

Abstract
The activation of AMP-activated protein kinase (AMPK) and phosphorylation/inhibition of acetyl-CoA carboxylase 2 (ACC2) is believed to be the principal pathway regulating fatty acid oxidation. However, during exercise AMPK activity and ACC Ser-221 phosphorylation does not always correlate with rates of fatty acid oxidation. To address this issue we have investigated the requirement for skeletal muscle AMPK in controlling aminoimidazole-4-carboxymide-1-β-d-ribofuranoside (AICAR) and contraction-stimulated fatty acid oxidation utilizing transgenic mice expressing a muscle-specific kinase dead (KD) AMPK α2. In wild-type (WT) mice, AICAR and contraction increased AMPK α2 and α1 activities, the phosphorylation of ACC2 and rates of fatty acid oxidation while tending to reduce malonyl-CoA levels. Despite no activation of AMPK in KD mice, ACC2 phosphorylation was maintained, malonyl-CoA levels were reduced and rates of fatty acid oxidation were comparable between genotypes. During treadmill exercise both KD and WT mice had similar values of respiratory exchange ratio. These studies suggested the presence of an alternative ACC2 kinase(s). Using a phosphoproteomics-based approach we identified 18 Ser/Thr protein kinases whose phosphorylation was increased by greater than 25% in contracted KD relative to WT muscle. Utilizing bioinformatics we predicted that extracellular regulated protein-serine kinase (ERK1/2), inhibitor of nuclear factor (NF)-κB protein-serine kinase β (IKKβ) and protein kinase D (PKD) may phosphorylate ACC2 at Ser-221 but during in vitro phosphorylation assays only AMPK phosphorylated ACC2. These data demonstrate that AMPK is not essential for the regulation of fatty acid oxidation by AICAR or muscle contraction.
Skeletal muscle is a dynamic tissue that preferentially utilizes fatty acids as a fuel source during postprandial conditions. Defects in skeletal muscle fatty acid oxidation contribute to the pathogenesis of insulin resistance and obesity (Savage et al. 2007), therefore an understanding of the signalling pathways mediating fatty acid oxidation may yield therapeutic targets for the treatment of insulin resistance and associated disorders. The AMP-activated protein kinase (AMPK) is thought to regulate fatty acid oxidation in response to energy demand, nutrients and hormones by directly phosphorylating the muscle-specific isoform of acetyl-CoA carboxylase-2 (ACC2) on Ser-221 (corresponding to Ser-79 in ACC1) (for review see Steinberg et al. 2006a). ACC2 resides on the mitochondrial membrane catalysing the carboxylation of acetyl CoA to malonyl CoA, an allosteric inhibitor of the mitochondrial long-chain fatty-acyl CoA shuttle, carnitine palmitoyltransferase-1 (CPT-1) (Abu-Elheiga et al. 2000). Phosphorylation of ACC reduces enzyme activity (Carling et al. 1987) causing a reduction in malonyl CoA levels, a release of the malonyl CoA-mediated inhibition of CPT-1, and an increase in fatty acid β-oxidation by mitochondria (McGarry et al. 1978). The importance of ACC2 in the regulation of skeletal muscle fatty acid metabolism is evident as mice lacking ACC2 have substantially enhanced rates of fatty acid oxidation, reductions in malonyl-CoA and remain lean despite hyperphagia (Abu-Elheiga et al. 2001). In both rodents and humans the activation of AMPK by aminoimidazole-4-carboxymide-1-β-d-ribofuranoside (AICAR) (Merrill et al. 1997; Koistinen et al. 2003; Steinberg et al. 2004), or muscle contractions (Vavvas et al. 1997; Winder et al. 1997; Chen et al. 2000; Fujii et al. 2000; Wojtaszewski et al. 2000) results in increased phosphorylation of ACC and reductions in malonyl-CoA. With obesity reduced skeletal muscle AMPK activity and ACC phosphorylation accompanied by increased levels of malonyl-CoA may contribute to the accumulation of intramuscular lipids (Bandyopadhyay et al. 2006; Steinberg et al. 2006b). However, it should be noted that reductions in AMPK signalling are not always observed in moderately obese humans (Steinberg et al. 2004) or in animals fed a high-fat diet (Martin et al. 2006; Watt et al. 2006a).
Despite the significant correlative evidence for a role of AMPK in regulating ACC2 phosphorylation/activity and fatty acid oxidation, direct genetic evidence supporting the role of AMPK in contraction-stimulated fatty acid oxidation has not been demonstrated. Indeed, several recent studies in both rodents and humans suggest that contraction-driven fatty acid oxidation is quite resilient to changes in AMPK signalling. One example is observed in LKB1-deficient mice who, despite no activation of AMPK α2 during muscle contraction, are able to maintain contraction-stimulated phosphorylation of ACC (Koh et al. 2006; Thomson et al. 2007b) and the ability to suppress malonyl-CoA (Thomson et al. 2007a) although it should be noted that this has not been observed in all studies (Sakamoto et al. 2005). Similarly, in mice with a deletion of the α2 (Jorgensen et al. 2003) or γ3 (Barnes et al. 2004) subunits, contraction-stimulated ACC phosphorylation is normal despite considerable reductions in AMPK phosphorylation in white muscle. It has also been reported that low intensity contraction can increase fatty acid oxidation independent of increased AMPK activity (Raney et al. 2005) and that AICAR and contraction may have additive effects on fatty acid oxidation (Smith et al. 2005). In humans, there are also several examples in which there is a dichotomy between AMPK activity, ACC phosphorylation and rates of fatty acid oxidation such as exercise training (McConell et al. 2005), high-intensity exercise (Chen et al. 2000) or exercise in women (Roepstorff et al. 2006). Similarly, intracellular concentrations of malonyl-CoA do not correlate with rates of fatty acid oxidation (Odland et al. 1996,1998; Roepstorff et al. 2005). The aim of this study was to determine the requirement of AMPK in contraction-stimulated fatty acid oxidation using mice over-expressing a muscle-specific kinase dead (KD) form of AMPK α2, and to identify possible alternative signalling pathways regulating skeletal muscle fatty acid oxidation during muscle contraction.
Methods
Ethical approval
All procedures were carried out and supervised in accordance with the St Vincent's Health Animal Ethics Committee as outlined by the Bureau of Animal Welfare (Victoria, Australia). For ex vivo and in situ contraction experiments mice were anaesthetized with sodium pentobarbital (6 mg (100 g body wt)−1) delivered i.p. before muscles were removed as described below. Mice were then killed by cervical dislocation. For treadmill running experiments mice were killed by cervical dislocation before the removal of muscles for analysis. For metabolic treadmill running experiments mice were placed back in their cage at the completion of the exercise bout. Ninety-five mice were used for these experiments.
