, and applied pairs of identical presynaptic voltage-clamp steps to +28 mV. At the beginning of each experiment, we varied the length of both steps with the aim of evoking a small first EPSC (range, 0.5–2 nA across cells; conditions of ‘low’ release probability). In addition to Ca2+ current facilitation, we also wished to assess the contribution of residual free [Ca2+]i to paired-pulse facilitation. We therefore imaged the spatially averaged, ‘residual’ Ca2+ signal with a low-affinity Ca2+ indicator, fura-6F (100 μm), that was added to the presynaptic patch-pipette solution alone, or together with 75 μm EGTA. Introducing a low concentration of EGTA should mimic the endogenous mobile Ca2+ buffer of calyces of Held (Müller et al. 2007), while fura-6F, due to its low Ca2+ affinity, should not add significantly to the Ca2+ buffering strength (κ) of the calyx of Held. Specifically, the Ca2+ buffering capacity of an exogenously added buffer (κB) can be approximated by
| (1) |
for [Ca2+]i≪Kd (Neher, 1998). Thus, the κ added by 100 μm fura-6F is only ∼6 (since the Kd of fura-6F is 15 μm; see Methods). It is seen that the added κB is small as compared to the endogenous Ca2+ binding capacity of calyces of Held (κS∼30–40; Helmchen et al. 1997).
Figure 3 shows a paired recording with 75 μm EGTA and 100 μm fura-6F. A pair of short (1.0 ms) presynaptic depolarizing steps given at a brief interval (10 ms) resulted in robust paired-pulse facilitation of the EPSCs (Fig. 3Aa, middle). At the same time, we observed a slight facilitation of the presynaptic whole-cell Ca2+ current (Fig. 3Aa, arrowhead). At longer interstimulus intervals, facilitation of the EPSCs and of the Ca2+ currents was smaller (not shown), and at the longest interstimulus interval tested here (400 ms), we commonly observed a slight paired-pulse depression of EPSCs and Ca2+ currents (Fig. 3Ab; Xu & Wu, 2005). In order to analyse the amount of transmitter release facilitation, we applied EPSC deconvolution to derive the transmitter release rates (Fig. 3Aa and b, bottom; Neher & Sakaba, 2001). In the cell shown in Fig. 3, transmitter release facilitation had a peak amplitude of 173 ± 17% of the control value, and decayed with a time constant of 24 ms (Fig. 3B, top). Ca2+ current facilitation was maximal at the shortest interstimulus interval (4 ms; 107 ± 1% of the control value), and decayed thereafter, showing paired-pulse depression at intervals of 100 ms or longer (to 94 ± 1% of control at 400 ms; Fig. 3B, bottom). On average, transmitter release facilitation and Ca2+ current facilitation were 227 ± 31% and 113 ± 4%, respectively (Fig. 3D, n = 4 cells). In the cell shown in Fig. 3A and B, the spatially averaged [Ca2+]i signal (Fig. 3C) peaked at 0.24 μm, and decayed with a fast and a slow decay time constant of 37 ms and 744 ms, respectively.
The results of similar experiments under three different presynaptic Ca2+-buffering conditions are summarized in Fig. 4. We will first compare the data obtained with fura-6F alone, with the cells recorded in the presynaptic presence of fura-6F and 75 μm EGTA (Fig. 4A–C; black and blue traces). Under both conditions, we observed robust paired-pulse facilitation of transmitter release at the shortest interval (4 ms; 243 ± 19% for fura-6F, n = 4; and 227 ± 31% for fura-6F plus EGTA, n = 4). Transmitter release facilitation decayed significantly faster in the presence of 75 μm EGTA (25 ± 9 ms) than with fura-6F alone (70 ± 18 ms; P = 0.04; see Fig. 4A, black and blue data points and fit lines). The [Ca2+]i transients had similar amplitudes (252 ± 18 nm and 255 ± 33 nm; fura-6F alone and fura-6F plus 75 μm EGTA, respectively), but the fast exponential componenent of the [Ca2+]i decay was significantly faster in the presence of EGTA (τfast, 26 ± 6 ms, n = 4) than in the absence of EGTA (τfast, 95 ± 18 ms; n = 4; P < 0.001; see Fig. 4C, black and blue traces). Thus, low concentrations of EGTA accelerated the decay of the spatially averaged [Ca2+]i transient and facilitation of transmitter release, in agreement with previous results (Atluri & Regehr, 1996; Müller et al. 2007).
The maximal Ca2+ current facilitation at the shortest interstimulus interval was not significantly different with and without EGTA, with amplitudes of 111 ± 2% (fura-6F; n = 4) and 113 ± 4% (fura-6F plus EGTA; n = 4; P = 0.72; Fig. 3B, black and blue data points). Also, when we fitted the decay of Ca2+ current facilitation with exponential functions, we found similar decay time constants (τ= 86 ± 12 ms and 71 ± 10 ms in the absence and presence of EGTA; P = 0.38). Nevertheless, in the presence of EGTA, there was slightly more Ca2+ current depression at an interval of 400 ms (91 ± 2% of control; n = 4 cells) than with fura-6F alone (96 ± 1%; n = 4 cells; P = 0.025; Fig. 4B). Thus, Ca2+ current facilitation decayed with a similar time constant in the two Ca2+-buffering conditions, but the absolute decrease of Ca2+ current facilitation was somewhat more pronounced in the presence of EGTA, probably because the balance between Ca2+ current facilitation and Ca2+ current depression was changed by EGTA. The finding that a low concentration of EGTA markedly accelerates the decay of paired-pulse facilitation (Fig. 4A), while leaving the decay of Ca2+ current facilitation largely unaffected (Fig. 4B) indicates that Ca2+ current facilitation has a limited contribution to facilitation of transmitter release. Below, we will analyse the contribution of Ca2+ current facilitation to paired-pulse facilitation of release in detail (see Figs 5 and 6).