Animals
Transgenic mice over-expressing a kinase dead (KD) form of the AMPK α2 protein (AMPK KD) under the muscle creatine kinase promoter have been previously described (Mu et al. 2001). AMPK (KD) and littermate controls (WT) were used for experiments at 8–12 weeks of age. Mice were fed a standard rodent chow ad libitum and maintained on a 12 h light/12 h dark cycle with lights on at 07.00 h.
Ex vivo determination of fatty acid oxidation and AMPK signalling in isolated EDL and SOL muscle
Extensor digitorum longus (EDL) and soleus (SOL) muscles were carefully dissected tendon to tendon for muscle incubations as described (Steinberg et al. 2002). Fatty acid metabolism experiments were conducted using procedures previously described (Steinberg & Dyck, 2000; Steinberg et al. 2002). For all experiments, isolated EDL and SOL muscles were placed in warmed (30°C) Krebs-Henseleit buffer pH 7.4 containing 2 mm pyruvate, 4% fatty acid-free bovine serum albumin (Bovogen, VIC, Australia) and 1 mm palmitic acid (Sigma, St Louis, MO, USA). For resting experiments with or without AICAR (Sigma) EDL and SOL muscle incubation volume was 2 ml while for contraction experiments, the proximal and distal tendons of isolated EDL and SOL muscles were tied with silk suture and mounted to a force transducer in a 15 ml incubation reservoir (Radnoti, Monrovia, CA, USA). After an initial incubation of 20 min, the incubation buffer was replaced with the same buffer described above supplemented with 0.5 μCi ml−1 of [1-14C]palmitate (Amersham BioSciences, Little Chalfont, UK). For resting experiments fatty acid metabolism was determined over 90 min in the presence or absence of 2 mm AICAR. In contraction experiments fatty acid metabolism was measured in fused tetani of EDL (50 Hz, 350 ms pulse duration, 6 tetani min−1) and SOL (30 Hz, 600 ms pulse duration, 18 tetani min−1) over 25 min. This contraction protocol was selected as it has previously been demonstrated to maximally stimulate fatty acid metabolism in isolated muscles (Dyck & Bonen, 1998). Force was calculated from the integral of force × time for one contraction at a given time point using Powerlab software (ADInstruments, Colorado Springs, CO, USA). In separate experiments to obtain muscles for AMPK signalling the identical protocol was employed but in the absence of radiolabelled [1-14C]palmitate. At the completion of the contraction protocol muscles were removed and snap-frozen in liquid nitrogen and stored at −80°C.
In situ muscle contractions
AMPK signalling was also determined in tibialis anterior (TA) muscles contracted in situ as previously described (Schertzer & Lynch, 2006). Briefly, the TA muscle was exposed by a single incision and the tendon tied securely to the lever arm of a dual-mode servomotor (Aurora Scientific, Richmond Hill, Ontario, Canada) and the foot was immobilized. The TA muscle was stimulated over 20 min by 10 V 0.2 ms square-wave pulses for 300 ms via two electrodes adjacent to the femoral nerve. A sham operation was performed on the contralateral leg which served as the resting control muscle. Following the contraction period both muscles were freeze clamped in liquid nitrogen and stored at −80°C.
Treadmill running
Prior to the exercise experiment, all mice were acclimatized to treadmill running at days −3 and −2 by 10 min at rest in the treadmill apparatus (Columbus Instruments International Exer4, USA) followed by running at a 10% slope for 5 min at 10 m min−1 and 1 min at 15 m min−1 as previously described (Jorgensen et al. 2005). On the experimental day, mice were randomly divided into a non-exercised basal group and a treadmill-exercised group which ran at a 10% slope for 10 min at 10 m min−1 followed by 15 m min−1 for 50 min. Muscles were quickly dissected and freeze clamped.
For determination of metabolic rate/fuel selection during treadmill exercise, mice were exercised at the same intensity as described above (10% slope for 10 min at 10 m min−1 followed by 15 m min−1 for 50 min). Expired gases were collected after steady state was reached between 15 and 20 min or 45 and 50 min using a Columbus Instruments Laboratory Animal Monitoring System (CLAMS) and sealed treadmill according to the manufacturer's recommendations.
Immunoblotting and AMPK activity
Protein lysates were prepared as described (Chen et al. 2000). Primary antibodies to detect AMPK α1, α2, total AMPK α, phosphorylation of AMPK at Thr-172 and phosphorylation of ACC2 on Ser-221 were used as previously described (Chen et al. 2000).
Liquid chromatography/mass spectrometry (LC/MS) analysis of malonyl CoA
Malonyl CoA was extracted from ∼20 mg of frozen TA muscle which was ground and powdered in liquid nitrogen using a mortar and pestle in 200 μl of 10% trichloroacetic acid. Each sample was supplemented with 100 pmol of propionyl CoA (Sigma) and prepared for analysis by solid phase extraction using 60 mg Oasis HLB cartridges (Waters). Standards containing 100 pmol proponyl CoA and 5–50 pmol malonyl CoA (Sigma) were prepared in an identical fashion. Malonyl CoA was eluted from the cartridges with 96% MeOH–4% NH4 and frozen in liquid nitrogen before being concentrated by freeze drying. Dried samples were stored at −80°C until they were resuspended in 20 mm ammonium formate shortly before LC/MS analysis. Malonyl CoA and propionyl CoA were injected onto a 1 mm × 150 mm Acclaim Pepmap C18 column (LC Packings) equilibrated with 20 mm ammonium formate at 50 μl min−1 and resolved on a 0–30% MeOH gradient over 8 min. Mass spectrometry was performed with a Turboionspray source at 5000 V and 500°C attached to QSTAR Pulsar-i mass spectrometer (Applied Biosystems). Malonyl CoA and proponyl CoA were detected using an MRM strategy: malonyl CoA q1 = 854.2 q3 = 347.1, proponyl CoA q1 = 824.1 q2 = 317.1 as previously described (Gao et al. 2007). Mass spectrometry data was manually integrated from the standard curve (malonyl CoApeak/proponyl CoApeak+ 0.0024)/0.004, R = 0.9986).
Kinexus antibody array
Tibialis anterior muscles from WT and AMPK KD mice were contracted in situ for 20 min as described above and prepared and analysed according to the methods of Kinexus Bioinformatic Incorporation (Vancouver, BC, Canada). The antibody-array utilized 350 pan antibodies detecting 190 protein kinases and 140 other proteins.
In vitro phosphorylation assays
Phosphorylation of recombinant baculovirus-expressed human ACC2 with a deletion of the first 20 amino acids (incorporating the mitochondrial membrane insertion region) was performed in kinase assay buffer (50 mm Hepes pH 7.5, 10 mm MgCl2, 5% glycerol, 0.05% Triton X-100) containing 1 mm DTT, 100 μm[γ32-P]ATP (10 000 c.p.m. pmol−1) (Amersham Biosciences, Little Chalfont, UK) and 50 μm AMP. Phosphorylation was initiated by addition of recombinant AMPK αβγ produced by baculovirus-driven expression and phosphorylated by CaMKKβ as previously described (Iseli et al. 2008) or by addition of recombinant IKKβ, ERK and PKD (Invitrogen, San Diego, CA, USA). The activity of all kinases was confirmed by examining the phosphorylation of myelin basic protein (AMPK, ERK and PKD), or in the case of IKKβ, autophosphorylation (data not shown). Samples were incubated at 30°C for 60 min with constant mixing. Phosphorylation was terminated by the addition of SDS sample buffer (67 mm Tris-HCl pH 6.8, 2% SDS, 2 mm EGTA, 0.07% bromophenol blue) followed by the heating of samples at 90°C for 5 min. Proteins were separated by SDS-PAGE and gels stained with Coomassie blue (Sigma) for 1 h before overnight de-staining (7% acetic acid, 12.5% ethanol) and autoradiography. Duplicate SDS-PAGE gels were used for ACC2 immunoblots.
Statistical analysis
All data are reported as mean ± standard error of the mean (s.e.m.). Results were analysed using Student's t test, paired t test or analysis of variance (ANOVA) procedures where appropriate using Graphpad Prism software. Tukey's post hoc test was used to test for significant differences revealed by ANOVA. Significance was accepted at P≤ 0.05. The quantity of palmitate oxidized was calculated using the specific activity of labelled palmitate in the incubation medium. Kinexus antibody microarray data were calculated as the difference in fold-change between resting and contracted muscles from WT relative to AMPK KD littermates.
Results
ACC phosphorylation is maintained with muscle contraction and AICAR in AMPK KD mice ex vivo
We investigated the importance of AMPK in the regulation of skeletal muscle fatty acid oxidation using transgenic muscle-specific AMPK α2 kinase dead (AMPK KD) mice (kindly provided by Professor Morris Birnbaum, University of Pennsylvania) and their wild-type (WT) littermates (Mu et al. 2001). In initial experiments glycolytic (extensor digitorum longus, EDL) and oxidative (soleus, SOL) muscles were removed and incubated ex vivo under basal/resting conditions, in the presence of AICAR (2 mm) or during electrically induced muscle contractions. Both AICAR and muscle contractions increased AMPK α2 activity in EDL and SOL muscles from wild-type mice (Fig. 1A). AMPK α1 activity was only increased with contraction in EDL muscle from wild-type animals (Fig. 1B). AMPK α2 activity was nearly eliminated and AMPK α1 was substantially suppressed at rest in AMPK KD SOL and EDL muscles, an effect that may have been due to the displacement of endogenous α1 from the βγ heterotrimer as has previously been suggested (Mu et al. 2001). Importantly neither AMPK α1 or α2 activities were increased in AMPK KD mice by AICAR or muscle contraction in either muscle type (Fig. 1A and B). These data indicate that AMPK activity is markedly suppressed and is not increased in AMPK KD mice in response to AICAR or muscle contractions.
Figure 1. AMPK activity, and the phosphorylation of ACC2 Ser-221 (S221) and HSL Ser-565 (S565) in isolated EDL and SOL muscle.

AMPK α2 activity (A) and AMPK α1 activity (B) in isolated EDL and SOL from WT and KD mice at rest, incubated with 2 mm AICAR, or contracted as described in Methods. C, representative Western blots and quantification of ACC S221 phosphorylation relative to total ACC. D, representative Western blots and quantification of HSL S565 phosphorylation relative to total HSL. All data are mean ±s.e.m., n = 9–12. *P < 0.05 versus WT rest, **P < 0.01 versus WT rest, ***P < 0.001 versus WT rest, #P < 0.05 versus KD rest, ##P < 0.01 versus KD rest, ###P < 0.001 versus KD rest, +P < 0.05 versus corresponding WT group, ++P < 0.01 versus corresponding WT group, +++P < 0.001 versus corresponding WT group.
We then examined the phosphorylation of ACC2 and hormone-sensitive lipase (HSL). Hormone-sensitive lipase is a substrate of AMPK that regulates diacylglycerol hydrolysis, and is phosphorylated at Ser-565 with AICAR and muscle contractions (Roepstorff et al. 2004; Watt et al. 2005). Under basal conditions the phosphorylation of ACC was lower in both AMPK KD EDL and SOL muscles (P < 0.05) (Fig. 1C). Both AICAR and contraction increased the phosphorylation of ACC to a similar degree in muscle from WT and AMPK KD littermates although the absolute degree of phosphorylation was modestly reduced in AMPK KD mice (Fig. 1C). In contrast to the effects on ACC phosphorylation, AICAR stimulated HSL Ser-565 phosphorylation in WT but not AMPK KD muscle. However, muscle contraction increased HSL Ser-565 phosphorylation to a similar extent in both genotypes (Fig. 1D).
AICAR and muscle contraction increase fatty acid oxidation in AMPK KD mice ex vivo
Increases in ACC phosphorylation by AMPK are reported to regulate fatty acid oxidation by reducing malonyl-CoA content thereby relieving inhibition of CPT-1 and enhancing the flux of fatty acyl-CoA into the mitochondria (Merrill et al. 1997). We therefore measured fatty acid oxidation in resting and AICAR-treated SOL and EDL using the same procedure as used for the examination of AMPK signalling. Consistent with previous reports, SOL muscle had higher rates of fatty acid oxidation than EDL under resting conditions (Fig. 2A) (Dyck et al. 1997); however, rates of fatty acid oxidation were not different between WT and KD littermates in either muscle type at rest. AICAR stimulated fatty acid oxidation in both WT and KD EDL but not SOL muscle (Fig. 2A). We hypothesized that since high levels of fatty acids increase oxidation through allosteric activation of AMPK (Watt et al. 2006b), that the effect of AICAR may be masked in SOL muscles due to higher rates of fatty acid uptake relative to EDL (Bonen et al. 1998). Therefore, we incubated SOL muscles with a lower concentration of fatty acid (0.25 mm palmitate) and despite lower absolute rates of fatty acid oxidation no increase in fatty acid oxidation was detected in SOL with AICAR (see Fig. 1 in online Supplemental material). During muscle contraction fatty acid oxidation was not different between wild-type and AMPK KD muscles (Fig. 2). There was no difference in force production between wild-type and AMPK KD EDL and SOL muscles (Supplemental Fig. 2A and B). In order to ensure that our assay system was sensitive enough to detect reductions in fatty acid oxidation during muscle contraction, isolated EDL and SOL muscles from WT mice were incubated in the presence of 50 μm etomoxir, an irreversible CPT-1 inhibitor. At rest, etomoxir had no significant effect on fatty acid oxidation; however, when etomoxir was included during muscle contraction fatty acid oxidation was reduced by ∼50 and ∼90% in EDL and SOL muscle, respectively (Fig. 2B) indicating the critical role of CPT-1 in the regulation of contraction-stimulated fatty acid oxidation.