In the presence of the high-affinity Ca2+ buffer fura-2 (50 μm; Kd∼0.2 μm, see Methods), the presynaptic [Ca2+]i transient was strongly suppressed (amplitude, 36 ± 5 nm, n = 4 cells) and prolonged (τ= 680 ± 106 ms; Fig. 4C, red trace). This was expected, because the exogenous Ca2+-buffering capacity (κB) introduced by 50 μm fura-2 (see eqn (1); κB of 50 μm fura-2 ∼250) largely overrides the relatively low endogenous Ca2+-buffering capacity of calyces of Held (κs∼30–40; Helmchen et al. 1997). With fura-2, transmitter release facilitation was reduced in amplitude (160 ± 12%) and decayed very rapidly (τ= 14 ± 5 ms; Fig. 4A, red data points), such that at intervals of 25 ms or longer, almost no facilitation remained. Ca2+ current facilitation was 103 ± 1% of control at the shortest interval (4 ms) (Fig. 4B, red data points), significantly smaller than with fura-6F alone (P = 0.009), and slightly smaller than with fura-6F and EGTA (P = 0.08). The smaller Ca2+ current facilitation seen with fura-2 indicates that this fast, and high-affinity buffer lowers [Ca2+]i close to the Ca2+ channels, and can thereby reduce the amount of Ca2+ current facilitation (Cuttle et al. 1998).
Figure 4D summarizes the average absolute values for the EPSC amplitudes (top) and the presynaptic Ca2+ current charges (bottom) recorded under the three Ca2+-buffering conditions tested here. Our approach of choosing the lengths of the presynaptic depolarizations such that the resulting EPSC amplitudes were 0.5–2 nA (conditions of reduced release probability; see above), resulted in EPSC amplitudes that were similar across the different Ca2+-buffering conditions (Fig. 4D, upper panel; range 1.3–1.6 nA on average). However, in the presence of fura-2, the average Ca2+ current charge was significantly larger than with fura-6F alone (P = 0.04; Fig. 4D, bottom). This indicates that we needed to inject a slightly larger Ca2+ current charge in the presence of fura-2 to reach EPSC amplitudes that were comparable to the other Ca2+-buffering conditions, suggesting that 50 μm fura-2 partially blocked release (see also Rozov et al. 2001). With fura-6F, or fura-6F and EGTA, the absolute Ca2+ current charge was 0.7–0.8 pC (Fig. 4D, bottom), slightly smaller than the Ca2+ charge that has been estimated to enter a calyx of Held during an AP at normal extracellular [Ca2+] (∼0.9–1 pC; Helmchen et al. 1997; Borst & Sakmann, 1998a). The slightly smaller value is expected, since we used short presynaptic depolarizations in order to obtain conditions of low initial release probability.
Taken together, the average data in Fig. 4 demonstrate a good correlation between the decay of paired-pulse facilitation, and the decay of the residual free [Ca2+]i transient under conditions of low Ca2+ buffering strength (fura-6F, with or without 75 μm EGTA). However, there was a mismatch between the time course of transmitter release facilitation and Ca2+ current facilitation in the presence of EGTA, which will be analysed in more detail below (Fig. 6B).
Quantitative estimate of the contribution of Ca2+ current facilitation and residual free [Ca2+]i to paired-pulse facilitation
We next sought to quantify the contribution of Ca2+ current facilitation and residual free [Ca2+]i elevation to paired-pulse facilitation of transmitter release. To do so, we analysed the measured transmitter release rates in the framework of a ‘five-site’ model of Ca2+ binding and transmitter release based on Ca2+ uncaging experiments (Schneggenburger & Neher, 2000), with parameters determined by Felmy et al. (2003). In order to analyse the effect of Ca2+ current facilitation, and of an increase in residual free [Ca2+]i on transmitter release during facilitation, we first back-calculated the ‘local’[Ca2+]i signal underlying the observed transmitter release rates.
Figure 5 shows an example of how we derived the local [Ca2+]i signal from the measured transmitter release rates. In this example, the first voltage-clamp stimulus elicited a presynaptic Ca2+ current with a charge of 0.95 pC (Fig. 5Aa, left), giving rise to a peak release rate of 67 vesicles ms−1 (Fig. 5Ac, left). To derive the ‘local’[Ca2+]i signal (Fig. 5Ab, blue trace) compatible with the measured transmitter release rate, we assumed that the local [Ca2+]i signal rises with the same speed as the recorded whole-cell Ca2+ current (Fig. 5Ab; the inverted and scaled Ca2+ current is shown as the black trace). We used this waveform as a starting estimate of the local [Ca2+]i signal (Fig. 5Ab, blue trace), and varied the decay of the local [Ca2+]i signal, its location on the abscissa, and its peak amplitude until the model output fitted the release rate measured in response to the first depolarization (Fig. 5Ac, blue and black trace). In this example, the back-calculated local [Ca2+]i had a 20–80% rise time of 0.087 ms (as constrained by the rise-time of the Ca2+ current; see above), a half-duration of 0.48 ms, and an amplitude of 12.7 μm (Fig. 5Ab, blue trace, right scale).
In order to describe the facilitated transmitter release during the second depolarization (Fig. 5Ac, black trace, right), we first scaled the inferred local [Ca2+]i signal by the relative amount of Ca2+ current facilitation measured during the second pulse (in this example 106% of control; scaling factor 1.06). Driving the model with such a scaled local [Ca2+]i waveform (Fig. 5Ab, red dotted trace, ‘Q’) predicted a transmitter release rate with a peak of 83 vesicles ms−1 (Fig. 5Ac, red dotted line), corresponding to a release facilitation of 124% of control. Thus, the predicted facilitation accounted for less than half of the observed transmitter release facilitation, which was 182% in this example (Fig. 5Ac, black trace, right).