Figure 2. Fatty acid oxidation is maintained in AMPK KD muscle at rest and with muscle contraction but is impaired in WT mice with etomoxir.

A, rates of palmitate oxidation in isolated EDL and SOL from WT and KD mice at rest, incubated with 2 mm AICAR, or contracted as described in Methods. All data are mean ±s.e.m., n = 9–12. B, isolated EDL and SOL muscles from WT mice were incubated at rest or contracted in the presence or absence of 50 μm etomoxir as described in Methods. Data are mean ±s.e.m., n = 4. *P < 0.05 versus WT rest, **P < 0.01 versus WT rest, ***P < 0.001 versus WT rest, #P < 0.05 versus KD rest, ##P < 0.01 versus KD rest, ###P < 0.001 versus KD rest, +P < 0.05 versus corresponding WT group, ++P < 0.01 versus corresponding WT group, +++P < 0.001 versus corresponding WT group.
In situ muscle contraction increases ACC phosphorylation and reduces muscle malonyl-CoA
In previous experiments EDL and SOL muscle were contracted ex vivo therefore we wanted to examine whether the maintenance of ACC phosphorylation in the absence of AMPK activation was also observed during muscle contractions performed in situ. In situ muscle contraction experiments were conducted by exposing the sciatic nerve in both hindlimbs and stimulating one tibialis anterior (TA) muscle over a 20 min time period while the TA muscle of the contralateral limb served as a resting control. Over the contraction period fatigue of the TA muscles was comparable between genotypes (Supplemental Fig. 3). AMPK α2 activity was significantly lower in the TA muscles from AMPK KD mice at rest but was not elevated with muscle contractions as observed in WT littermates (Fig. 3A). Contraction did not increase AMPK α1 activity in either wild-type or AMPK KD muscles (Fig. 3B). However, during contraction, increases in ACC Ser-221 phosphorylation in AMPK KD TA were similar to increases observed in WT mice (Fig. 3C).
Figure 3. AMPK signalling and malonyl-CoA during in situ muscle contraction.

AMPK α2 (A) and α1 (B) activity in TA muscle from WT and KD mice at rest, or contracted in situ as described in Methods. C, representative Western blots and quantification of ACC S221 phosphorylation relative to total ACC from TA muscle. D, malonyl CoA levels in TA muscle from WT and KD mice at rest, or contracted in situ as described in Methods. All data are mean ±s.e.m., n = 8. *P < 0.05 versus rest, **P < 0.01 versus rest, +P < 0.05 versus corresponding WT group, +++P < 0.001 versus corresponding WT group.
The phosphorylation of ACC2 at Ser-221 inhibits its activity (Davies et al. 1990) leading to reductions in malonyl-CoA which is a potent inhibitor of CPT-1 (McGarry et al. 1978). Surprisingly, muscle malonyl-CoA levels were lower in AMPK KD mice than in wild-type littermates under resting conditions (P < 0.05). With muscle contraction the malonyl-CoA content tended to be reduced by ∼25% in both wild-type (P = 0.07) and KD mice (P = 0.08) (Fig. 3D). These findings support studies suggesting a critical role for malonyl-CoA in the regulation of fatty acid oxidation (Winder & Hardie, 1996; Ruderman et al. 1999) and are consistent with the maintenance of ACC2 Ser-221 phosphorylation in AMPK KD mice during muscle contraction.
Fatty acid oxidation is maintained during treadmill exercise in AMPK KD mice
We examined ACC phosphorylation and fatty acid oxidation in muscle from AMPK KD mice during treadmill running. Pilot experiments were conducted to ensure that WT and AMPK KD mice were capable of performing equal amounts of work since it has been reported that AMPK KD mice have reduced exercise tolerance/capacity (Mu et al. 2003). Both wild-type and KD littermates either rested in a cage beside the treadmill or ran for 50 min at 15 m min−1 and 10% gradient. AMPK α2 activity was increased in gastrocnemius muscle with treadmill running relative to resting wild-type controls, an effect that was not observed in gastrocnemius muscle from AMPK KD mice (Fig. 4A). AMPK α1 activity was not different between genotypes or with exercise (Fig. 4B). The increase in AMPK α2 in treadmill-exercised wild-type mice was relatively modest and did not lead to increases in ACC phosphorylation (Fig. 4C) probably due to the relatively low workload/intensity which was required due to the impaired exercise capacity of AMPK KD mice. Importantly despite a lack of activation of AMPK α2 activity in KD mice, ACC phosphorylation was increased with treadmill running (Fig. 4C). Fatty acid and glucose oxidation during treadmill running was assessed in wild-type and AMPK KD mice by collecting expired respiratory gases, which during submaximal endurance exercise is indicative of muscle metabolism. Oxygen uptake 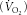 and the respiratory exchange ratio (RER) (Table 1) were not different between genotypes during treadmill exercise, indicating that the proportion of fat and carbohydrate oxidized was comparable between wild-type and KD mice (Fig. 4D).
and the respiratory exchange ratio (RER) (Table 1) were not different between genotypes during treadmill exercise, indicating that the proportion of fat and carbohydrate oxidized was comparable between wild-type and KD mice (Fig. 4D).
Figure 4. Fatty acid oxidation is not altered in AMPK KD mice during submaximal treadmill running.

AMPK α2 (A) and α1 (B) activity in gastrocnemius muscle following treadmill running. C, representative Western blots and quantification of ACC S221 phosphorylation relative to total ACC in gastrocnemius muscle following treadmill running. D, estimated percentage fat or carbohydrate utilization determined at 15 or 45 min during treadmill exercise. All data are mean ±s.e.m., n = 6–8. *P < 0.05 versus rest, **P < 0.01 versus rest, +++P < 0.001 versus corresponding WT group.
Table 1.
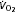 and respiratory exchange ratio (RER) during treadmill exercise at 15 m min−1 and 10% gradient in WT and AMPK KD mice measured at 15 and 45 min of exercise
and respiratory exchange ratio (RER) during treadmill exercise at 15 m min−1 and 10% gradient in WT and AMPK KD mice measured at 15 and 45 min of exercise
| 15 min | 45 min | |||
|---|---|---|---|---|
| WT | AMPK KD | WT | AMPK KD | |
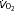 (ml kg−1 h−1) (ml kg−1 h−1) |
5490 ± 360 | 4760 ± 195 | 4905 ± 205 | 5080 ± 85 |
RER (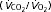 ) ) |
0.75 ± 0.01 | 0.77 ± 0.02 | 0.71 ± 0.0 | 0.71 ± 0.01 |
All data are mean ±s.e.m., n = 6.
Taken together these data demonstrate that muscle contractions performed ex vivo, in situ or during treadmill exercise activates an alternative kinase capable of phosphorylating ACC Ser-221 and stimulating fatty acid oxidation in the absence of increases in AMPK activity.
Screening for alternative ACC kinases in contracting muscles
To identify alternative ACC2 kinase(s) we performed an antibody microarray on muscle from resting and in situ contracted WT and AMPK KD mice. We hypothesized that an alternative ACC2 kinase capable of substituting for AMPK would be activated to a greater degree during muscle contraction in AMPK KD mice compared with WT littermates. We identified 18 Ser/Thr kinases whose phosphorylation was up-regulated by greater than 25% in contracted muscles from AMPK KD mice (Table 2). Of these candidates we then utilized in silico screening using both ScanSite (Obenauer et al. 2003) and Predikin (Brinkworth et al. 2003) prediction-based software, which utilize substrate-specificity databases and structural databases, respectively, to assess which of these protein kinases might phosphorylate ACC2 Ser-221. The results of this survey suggested three possible candidates: extracellular regulated protein-serine kinase (ERK) 1/2 (also known as p44/p42 MAP kinases), inhibitor of NF-κB protein-serine kinase α/β (IKKα/β) and protein kinase D (PKD-formerly known as PKCμ) all of which have previously been demonstrated to be activated in skeletal muscle during muscle contraction (Sherwood et al. 1999; Ryder et al. 2000; Turcotte et al. 2005; Luiken et al. 2008).
Table 2.
Summary of Ser/Thr kinases up-regulated by greater than 25% in contracted AMPK KD skeletal muscle
| Accession | Name | Short name | Phosphorylation site | Fold change |
|---|---|---|---|---|
| P27361 | Extracellular regulated protein-serine kinase 1/2 (p44/p42 MAP kinases) | ERK1/2 | T202 + Y204/T185 + Y187 | 3.46 |
| P46734 | MAP kinase protein-serine kinase 3/6 (MKK3/6) | MEK3/6 (MAP2K3/MAP2K6) | S189/S207 | 3.06 |
| O15111 | Inhibitor of NF-κB protein-serine kinase α (CHUK)/β | IKKα/β | S180/S181 | 2.22 |
| P23443 | p70 ribosomal protein-serine S6 kinase alpha/p85 ribosomal protein-serine S6 kinase 2 | S6Ka (p70 S6Ka)/S6K2 (p85 S6K2) | T229/T252 | 2.03 |
| P49137 | Mitogen-activated protein kinase-activated protein kinase 2 | MAPKAPK2 | T334 | 2.02 |
| P13861 | cAMP-dependent protein-serine kinase regulatory type 2 subunit α | PKA R2α | S98 | 1.71 |
| P49841 | Glycogen synthase-serine kinase 3 α/β | GSK3α/β | Y279/Y216 | 1.71 |
| Q15139 | Protein-serine kinase C μ (protein kinase D) | PKCμ (PKD) | S910 | 1.66 |
| P17252 | Protein-serine kinase C α | PKCα | S657 | 1.66 |
| P24723 | Protein-serine kinase C θ | PKCθ | S674 | 1.64 |
| Q15418 | Ribosomal S6 protein-serine kinase 1/2 | RSK1/2 | S380/S386 | 1.60 |
| P31749 | Protein-serine kinase B α | PKBα (Akt1) | S473 | 1.47 |
| Q02750 | MAPK/ERK protein-serine kinase 1 (MKK1) | MEK1 (MAP2K1) | T291 | 1.43 |
| O75582 | Mitogen and stress-activated protein-serine kinase 1 | Msk1 | S376 | 1.35 |
| P41743 | Protein-serine kinase C λ/ι | PKCl λ/ι | T555 | 1.31 |
| P36507 | MAPK/ERK protein-serine kinase 2 (MKK2) | MEK2 (MAP2K2) | T394 | 1.30 |
| Q02156 | Protein-serine kinase C ɛ | PKCɛ | S729 | 1.29 |
| Q15139 | Protein-serine kinase C μ (protein kinase D) | PKCμ (PKD) | S738 + S742 | 1.26 |
| P05129 | Protein-serine kinase C γ | PKCγ | T514 | 1.25 |
To test if these kinases could phosphorylate ACC2 we conducted in vitro experiments utilizing recombinant baculovirus-expressed ACC2 as a substrate for active recombinant AMPK, ERK2, IKKβ and PKD. AMPK markedly stimulated [32P]ATP-mediated phosphorylation of ACC2. PKD caused a minor stimulation of ACC2 phosphorylation (only 5–10% of that caused by AMPK) while neither IKKβ nor ERK2 increased 32P incorporation into ACC2. Importantly, only AMPK was able to increase ACC2 Ser-221 phosphorylation indicating that ERK2, IKKβ or PKD are not ACC2 kinases (Supplemental Fig. 4).
Discussion
Despite the well-recognized clinical importance of exercise in the prevention and treatment of insulin resistance and type 2 diabetes, the molecular mechanisms mediating the positive effects of exercise have remained elusive (Diabetes Prevention Program Research Group, 2002). The discovery that AMPK is activated by exercise in both rodents (Winder & Hardie, 1996; Vavvas et al. 1997) and humans (Chen et al. 2000; Fujii et al. 2000; Wojtaszewski et al. 2000) provided a possible molecular mechanism explaining the beneficial effects of exercise on metabolism. In the current study we demonstrate that the activation of AMPK by both AICAR and muscle contraction is not required for stimulating fatty acid oxidation in skeletal muscle, suggesting that alternative pathways exist that are capable of compensating for AMPK in the control of fatty acid oxidation.
During exercise, increased ATP demands by contracting sarcomeres and Ca2+ pumps in the sarcoplasmic reticulum can increase ATP turnover by in excess of 100-fold (Spriet & Dyck, 1996). This dramatic increase in ATP demand is tightly matched by the rapidly increasing oxidative and non-oxidative metabolism of substrates derived from intra- and extracellular lipid and carbohydrate stores (Spriet & Dyck, 1996). Studies in AMPK α2 (Jorgensen et al. 2005) and LKB1 null mice (Sakamoto et al. 2005) have highlighted the importance of AMPK signalling in the maintenance of energy charge as both models have an elevated muscle AMP: ATP ratio during treadmill running and ex vivo muscle contractions, respectively. During endurance exercise fatty acid oxidation is the predominant substrate for working skeletal muscle. Therefore, our findings that muscle fatty acid oxidation was not impaired during muscle contraction both ex vivo or during endurance treadmill exercise suggests that this is not the primary defect contributing to a reduced ability to buffer ATP in the absence of AMPK. Indeed we also found that carbohydrate oxidation was also maintained during treadmill exercise supporting previous findings demonstrating that glucose uptake is not impaired in skeletal muscle of mice with AMPK deficiency (Jorgensen et al. 2003; Barnes et al. 2004; Fujii et al. 2005; Thomson et al. 2007b). Taken together these data support the concept that reduced exercise capacity in AMPK-deficient mice (Jorgensen et al. 2003; Thomson et al. 2007b) may be due to secondary effects on cardiac metabolism which reduces cardiac output or perfusion (Mu et al. 2003).