We next asked whether taking into account a linear summation of the measured residual free [Ca2+]i (in this cell 0.24 μm) to the local [Ca2+]i signal (12.7 μm in this example) would yield a closer prediction of the observed release facilitation. This, however, only led to a small increase in the predicted facilitation (Fig. 5Ac, red trace, ‘Q+R’; predicted facilitation: 132% of control). On average, Ca2+ current facilitation together with a direct effect of residual free [Ca2+]i on the Ca2+ sensor for vesicle fusion explained only 51% (fura-6F alone; n = 4 cells) and 64% (fura-6F plus 75 μm EGTA; n = 4 cells) of the release facilitation observed at an interstimulus interval of 10 ms. Hence, Ca2+ current facilitation, together with linear summation of free residual [Ca2+]i and the local [Ca2+]i signal, could not explain the full amount of transmitter release facilitation detected under the two Ca2+-buffering conditions.
As suggested before for transmitter release facilitation induced by long (100 ms) conditioning depolarizations (Felmy et al. 2003), one possibility to explain the remaining facilitation is to assume a supra-linear summation of the residual [Ca2+]i and the local [Ca2+]i, which might be caused by Ca2+-buffer saturation (Neher, 1998; Maeda et al. 1999). In order to assess the degree of such a possible supra-linearity needed to fully explain paired-pulse facilitation, we further scaled the inferred local [Ca2+]i signal prior to adding the measured residual [Ca2+]i, until the prediction matched the observed release rate during facilitation (Fig. 5Ac, green dotted trace, ‘Q+R+S’). On average, an additional increment of 6% (in the case of 100 μm fura-6F and 75 μm EGTA) and 10% (in the case of 100 μm fura-6F) above the local [Ca2+]i signal calculated for Ca2+ current facilitation and linear summation of the free [Ca2+]i values could explain facilitation. Thus, the additional [Ca2+]i increment that needs to be assumed to fully explain facilitation is small (see also Felmy et al. 2003). However, it remains equally possible that the Ca2+ current-independent facilitation of release is mediated by other facilitation mechanisms, such as the ‘facilitation site model’ (Tang et al. 2000; see Discussion).
The simulations shown in Fig. 5A predict the effect of small increases in the ‘local’[Ca2+]i signal on transmitter release output, based on the five-site model of Ca2+ binding and transmitter release (Schneggenburger & Neher, 2000). What is the Ca2+ release cooperativity predicted by this model? To analyse the cooperativity, we scaled the local [Ca2+]i waveform found for the cell shown in Fig. 5 (see Fig. 5Ab, left, blue trace) to n = 20 peak [Ca2+]i values in the range of 1–100 μm. We then plotted the predicted peak release rates as a function of the [Ca2+]i transient amplitude (Fig. 5B, continuous black line), and fitted adjacent points of the resulting dose–response curve in double-logarithmic coordinates in order to obtain the slope values (Fig. 5B, red symbols connected by red lines). As can be seen, the five-site model of Ca2+ binding and vesicle fusion, with the parameters of Felmy et al. (2003) used here, predicts peak release rates of 30–130 vesicles ms−1 for [Ca2+]i transients with peak amplitudes of 10–15 μm (Fig. 5B, continuous black trace). In this range of [Ca2+]i, the slope was 3.6 in double-logarithmic coordinates (Fig. 5B, red circles connected by a red line). Interestingly, this cooperativity value agrees well with the Ca2+ current–EPSC cooperativity measured in various studies (∼3: Fig. 2B; ∼3.5: Schneggenburger et al. 1999; Xu & Wu, 2005; Kochubey, Han & R. S., in preparation). A cooperativity value close to 5, which is expected for the ‘five-site’ model in the limit of low [Ca2+]i, is only reached with [Ca2+]i transients of exceedingly low amplitude (cooperativity of 4.8 at 1–1.5 μm[Ca2+]i; Fig. 5B, red squares connected by a red line). Thus, the five-site model of Ca2+ binding and vesicle fusion predicts that the release machinery has a cooperativity of ∼3.5 for [Ca2+]i transients in the physiologically relevant range of 10–15 μm.
Similar results were obtained when we modelled release with the more recently developed ‘allosteric model’ (Lou et al. 2005; Fig. 5B, dashed line), which predicted a cooperativity of 3.4 for [Ca2+]i transients of 10–15 μm. Lower values of cooperativity at low [Ca2+]i, which are a feature of the allosteric model, were only reached for [Ca2+]i transients below ∼4 μm (see Fig. 5B, dashed line). Since the allosteric- and the five-site model predicted similar Ca2+ cooperativities in the relevant range of [Ca2+]i (10–15 μm), both models also yielded similar predictions of the influence of residual [Ca2+]i and Ca2+ current facilitation on release facilitation (simulations not shown).
The time course of paired-pulse facilitation predicted by Ca2+ current facilitation and residual Ca2+
We next applied the simulation approach shown in Fig. 5A to all interstimulus intervals (4–400 ms), to assess the time course of release facilitation predicted by Ca2+ current facilitation, and by linear summation of the residual [Ca2+]i and the local [Ca2+]i. The local [Ca2+]i waveforms that were used to drive the release model were based on the average parameters obtained from the inferred local [Ca2+]i of individual cells under each Ca2+ buffering condition.
Figure 6A shows the average paired-pulse facilitation of transmitter release observed in the presence of 100 μm fura-6F (black squares; data re-plotted from Fig. 4A). Simulations based on the average observed Ca2+ current facilitation predicted a release rate facilitation of 148% of control at the shortest interstimulus interval (4 ms), which decayed to 86% (net depression) at the longest interval of 400 ms (Fig. 6A, open red squares), caused by Ca2+ current inactivation. Simulations incorporating Ca2+ current facilitation and linear summation of the local and the residual [Ca2+]i signals (Fig. 6A, red circles; ‘Q+R’) resulted in a maximal release facilitation of 164% of control, which decayed to 86% at an interval of 400 ms with a time constant of 73 ms. Thus, similar to the example cell shown in Fig. 5A, the combined contribution of Ca2+ current facilitation and a linear summation of the free [Ca2+]i signals explained only about half of the observed release facilitation, at all interstimulus intervals tested. In contrast to the difference in amplitude, the time course between the predicted (τ= 73 ms) and the observed release facilitation (τ= 70 ± 18 ms) matched well.
We next tested how much supra-linearity in the summation of residual free [Ca2+]i and local [Ca2+]i would be needed to explain the maximal amplitude of paired-pulse facilitation of release by the modelling approach outlined in Fig. 5 (Fig. 5Ab, green dotted trace; see also Felmy et al. 2003). We found that a supra-linearity of ∼10% in the summation of the [Ca2+]i signals, in combination with the effect of Ca2+ current facilitation, was sufficient to describe the observed transmitter release facilitation (Fig. 6A; green open symbols; in this case the [Ca2+]i signal was scaled until the simulated release facilitation matched the mean facilitation observed at the intervals of 4 ms and 10 ms). In the lower panel of Fig. 6A, the relative increase of the ‘local’[Ca2+]i signal with respect to the unscaled control [Ca2+]i waveform is shown for all interstimulus intervals. The relative increase in the amplitude of the local [Ca2+]i signal caused by Ca2+ current facilitation alone (red open data points, Fig. 6A, bottom) simply corresponds to the average Ca2+ current facilitation (see Fig. 4B, black symbols), since we assume that an increased measured Ca2+ current causes a linear increase of the local [Ca2+]i seen by readily releasable vesicles (see below, and Discussion for the validity of this assumption). The effect of an additional linear summation of the residual [Ca2+]i to the local [Ca2+]i signal was only minimally different from the case of Ca2+ current facilitation alone, and is not shown in Fig. 6A (bottom) for clarity. The relative increase of the peak local [Ca2+]i that was needed to fully explain facilitation is shown by the green symbols. It is seen that the [Ca2+]i increment caused by an assumed supra-linearity in the summation of residual and ‘local’[Ca2+]i signals roughly equals the increment in the local [Ca2+]i signal produced by Ca2+ current facilitation alone. This reflects the fact that Ca2+ current facilitation alone only explains about half of the observed transmitter release facilitation.
Figure 6B shows the corresponding analysis for the experiments performed in the presence of fura-6F and 75 μm EGTA. As reported above, in the presence of 75 μm EGTA (Fig. 4A and B), paired-pulse facilitation of release decayed significantly faster (τ= 25 ms) than Ca2+ current facilitation (τ= 71 ms). As a consequence, the analysis in Fig. 6B yields an unsatisfactory fit at intermediate intervals (∼50 ms), since the time course of the simulated release facilitation is influenced by the slow decay of the Ca2+ current facilitation. At short intervals, Ca2+ current facilitation alone, or in combination with a linear summation of the residual [Ca2+]i and the local [Ca2+]i signal, predicted ∼45%, or ∼60% of the observed transmitter release facilitation, respectively (Fig. 6B, top, red open and filled symbols, respectively). As the predicted release facilitation was smaller than the observed one, we again assessed the degree of supra-linearity in the summation of residual [Ca2+]i and local [Ca2+]i that would be necessary to fully explain facilitation (Fig. 6B, green symbols). Similar to the data obtained with fura-6F alone (Fig. 6A), we found that an additional increase of the local [Ca2+]i transient by ∼8% was needed to explain transmitter release facilitation at the shortest intervals (Fig. 6B, bottom, green symbols). Taken together, this analysis shows that Ca2+ current facilitation, together with a linear process of [Ca2+]i summation explains only about half of the observed transmitter release facilitation.
The contribution of Ca2+ current facilitation is unchanged under block of presynaptic N- and R-type Ca2+ channels
Our analysis assumes that the Ca2+ current facilitation measured in presynaptic whole-cell recordings is equal to the facilitation of Ca2+ channels located at the active zone, where transmitter release takes place. A possible complication for this assumption arises because calyces of Held of young rats (postnatal days (P)8–10), such as the ones studied here, do not only express P/Q-type channels (∼80% of all Ca2+ channels), but also N- and R-type Ca2+ channels (Wu et al. 1999; Iwasaki et al. 2000). Ca2+ current facilitation is specific for P/Q-type channels, whereas presynaptic N-type channels do not show facilitation (Inchauspe et al. 2004; Ishikawa et al. 2005). In addition, N- and R-type channels drive release at the calyx less efficiently than P/Q-type channels (Wu et al. 1999). This combination of effects might lead to an underestimation of the impact of Ca2+ current facilitation on transmitter release facilitation. To address this possibility, we blocked N- and R-type Ca2+ channels by ω-conotoxin and SNX-482, respectively, and measured the amount of transmitter release facilitation and Ca2+ current facilitation for the remaining P/Q-type Ca2+ currents alone (Fig. 7).
We first evoked release by applying double pulses of brief (0.8–1.6 ms) presynaptic depolarizations under control conditions. The width of the presynaptic depolarizations was chosen such that the resulting EPSCs had amplitudes of 2–3 nA. After establishing baseline amplitudes for presynaptic Ca2+ currents and EPSCs (Fig. 7A), we then applied 3 μmω-conotoxin, and 0.5 μm SNX-482. In the example of Fig. 7, the toxins reduced the Ca2+ current charge (QCa) from 1.73 to 1.46 pC, and thus, to 84% of its control value. The first EPSC amplitude was reduced from 3.3 to 1.8 nA (54% of control). On average, QCa and EPSC amplitudes were reduced to 78 ± 7% and 69 ± 14%, respectively (Fig. 7C, n = 3 cell pairs). The amount of block of QCa indicates that ∼20% of all presynaptic Ca2+ channels at this developmental stage are N- and R-type Ca2+ channels, in agreement with previous results (Wu et al. 1999; Iwasaki et al. 2000). In addition, the finding that the EPSC block was only slightly stronger than the block of the Ca2+ charge (Fig. 7C) indicates that there is only a slight supra-linear relationship between Ca2+ entry through R- and N-type channels and release, confirming previous work (Wu et al. 1999).