ACC was one of the first substrates of AMPK identified and is commonly used as a surrogate marker of in vivo AMPK activity (Carling et al. 1987). Activation of AMPK by muscle contraction is associated with increased phosphorylation and deactivation of ACC2 (Winder, 1996; Vavvas et al. 1997). Surprisingly, the phosphorylation of ACC on Ser-221 was significantly increased in KD muscles with both AICAR and contraction despite no detectable increase in AMPK activity. Although it cannot be ruled out completely, we consider that residual AMPK activity in KD mice is not responsible for the phosphorylation of ACC during AICAR treatment or contraction since no increase in AMPK α1 or α2 activity could be detected under these conditions. In particular, AMPK α2 activity, the isoform predominantly activated by contraction and AICAR, was severely blunted in kinase dead mice. In addition, in the same muscles in which ACC Ser-221 phosphorylation was examined, the phosphorylation of HSL was impaired in AMPK KD muscle following AICAR treatment, but not contraction. This suggests that the maintenance of ACC phosphorylation following both AICAR and contraction was not due to residual AMPK activity as it would be expected that HSL would have been phosphorylated following AICAR treatment in AMPK KD mice if this was the case.
In addition to control by ACC2, malonyl-CoA levels are also regulated by malonyl-CoA decarboxylase (MCD) (Dyck et al. 1998). The depletion of MCD in muscle increases malonyl-CoA and reduces fatty acid oxidation (Bouzakri et al. 2008; Koves et al. 2008). MCD activity is increased in response to AICAR and contraction in skeletal muscle (Saha et al. 2000; Park et al. 2002) but this regulation appears to be indirect as AMPK does not phosphorylate MCD (Habinowski et al. 2001). Our data demonstrating reduced levels of malonyl-CoA in AMPK KD mice, both at rest and following muscle contraction, could be due to MCD activation but by a mechanism independent of AMPK. Clearly, further studies examining the regulation of malonyl-CoA and MCD with AICAR and muscle contraction are warranted.
In conclusion these studies demonstrate that AMPK is dispensable for the regulation of skeletal muscle fatty acid oxidation in response to muscle contraction and treadmill exercise. Importantly, we demonstrate that AICAR can also regulate fatty acid oxidation independent of AMPK activation highlighting the need for more specific activators of AMPK. Future studies investigating the entire muscle kinome or genetic evidence examining the role of ACC2 phosphorylation will be important to identify novel pathways regulating muscle fatty acid oxidation.
Acknowledgments
We thank Professor Morris Birnbaum for providing AMPK KD mice. These studies were supported by grants from the National Health and Medical Research Council (G.R.S., M.J.W., B.E.K., G.S.L.) and Diabetes Australia Research Trust (G.R.S.). N.D. was supported by a National Heart Foundation scholarship. S.B.J. was supported by a Danish Research Council of Health and Diseases postdoctoral fellowship. B.E.K. is an Australian Research Council Federation Fellow. S.W. is a Peter Doherty Fellow, M.J.W. is a R. Douglas Wright Fellow and G.R.S. a National Health and Medical Research Council Senior Research Fellow.
Supplemental material
Online supplemental material for this paper can be accessed at:
http://jp.physoc.org/cgi/content/full/jphysiol.2008.159814/DC1
References
- Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ. The subcellular localization of acetyl-CoA carboxylase 2. Proc Natl Acad Sci U S A. 2000;97:1444–1449. doi: 10.1073/pnas.97.4.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes. 2006;55:2277–2285. doi: 10.2337/db06-0062. [DOI] [PubMed] [Google Scholar]
- Barnes BR, Marklund S, Steiler TL, Walter M, Hjalm G, Amarger V, Mahlapuu M, Leng Y, Johansson C, Galuska D, Lindgren K, Abrink M, Stapleton D, Zierath JR, Andersson L. The AMPK γ3 isoform has a key role for carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem. 2004;279:38441–38447. doi: 10.1074/jbc.M405533200. [DOI] [PubMed] [Google Scholar]
- Bonen A, Luiken JJFP, Liu S, Dyck DJ, Kiens B, Kristiansen S, Turcotte LP, Van Der Vusse GJ, Glatz J. Palmitate transport and fatty acid transporters in red and white muscles. Am J Physiol Endocrinol Metab. 1998;275:E471–E478. doi: 10.1152/ajpendo.1998.275.3.E471. [DOI] [PubMed] [Google Scholar]
- Bouzakri K, Austin R, Rune A, Lassman ME, Garcia-Roves PM, Berger JP, Krook A, Chibalin AV, Zhang BB, Zierath JR. Malonyl coenzyme A decarboxylase regulates lipid and glucose metabolism in human skeletal muscle. Diabetes. 2008;57:1508–1516. doi: 10.2337/db07-0583. [DOI] [PubMed] [Google Scholar]
- Brinkworth RI, Breinl RA, Kobe B. Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc Natl Acad Sci U S A. 2003;100:74–79. doi: 10.1073/pnas.0134224100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol synthesis. FEBS Lett. 1987;223:217–222. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- Chen X-P, McConell GK, Michell BJ, Snow RJ, Canny BJ, Kemp BE. AMPK signaling in contracting human skeletal muscle: aceyl-CoA carboxylase and NO synthase phosphorylation. Am J Physiol Endocrinol Metab. 2000;279:E1202–E1206. doi: 10.1152/ajpendo.2000.279.5.E1202. [DOI] [PubMed] [Google Scholar]
- Davies SP, Sim AT, Hardie DG. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur J Biochem. 1990;187:183–190. doi: 10.1111/j.1432-1033.1990.tb15293.x. [DOI] [PubMed] [Google Scholar]
- Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyck DJ, Bonen A. Muscle contraction increases palmitate esterification and oxidation and triacylglycerol oxidation. Am J Physiol Endocrinol Metab. 1998;275:E888–E896. doi: 10.1152/ajpendo.1998.275.5.E888. [DOI] [PubMed] [Google Scholar]
- Dyck DJ, Peters SJ, Glatz J, Gorski J, Keizer H, Kiens B, Liu S, Richter EA, Spriet LL, Van Der Vusse GJ, Bonen A. Functional differences in lipid metabolism in resting skeletal muscle of various fiber types. Am J Physiol Endocrinol Metab. 1997;272:E340–E351. doi: 10.1152/ajpendo.1997.272.3.E340. [DOI] [PubMed] [Google Scholar]