After the blocking effect of ω-conotoxin and SNX had stabilized (Fig. 7B), we measured paired-pulse facilitation of transmitter release using a double-pulse protocol with varying interstimulus intervals (Fig. 7D). In the cell shown in Fig. 7, the EPSC facilitation was 275 ± 33% at the shortest interval (4 ms), and facilitation decayed with a time constant of 28 ms (Fig. 7D, top; 75 μm EGTA in the presynaptic pipette). Ca2+ current facilitation was 106% of control at the shortest interval (Fig. 7D, bottom), and decayed with similar kinetics as under control conditions (Fig. 3B). On average, Ca2+ current facilitation and transmitter release facilitation were 108 ± 1 and 219 ± 17%, and thus, similar as under control conditions in the same cells (P = 0.3 and 0.5, respectively). Assuming a power relationship with an exponent of 3.5 between the EPSC and the Ca2+ current, Ca2+ current facilitation alone causes a release facilitation of 131%, corresponding to 26% of the total release facilitation. This value is similar, or even lower than our estimate obtained under control conditions (∼45%; see Fig. 3D). Thus, we conclude that the presence of a small (∼20%) component of Ca2+ current mediated by N- and R-type channels in our experiments did not influence our estimate of the contribution of Ca2+ current facilitation to release facilitation.
Release facilitation independent of Ca2+ current facilitation shown in voltage-clamp titration experiments
The experiments and analyses in Figs 3–7 suggest that about one-half of the observed transmitter release facilitation cannot be explained by Ca2+ current facilitation (Figs 5 and 6). We therefore finally aimed to isolate transmitter release facilitation that is independent of Ca2+ current facilitation. To do so, we performed a voltage-clamp ‘titration’ of the Ca2+ charge entering during the second pulse, by applying second pulses with somewhat shorter durations than the first ones, with the aim of compensating for Ca2+ current facilitation (Fig. 8). In each experiment, we first applied two identical presynaptic voltage-clamp stimuli at an interval of 10 ms (Fig. 8A). As expected, this led to a facilitation of the presynaptic Ca2+ current (Fig. 8A, middle), and the EPSC (bottom). In the same cell pair, we then decreased the length of the second depolarization (Fig. 8B, top). In the example shown in Fig. 8B, the first depolarization with a length of 0.8 ms produced a Ca2+ current with a charge of 1.01 pC, and the second step with a length 0.78 ms elicited a Ca2+ current charge comparable to the first one (1.0 pC; Fig. 8B, middle panel). Interestingly, the EPSC still facilitated to 151% of control (Fig. 8B, bottom) despite the fact that the Ca2+ current facilitation was compensated by the shorter second voltage-clamp step.
In Fig. 8C, EPSC facilitation is plotted as a function of the relative Ca2+ current charge during the second pulse for the same cell pair as shown in Fig. 8A and B. In this plot, data resulting from voltage-clamp steps with equal duration are shown as filled black symbols, whereas the data for shorter second steps are shown as open symbols. In this recording, we also applied one second pulse with a longer duration than the first one (Fig. 8C, dark grey open symbol). A subset of the data in which the Ca2+ current facilitation was almost perfectly compensated is shown as grey filled symbols (98–100% of the charge entering during the first pulse; Fig. 8C). In this range of relative Ca2+ entry, EPSC facilitation was still present (∼140% of the control EPSC; Fig. 8C, grey data points). This shows that transmitter release facilitation has a component that is independent of Ca2+ current facilitation. The data in Fig. 8C were fitted with a polynomial function (grey line). For comparison, we also added a simple prediction of release facilitation mediated by Ca2+ current facilitation alone, by taking the 3.5th power of the relative Ca2+ current charge (Fig. 8C, dashed line). The prediction passes through the origin, whereas the data show a nearly constant y-offset for all values of Ca2+ current facilitation. This again shows that there must be an additional facilitation mechanism beyond Ca2+ current facilitation.
To analyse these experiments across various cell-pairs, we compared the facilitation of EPSCs recorded with two identical voltage-clamp steps (i.e. in the presence of Ca2+ current facilitation) with a group of data in which the reduced duration of the second pulse resulted in the absence of net Ca2+ current facilitation (second Ca2+ charge: 98–102% of control). As shown in Fig. 8D (left), the average EPSC facilitation and Ca2+ current facilitation seen with identical stimuli in n = 5 cells was 220 ± 9% of control, and 110 ± 6% of control, respectively (open and filled bars outlined in black, respectively), in good agreement with the results shown in Figs 3 and 7. When the Ca2+ current charge during the second pulse was equal to the one during the first pulse (range, 98–102%; average, 100 ± 0.4%, n = 4 cells), the EPSCs still facilitated to 171 ± 11% of control (Fig. 8D, open and filled bars outlined in grey, right). In good agreement with our analysis (Figs 5 and 6), this experiment shows that a large part of the measured release facilitation (∼60%) can be observed in the absence of net Ca2+ current facilitation, and is therefore independent of Ca2+ current facilitation.
Discussion
Ca2+ current facilitation does not fully account for paired-pulse facilitation
The calyx of Held shows depression under conditions of normal release probability (Borst et al. 1995; von Gersdorff et al. 1997; Wang & Kaczmarek, 1998). However, when the release probability is lowered, depression is converted to paired-pulse facilitation, similar to observations at other synapses (Zucker & Regehr, 2002 and references therein). We have previously reported that with afferent fibre stimulation, when the first EPSC amplitude was reduced to ∼1–2 nA by lowering the extracellular [Ca2+], paired-pulse facilitation was ∼200% of the first EPSC amplitude (see Fig. 3 of Müller et al. 2007). This value of paired-pulse facilitation is very similar to paired pulse facilitation observed here under presynaptic voltage clamp, using conditions of low release probability imposed by sufficiently short presynaptic depolarizations. Specifically, with presynaptic pipette solutions with low Ca2+-buffering strength (fura-6F, with or without 75 μm EGTA), paired-pulse facilitation of transmitter release was in the range of 180–250% (Fig. 4A), for first EPSC amplitudes in the range of 1.3–1.6 nA (Fig. 4D). The similarity of paired-pulse facilitation between patch-clamped calyces of Held and unperturbed calyces of Held indicates that facilitation of transmitter release is not disrupted by presynaptic whole-cell recordings, unlike post-tetanic potentiation (Korogod et al. 2005). Also, the finding that facilitation is similar under presynaptic current clamp (Fig. 1), and under presynaptic voltage clamp using identical voltage-clamp stimuli, indicates that small changes in the presynaptic AP waveform that occur during paired-pulse stimulation do not mediate paired-pulse facilitation of release.
A prerequisite for estimating the contribution of Ca2+ current facilitation to the facilitation of transmitter release is to correctly measure the amount of Ca2+ current facilitation. At an interstimulus interval of 10 ms, we observed an average Ca2+ current facilitation of ∼106% of control using AP waveforms as voltage-clamp commands (Fig. 2), and of ∼110% with brief presynaptic voltage-clamp steps to +28 mV (Figs 3 and 4). This amount of Ca2+ current facilitation agrees well with the Ca2+ current facilitation observed previously in response to two identical AP waveform voltage-clamp commands in rat calyces of Held (Borst & Sakmann, 1998b). In contrast, higher values of Ca2+ current facilitation of ∼120% of control were found by Cuttle et al. (1998) and Tsujimoto et al. (2002) in rat calyces of Held, and by Ishikawa et al. (2005) in mouse calyces of Held. In these latter studies, however, steps to lower voltages of −10 or 0 mV were used. Since Ca2+ current facilitation is mediated by an acceleration of the activation kinetics of Ca2+ channels, which is more predominant at negative membrane potentials (Borst & Sakmann, 1998b; Cuttle et al. 1998), steps to lower voltages are expected to show larger Ca2+ current facilitation than that observed during AP-like waveforms. Thus, the amount of Ca2+ current facilitation relevant for AP-evoked transmitter release is most likely in the order of 106–112% (Figs 2, 3, and 4; and Borst & Sakmann, 1998b).
In order to quantitatively estimate the effect of Ca2+ current facilitation on facilitation of transmitter release, we analysed transmitter release rates with a kinetic model of Ca2+ binding and vesicle fusion (the five-site model) that was developed previously based on the intracellular Ca2+ sensitivity of release found by Ca2+ uncaging (Schneggenburger & Neher, 2000; Felmy et al. 2003; see also Bollmann et al. 2000). The simulations, which model a release machinery with an effective Ca2+ cooperativity of ∼3.5 in the relevant range of [Ca2+]i (Fig. 5B), showed that the measured Ca2+ current facilitation can account for a transmitter release facilitation of only ∼150% of control (Fig. 6A), which corresponds to roughly 40% of the total amount of facilitation. By scaling the ‘local’[Ca2+]i transient with the amount of Ca2+ current facilitation measured in whole-cell recordings, we implicitly assume that Ca2+ channels located at different distances with respect to the readily releasable vesicles exhibit the same amount of Ca2+ current facilitation. We have excluded the possibility that N- and R-type Ca2+ channels, which are thought to couple to release more loosely than P/Q-type channels (Wu et al. 1999), and which do not show Ca2+ current facilitation (Inchauspe et al. 2004; Ishikawa et al. 2005), could have influenced our estimate of the contribution of Ca2+ current facilitation to release facilitation (Fig. 7). Nevertheless, it remains possible that not all P/Q-type channels recorded during a whole-cell recording are located at equal distances from readily releasable vesicles. If, in addition, it is assumed that the degree of Ca2+ current facilitation of P/Q-type Ca2+ channels would depend on their localization close to readily releasable vesicles, maybe mediated by interactions of molecular components of the release machinery with Ca2+ channel subunits, then a misestimate of the impact of Ca2+ current facilitation on transmitter release would result. However, it is unknown whether Ca2+ current facilitation indeed depends on the proximity of Ca2+ channels to the release apparatus. In addition, several independent lines of evidence show that the contribution of Ca2+ current facilitation to transmitter release facilitation is limited. Most importantly, compensating Ca2+ current facilitation during the second pulse clearly showed a substantial amount of release facilitation that remained in the absence of net Ca2+ current facilitation (Fig. 8). In addition, the finding that low concentrations of EGTA significantly speeded up the decay of transmitter release facilitation, without substantially accelerating the decay of Ca2+ current facilitation (Fig. 4) again indicates that Ca2+ current facilitation alone cannot fully explain facilitation of transmitter release.
The mechanism of Ca2+ current-independent facilitation
What is the mechanism that causes facilitation of transmitter release that is independent of Ca2+ current facilitation? When we modelled the combined effect of Ca2+ current facilitation, and of an assumed linear summation of the measured residual [Ca2+]i and the derived ‘local’[Ca2+]i, we could still only predict about half of the observed transmitter release facilitation (Fig. 6A, filled red symbols). Thus, a linear addition of residual free [Ca2+]i to the local [Ca2+]i only accounted for a small fraction of the observed facilitation (∼10%; Figs 5 and 6). This is because the increment in residual free [Ca2+]i (∼250 nm under our conditions of reduced release probability; Fig. 4) is relatively small compared to the peak ‘local’[Ca2+]i (∼12 μm). This confirms the generally held view that a direct effect of residual [Ca2+]i on the Ca2+ sensor for vesicle fusion is small, because of the large difference between the amplitudes of the local and the residual [Ca2+]i signal (Zucker & Regehr, 2002).
In addition to Ca2+ current facilitation and a linear summation of free [Ca2+]i signals, there are three widely discussed facilitation mechanisms that could mediate Ca2+ current-independent facilitation (see Introduction). The ‘bound Ca2+ model’ assumes that residual Ca2+ remains bound to a presumed high-affinity site that forms part of the Ca2+ sensor for vesicle fusion (Bertram et al. 1996; Matveev et al. 2006). However, this model predicts that the intracellular Ca2+ sensitivity of release is enhanced during facilitation (Bertram et al. 1996); a prediction that was not confirmed experimentally at the calyx of Held (Felmy et al. 2003). Based on our results, it is more difficult to distinguish between the ‘facilitation site model’ (Tang et al. 2000; Matveev et al. 2002), and Ca2+-buffer saturation (Blatow et al. 2003; Felmy et al. 2003) as an explanation for the Ca2+ current-independent facilitation. An argument against Ca2+-buffer saturation at the calyx of Held is that small concentrations of a high-affinity Ca2+ buffer with fast binding kinetics (fura-2, a BAPTA-like buffer) does not cause, but rather suppresses paired-pulse facilitation (Fig. 4). At the same time, small amounts of fura-2 strongly decrease the amplitude of the residual free [Ca2+]i transient (Helmchen et al. 1997; Müller et al. 2007). This, together with the finding that paired-pulse facilitation is correlated with the decay of residual [Ca2+]i (Fig. 4; Atluri & Regehr, 1996; Müller et al. 2007), shows that facilitation depends on the residual free[Ca2+]i. This, at first hand, seems to be more compatible with the facilitation site model, which assumes that the facilitation site is in rapid equilibrium with the fast decaying residual free [Ca2+]i transient, but it does not exclude a Ca2+-buffer saturation mechanism.