- Dyck JR, Barr AJ, Barr RL, Kolattukudy PE, Lopaschuk GD. Characterization of cardiac malonyl-CoA decarboxylase and its putative role in regulating fatty acid oxidation. Am J Physiol Heart Circ Physiol. 1998;275:H2122–H2129. doi: 10.1152/ajpheart.1998.275.6.H2122. [DOI] [PubMed] [Google Scholar]
- Fujii N, Hayashi T, Hirshman MF, Smith JT, Habinowski SA, Kaijser L, Mu J, Ljungqvist O, Birnbaum MJ, Witters LA, Thorell A, Goodyear LJ. Exercise induces isoform-specific increase in 5′AMP-activated protein kinase activity in human skeletal muscle. Biochem Biophys Res Commun. 2000;273:1150–1155. doi: 10.1006/bbrc.2000.3073. [DOI] [PubMed] [Google Scholar]
- Fujii N, Hirshman MF, Kane EM, Ho RC, Peter LE, Seifert MM, Goodyear LJ. AMP-activated protein kinase α2 activity is not essential for contraction- and hyperosmolarity-induced glucose transport in skeletal muscle. J Biol Chem. 2005;280:39033–39041. doi: 10.1074/jbc.M504208200. [DOI] [PubMed] [Google Scholar]
- Gao L, Chiou W, Tang H, Cheng X, Camp HS, Burns DJ. Simultaneous quantification of malonyl-CoA and several other short-chain acyl-CoAs in animal tissues by ion-pairing reversed-phase HPLC/MS. J Chromatogr B Anal Technol Biomed Life Sci. 2007;853:303–313. doi: 10.1016/j.jchromb.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Habinowski SA, Hirshman M, Sakamoto K, Kemp BE, Gould SJ, Goodyear LJ, Witters LA. Malonyl-CoA decarboxylase is not a substrate of AMP-activated protein kinase in rat fast-twitch skeletal muscle or an islet cell line. Arch Biochem Biophys. 2001;396:71–79. doi: 10.1006/abbi.2001.2589. [DOI] [PubMed] [Google Scholar]
- Iseli TJ, Oakhill JS, Bailey MF, Wee S, Walter M, van Denderen BJ, Castelli LA, Katsis F, Witters LA, Stapleton D, Macaulay SL, Michell BJ, Kemp BE. AMP-activated protein kinase subunit interactions: β1: γ1 association requires β1 Thr-263 and Tyr-267. J Biol Chem. 2008;283:4799–4807. doi: 10.1074/jbc.M708298200. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JFP. Knockout of the α-2 but not α-1 5′AMP-activated protein kinase isoform abolishes AICAR- but not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2003;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, Pilegaard H. Effects of α-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. 2005;19:1146–1148. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li Y, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR, Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006;26:8217–8227. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinen HA, Galuska D, Chibalin AV, Yang J, Zierath JR, Holman GD, Wallberg-Henriksson H. 5-Amino-imidazole carboxamide riboside increases glucose transport and cell-surface GLUT4 content in skeletal muscle from subjects with type 2 diabetes. Diabetes. 2003;52:1066–1072. doi: 10.2337/diabetes.52.5.1066. [DOI] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Vertommen D, Coort SL, Habets DD, El Hasnaoui M, Pelsers MM, Viollet B, Bonen A, Hue L, Rider MH, Glatz JF. Identification of protein kinase D as a novel contraction-activated kinase linked to GLUT4-mediated glucose uptake, independent of AMPK. Cell Signal. 2008;20:543–556. doi: 10.1016/j.cellsig.2007.11.007. [DOI] [PubMed] [Google Scholar]
- McConell GK, Lee-Young RS, Chen ZP, Stepto NK, Huynh NN, Stephens TJ, Canny BJ, Kemp BE. Short-term exercise training in humans reduces AMPK signalling during prolonged exercise independent of muscle glycogen. J Physiol. 2005;568:665–676. doi: 10.1113/jphysiol.2005.089839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry JD, Stark MJ, Foster DW. Hepatic malonyl-CoA levels of fed, fasted and diabetic rats as measured using a simple radioisotopic assay. J Biol Chem. 1978;253:8291–8293. [PubMed] [Google Scholar]
- Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem. 2006;281:18933–18941. doi: 10.1074/jbc.M512831200. [DOI] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol Endocrinol Metab. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- Mu J, Barton ER, Birnbaum MJ. Selective suppression of AMP-activated protein kinase in skeletal muscle: update on ‘lazy mice’. Biochem Soc Trans. 2003;31:236–241. doi: 10.1042/bst0310236. [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odland LM, Heigenhauser GJF, Lopaschuk GD, Spriet LL. Human skeletal muscle malonyl-CoA at rest and during prolonged submaximal exercise. Am J Physiol Endocrinol Metab. 1996;270:E541–E544. doi: 10.1152/ajpendo.1996.270.3.E541. [DOI] [PubMed] [Google Scholar]
- Odland LM, Howlett RA, Heigenhauser GJF, Hultman E, Spriet LL. Skeletal muscle malonyl-CoA content at the onset of exercise at varying power outputs. Am J Physiol Endocrinol Metab. 1998;274:E1080–E1085. doi: 10.1152/ajpendo.1998.274.6.E1080. [DOI] [PubMed] [Google Scholar]
- Park H, Kaushik VK, Constant S, Prentki M, Przybytkowski E, Ruderman NB, Saha AK. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277:32571–32577. doi: 10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]
- Raney MA, Yee AJ, Todd MK, Turcotte LP. AMPK activation is not critical in the regulation of muscle FA uptake and oxidation during low-intensity muscle contraction. Am J Physiol Endocrinol Metab. 2005;288:E592–E598. doi: 10.1152/ajpendo.00301.2004. [DOI] [PubMed] [Google Scholar]
- Roepstorff C, Halberg N, Hillig T, Saha AK, Ruderman NB, Wojtaszewski JF, Richter EA, Kiens B. Malonyl-CoA and carnitine in regulation of fat oxidation in human skeletal muscle during exercise. Am J Physiol Endocrinol Metab. 2005;288:E133–E142. doi: 10.1152/ajpendo.00379.2004. [DOI] [PubMed] [Google Scholar]
- Roepstorff C, Thiele M, Hillig T, Pilegaard H, Richter EA, Wojtaszewski JF, Kiens B. Higher skeletal muscle α2AMPK activation and lower energy charge and fat oxidation in men than in women during submaximal exercise. J Physiol. 2006;574:125–138. doi: 10.1113/jphysiol.2006.108720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepstorff C, Vistisen B, Donsmark M, Nielsen JN, Galbo H, Green KA, Hardie DG, Wojtaszewski JFP, Richter EA, Kiens B. Regulation of hormone sensitive lipase activity and Ser563 and Ser565 phosphorylation in human skeletal muscle during exercise. J Physiol. 2004;560:551–562. doi: 10.1113/jphysiol.2004.066480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol Endocrinol Metab. 1999;276:E1–E18. doi: 10.1152/ajpendo.1999.276.1.E1. [DOI] [PubMed] [Google Scholar]