Evidence for a fast-binding, saturable Ca2+ buffer as postulated by the Ca2+-buffer saturation model (Neher, 1998; Matveev et al. 2004) has been scarce at calyces of Held (Habets & Borst, 2006; Müller et al. 2007). At the young stages of development investigated here (P8–P10), calyces contain an endogenous mobile Ca2+ buffer with slow Ca2+-binding kinetics, probably represented by parvalbumin (Müller et al. 2007). Calretinin, a candidate for a fast-binding Ca2+ buffer (Edmonds et al. 2000) is only expressed from about P14 onwards (Felmy & Schneggenburger, 2004), and it is difficult to study its functional role at the calyx of Held, because in mice, only a small fraction of all calyces of Held express calretinin (Felmy & Schneggenburger, 2004). In addition, a recent in vitro study has ascribed only moderately fast average Ca2+ binding rates to calretinin (Faas et al. 2007). An alternative to the action of mobile Ca2+ buffers might be the possibility that saturation of an immobile Ca2+ buffer located close to the sites of vesicle fusion contributes to the postulated supra-linearity in the summation of [Ca2+]i signals (Fig. 6). Such a possibility, however, needs to be explored by detailed modelling (see also Matveev et al. 2004). Taken together, both a facilitation site, as well as Ca2+-buffer saturation could contribute to facilitation of transmitter release that is independent of Ca2+ current facilitation.
Physiological implications
What might be the physiological role of short-term facilitation in a synapse like the calyx of Held that shows net depression of EPSCs under most conditions of repetitive activity? In general, the presence of facilitation mechanisms could transiently oppose the effect of synaptic depression, before mechanisms like depletion of readily releasable vesicle pools, or postsynaptic desensitization become overwhelming. Indeed, facilitation of EPSCs has been observed at the onset of high-frequency trains at calyx of Held synapses with a relatively small first EPSC amplitude (Schneggenburger et al. 1999), or when AMPA receptor desensitization was prevented with rapid off-antagonists (Wong et al. 2003), showing that facilitation also acts during conditions of normal release probability. Since the decay of facilitation is fast, tracking the rapid decay of residual free [Ca2+]i (τ∼30–50 ms under unperturbed Ca2+ buffering conditions; Müller et al. 2007), stimulus trains at frequencies lower than ∼30 Hz are expected to spare the facilitation mechanisms. However, high frequency trains should lead to facilitation of the release probability with ongoing stimulation in parallel to net depression of release, as has been suggested recently by a quantitative analysis of pool depletion (Hosoi et al. 2007). Thus, it is possible that facilitation of the release probability of any given vesicle remaining in the readily releasable pool contributes to determining the steady-state EPSC amplitude under conditions of high-frequency stimulation. This possibility needs to be addressed in future studies.
Acknowledgments
We thank Erwin Neher for critical comments on a previous version of this paper. This work was supported by the Deutsche Forschungsgemeinschaft (SFB-406), and by the Swiss National Science Foundation (3100A0-114069).
References
- Abbott LF, Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram R, Sherman A, Stanley EF. Single-domain/bound calcium hypothesis of transmitter release and facilitation. J Neurophysiol. 1996;75:1919–1931. doi: 10.1152/jn.1996.75.5.1919. [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Geiger JR, Jonas P. Timing and efficacy of Ca2+ channel activation in hippocampal mossy fiber boutons. J Neurosci. 2002;22:10593–10602. doi: 10.1523/JNEUROSCI.22-24-10593.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatow M, Caputi A, Burnashev N, Monyer H, Rozov A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron. 2003;38:79–88. doi: 10.1016/s0896-6273(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B, Borst JG. Calcium sensitivity of glutamate release in a calyx-type terminal. Science. 2000;289:953–957. doi: 10.1126/science.289.5481.953. [DOI] [PubMed] [Google Scholar]
- Borst JG, Helmchen F, Sakmann B. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J Physiol. 1995;489:825–840. doi: 10.1113/jphysiol.1995.sp021095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Calcium current during a single action potential in a large presynaptic terminal of the rat brainstem. J Physiol. 1998a;506:143–157. doi: 10.1111/j.1469-7793.1998.143bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. J Physiol. 1998b;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton MP, Smith SJ, Zucker RS. Role of presynaptic calcium ions and channels in synaptic facilitation and depression at the squid giant synapse. J Physiol. 1982;323:173–193. doi: 10.1113/jphysiol.1982.sp014067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol. 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Edmonds B, Reyes R, Schwaller B, Roberts WM. Calretinin modifies presynaptic calcium signaling in frog saccular hair cells. Nat Neurosci. 2000;3:786–790. doi: 10.1038/77687. [DOI] [PubMed] [Google Scholar]
- Faas GC, Schwaller B, Vergara JL, Mody I. Resolving the fast kinetics of cooperative binding: Ca2+ buffering by calretinin. PLoS Biol. 2007;5:e311. doi: 10.1371/journal.pbio.0050311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY. Developmental transformation of the release modality at the calyx of Held synapse. J Neurosci. 2005;25:4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmy F, Neher E, Schneggenburger R. Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron. 2003;37:801–811. doi: 10.1016/s0896-6273(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Felmy F, Schneggenburger R. Developmental expression of the Ca2+-binding proteins calretinin and parvalbumin at the calyx of Held of rats and mice. Eur J Neurosci. 2004;20:1473–1482. doi: 10.1111/j.1460-9568.2004.03604.x. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Habets RLP, Borst JGG. An increase in calcium influx contributes to post-tetanic potentiation at the rat calyx of Held synapse. J Neurophysiol. 2006;96:2868–2876. doi: 10.1152/jn.00427.2006. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Borst JG, Sakmann B. Calcium dynamics associated with a single action potential in a CNS presynaptic terminal. Biophys J. 1997;72:1458–1471. doi: 10.1016/S0006-3495(97)78792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi N, Sakaba T, Neher E. Quantitative analysis of calcium-dependent vesicle recruitment and its functional role at the calyx of Held synapse. J Neurosci. 2007;27:14286–14298. doi: 10.1523/JNEUROSCI.4122-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inchauspe CG, Forsythe ID, Uchitel OD. Changes in synaptic transmission properties due to the expression of N-type calcium channels at the calyx of Held synapse of mice lacking P/Q-type calcium channels. J Physiol. 2007;584:835–851. doi: 10.1113/jphysiol.2007.139683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inchauspe CG, Martini FJ, Forsythe ID, Uchitel OD. Functional compensation of P/Q by N-type channels blocks short-term plasticity at the calyx of Held presynaptic terminal. J Neurosci. 2004;24:10379–10383. doi: 10.1523/JNEUROSCI.2104-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Kaneko M, Shin HS, Takahashi T. Presynaptic N-type and P/Q-type Ca2+ channels mediating synaptic transmission at the calyx of Held of mice. J Physiol. 2005;568:199–209. doi: 10.1113/jphysiol.2005.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Momiyama A, Uchitel OD, Takahashi T. Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci. 2000;20:59–65. doi: 10.1523/JNEUROSCI.20-01-00059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB, Konnerth A, Augustine GJ. Action potential broadening and frequency-dependent facilitation of calcium signals in pituitary nerve terminals. Proc Natl Acad Sci U S A. 1991;88:380–384. doi: 10.1073/pnas.88.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of Held. J Neurosci. 2005;25:5127–5137. doi: 10.1523/JNEUROSCI.1295-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Schwaller B, Neher E. Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells: implications for [Ca2+] transients of neuronal dendrites. J Physiol. 2000;525:419–432. doi: 10.1111/j.1469-7793.2000.t01-2-00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Scheuss V, Schneggenburger R. Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature. 2005;435:497–501. doi: 10.1038/nature03568. [DOI] [PubMed] [Google Scholar]
- Maeda H, Ellis-Davies GC, Ito K, Miyashita Y, Kasai H. Supralinear Ca2+ signaling by cooperative and mobile Ca2+ buffering in Purkinje neurons. Neuron. 1999;24:989–1002. doi: 10.1016/s0896-6273(00)81045-4. [DOI] [PubMed] [Google Scholar]
- Matveev V, Bertram R, Sherman A. Residual bound Ca2+ can account for the effects of Ca2+ buffers on synaptic facilitation. J Neurophysiol. 2006;96:3389–3397. doi: 10.1152/jn.00101.2006. [DOI] [PubMed] [Google Scholar]
- Matveev V, Sherman A, Zucker RS. New and corrected simulations of synaptic facilitation. Biophys J. 2002;83:1368–1373. doi: 10.1016/S0006-3495(02)73907-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveev V, Zucker RS, Sherman A. Facilitation through buffer saturation: constraints on endogenous buffering properties. Biophys J. 2004;86:2691–2709. doi: 10.1016/S0006-3495(04)74324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AC, Neher E, Schneggenburger R. Estimation of quantal size and number of functional active zones at the calyx of Held synapse by nonstationary EPSC variance analysis. J Neurosci. 2001;21:7889–7900. doi: 10.1523/JNEUROSCI.21-20-07889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Few AP, Scheuer T, Catterall WA. Regulation of presynaptic Cav2.1 channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron. 2008;57:210–216. doi: 10.1016/j.neuron.2007.11.036. [DOI] [PubMed] [Google Scholar]
- Müller M, Felmy F, Schwaller B, Schneggenburger R. Parvalbumin is a mobile presynaptic Ca2+ buffer in the calyx of Held that accelerates the decay of Ca2+ and short-term facilitation. J Neurosci. 2007;27:2261–2271. doi: 10.1523/JNEUROSCI.5582-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Usefulness and limitations of linear approximations to the understanding of Ca2+ signals. Cell Calcium. 1998;24:345–357. doi: 10.1016/s0143-4160(98)90058-6. [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Combining deconvolution and noise analysis for the estimation of transmitter release rates at the calyx of Held. J Neurosci. 2001;21:444–461. doi: 10.1523/JNEUROSCI.21-02-00444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol. 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Tang Y, Schlumpberger T, Kim T, Lueker M, Zucker RS. Effects of mobile buffers on facilitation: experimental and computational studies. Biophys J. 2000;78:2735–2751. doi: 10.1016/s0006-3495(00)76819-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto T, Jeromin A, Saitoh N, Roder JC, Takahashi T. Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science. 2002;295:2276–2279. doi: 10.1126/science.1068278. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E. Presynaptic depression at a calyx synapse: The small contribution of metabotropic glutamate receptors. J Neurosci. 1997;17:8137–8146. doi: 10.1523/JNEUROSCI.17-21-08137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L-Y, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wong AYC, Graham BP, Billups B, Forsythe ID. Distinguishing between presynaptic and postsynaptic mechanisms of short-term depression during action potential trains. J Neurosci. 2003;23:4868–4877. doi: 10.1523/JNEUROSCI.23-12-04868.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JGG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, He L, Wu LG. Role of Ca2+ channels in short-term synaptic plasticity. Curr Opin Neurobiol. 2007;17:352–359. doi: 10.1016/j.conb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Xu J, Wu LG. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Yamada WM, Zucker RS. Time course of transmitter release calculated from simulations of a calcium diffusion model. Biophys J. 1992;61:671–682. doi: 10.1016/S0006-3495(92)81872-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]