- Ryder JW, Fahlman R, Wallberg-Henriksson H, Alessi DR, Krook A, Zierath JR. Effect of contraction on mitogen-activated protein kinase signal transduction in skeletal muscle. Involvement of the mitogen- and stress-activated protein kinase 1. J Biol Chem. 2000;275:1457–1462. doi: 10.1074/jbc.275.2.1457. [DOI] [PubMed] [Google Scholar]
- Saha AK, Schwarsin AJ, Roduit R, Masse F, Kaushik V, Tornheim K, Prentki M, Ruderman NB. Activation of malonyl-CoA decarboxylase in rat skeletal muscle by contraction and the AMP-activated protein kinase activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside. J Biol Chem. 2000;275:24279–24283. doi: 10.1074/jbc.C000291200. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87:507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schertzer JD, Lynch GS. Comparative evaluation of IGF-I gene transfer and IGF-I protein administration for enhancing skeletal muscle regeneration after injury. Gene Ther. 2006;13:1657–1664. doi: 10.1038/sj.gt.3302817. [DOI] [PubMed] [Google Scholar]
- Sherwood DJ, Dufresne SD, Markuns JF, Cheatham B, Moller DE, Aronson D, Goodyear LJ. Differential regulation of MAP kinase, p70 (S6K), and Akt by contraction and insulin in rat skeletal muscle. Am J Physiol Endocrinol Metab. 1999;276:E870–E878. doi: 10.1152/ajpendo.1999.276.5.E870. [DOI] [PubMed] [Google Scholar]
- Smith AC, Bruce CR, Dyck DJ. AICAR further increases fatty acid oxidation and blunts triacylglycerol hydrolysis in contracting rat soleus muscle. J Physiol. 2005;565:547–553. doi: 10.1113/jphysiol.2004.081687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriet LL, Dyck DJ. The glucose-fatty acid cycle in skeletal muscle at rest and during exercise. In: Maughan RJ, Shirreffs SM, editors. Biochemistry of Exercise. Windsor, Ontario: Human Kinetics; 1996. pp. 127–157. [Google Scholar]
- Steinberg GR, Bonen A, Dyck DJ. Fatty acid oxidation and triacylglycerol hydrolysis are enhanced following chronic leptin treatment in rats. Am J Physiol Endocrinol Metab. 2002;282:E593–E600. doi: 10.1152/ajpendo.00303.2001. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Dyck DJ. Development of leptin resistance in rat soleus muscle in response to high-fat diets. Am J Physiol Endocrinol Metab. 2000;279:E1374–E1382. doi: 10.1152/ajpendo.2000.279.6.E1374. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Macaulay SL, Febbraio MA, Kemp BE. AMP-activated protein kinase – the fat controller of the energy railroad. Can J Physiol Pharmacol. 2006a;84:655–665. doi: 10.1139/y06-005. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Michell BJ, van Denderen BJ, Watt MJ, Carey AL, Fam BC, Andrikopoulos S, Proietto J, Gorgun CZ, Carling D, Hotamisligil GS, Febbraio MA, Kay TW, Kemp BE. Tumor necrosis factor α-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006b;4:465–474. doi: 10.1016/j.cmet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Smith AC, van Denderen BJW, Chen Z, Murthy S, Campbell DJ, Heigenhauser GJF, Dyck DJ, Kemp BE. AMP-activated protein kinase is not down-regulated in human skeletal muscle of obese females. J Clin Endocrinol Metab. 2004;89:4575–4580. doi: 10.1210/jc.2004-0308. [DOI] [PubMed] [Google Scholar]
- Thomson DM, Brown JD, Fillmore N, Condon BM, Kim HJ, Barrow JR, Winder WW. LKB1 and the regulation of malonyl-CoA and fatty acid oxidation in muscle. Am J Physiol Endocrinol Metab. 2007a;293:E1572–E1579. doi: 10.1152/ajpendo.00371.2007. [DOI] [PubMed] [Google Scholar]
- Thomson DM, Porter BB, Tall JH, Kim HJ, Barrow JR, Winder WW. Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am J Physiol Endocrinol Metab. 2007b;292:E196–E202. doi: 10.1152/ajpendo.00366.2006. [DOI] [PubMed] [Google Scholar]
- Turcotte LP, Raney MA, Todd MK. ERK1/2 inhibition prevents contraction-induced increase in plasma membrane FAT/CD36 content and FA uptake in rodent muscle. Acta Physiol Scand. 2005;184:131–139. doi: 10.1111/j.1365-201X.2005.01445.x. [DOI] [PubMed] [Google Scholar]
- Vavvas D, Apazidis A, Saha AK, Gamble J, Patel A, Kemp BE, Witters LA, Ruderman NB. Contraction-induced changes in acetyl-CoA carboxylase and 5′-AMP-activated kinase in skeletal muscle. J Biol Chem. 1997;272:13255–13261. doi: 10.1074/jbc.272.20.13255. [DOI] [PubMed] [Google Scholar]
- Watt MJ, Dzamko N, Thomas WG, Rose-John S, Ernst M, Carling D, Kemp BE, Febbraio MA, Steinberg GR. CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat Med. 2006a;12:541–548. doi: 10.1038/nm1383. [DOI] [PubMed] [Google Scholar]
- Watt MJ, Holmes AG, Pinnamaneni SK, Garnham AP, Steinberg GR, Kemp BE, Febbraio MA. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab. 2005 doi: 10.1152/ajpendo.00361.2005. [DOI] [PubMed] [Google Scholar]
- Watt MJ, Steinberg GR, Chen ZP, Kemp BE, Febbraio MA. Fatty acids stimulate AMP-activated protein kinase and enhance fatty acid oxidation in L6 myotubes. J Physiol. 2006b;574:139–147. doi: 10.1113/jphysiol.2006.107318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder WW. Malonyl-CoA as a metabolic regulator. In: Maughan RJ, Shirreffs SM, editors. Biochemistry of Exercise IX. Champaign: Human Kinetics, IL, USA; 1996. [Google Scholar]
- Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol Endocrinol Metab. 1996;270:E299–E304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- Winder WW, Wilson HA, Hardie DG, Rasmussen BB, Hutber CA, Call GB, Clayton RD, Conley LM, Yoon S, Zhou B. Phosphorylation of rat muscle acetyl-CoA carboxylase by AMP-activated protein kinase and protein kinase A. J Appl Physiol. 1997;82:219–225. doi: 10.1152/jappl.1997.82.1.219. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B. Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J Physiol. 2000;528:221–226. doi: 10.1111/j.1469-7793.2000.t01-1-00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.