

Abstract
Hypoxia is a major cause of pulmonary hypertension. Gene expression activated by the transcription factor hypoxia-inducible factor (HIF) is central to this process. The oxygen-sensing iron-dependent dioxygenase enzymes that regulate HIF are highly sensitive to varying iron availability. It is unknown whether iron similarly influences the pulmonary vasculature. This human physiology study aimed to determine whether varying iron availability affects pulmonary arterial pressure and the pulmonary vascular response to hypoxia, as predicted biochemically by the role of HIF. In a controlled crossover study, 16 healthy iron-replete volunteers undertook two separate protocols. The ‘Iron Protocol’ studied the effects of an intravenous infusion of iron on the pulmonary vascular response to 8 h of sustained hypoxia. The ‘Desferrioxamine Protocol’ examined the effects of an 8 h intravenous infusion of the iron chelator desferrioxamine on the pulmonary circulation. Primary outcome measures were pulmonary artery systolic pressure (PASP) and the PASP response to acute hypoxia (ΔPASP), assessed by Doppler echocardiography. In the Iron Protocol, infusion of iron abolished or greatly reduced both the elevation in baseline PASP (P < 0.001) and the enhanced sensitivity of the pulmonary vasculature to acute hypoxia (P = 0.002) that are induced by exposure to sustained hypoxia. In the Desferrioxamine Protocol, desferrioxamine significantly elevated both PASP (P < 0.001) and ΔPASP (P = 0.01). We conclude that iron availability modifies pulmonary arterial pressure and pulmonary vascular responses to hypoxia. Further research should investigate the potential for therapeutic manipulation of iron status in the management of hypoxic pulmonary hypertensive disease.
Pulmonary hypertensive disorders frequently complicate hypoxic lung disease and worsen patient survival (Barbera et al. 2003; Chaouat et al. 2005; McLaughlin & McGoon, 2006). For example, up to 90% of patients with advanced chronic obstructive pulmonary disease (COPD) have moderate to severe pulmonary hypertension that worsens survival despite maximal therapy (Oswald-Mammosser et al. 1995; Scharf et al. 2002; Mal, 2007). Hypoxia-induced pulmonary hypertension is also a major cause of morbidity among high altitude residents, while in non-residents it directly precipitates high altitude pulmonary oedema, the most common cause of death related to high altitude (Hackett & Roach, 2001; Penaloza & Arias-Stella, 2007). Hypoxia causes pulmonary hypertension through hypoxic pulmonary vasoconstriction and vascular remodelling (Barbera et al. 2003). It has recently become apparent that these processes are regulated at least in part by the hypoxia-inducible factor (HIF) family of transcription factors, which coordinate intracellular responses to hypoxia by directly or indirectly regulating the expression of several hundred genes (Semenza, 2004; Smith et al. 2008). In mice, heterozygous deficiency for functional HIF genes markedly inhibits the development of hypoxia-induced pulmonary hypertension and vascular remodelling (Yu et al. 1999; Brusselmans et al. 2003), and impairs hypoxic responses in the pulmonary arterial myocytes responsible for these processes (Shimoda et al. 2001). In patients diagnosed with the rare recessive disease Chuvash polycythaemia, HIF-mediated gene activation is pathologically increased and is associated with pulmonary hypertension (Bushuev et al. 2006; Smith et al. 2006) and a grossly exaggerated pulmonary vasoconstrictive response to hypoxia (Smith et al. 2006). Other rare HIF-activating mutations have also been linked to the development of pulmonary hypertension (Gale et al. 2008).
HIF is synthesized continuously and is primarily regulated through oxygen-dependent proteosomal degradation. HIF is targeted for proteolysis by hydroxylation of specific residues in the HIF-α subunit (Ivan et al. 2001; Jaakkola et al. 2001). This crucial oxygen-sensitive step is catalysed by HIF hydroxylase enzymes belonging to the iron- and 2-oxoglutarate-dependent dioxygenase superfamily, and requires iron as an obligate cofactor. In cultured cells HIF degradation is both inhibited by iron chelation with desferrioxamine (DFO) and potentiated by supraphysiological iron supplementation (Wang & Semenza, 1993; Knowles et al. 2003), raising the question of what effect variations in iron availability have on HIF-signalling at the systemic level. Previous studies in humans have demonstrated that DFO stimulates production of the HIF-regulated hormone erythropoietin (Ren et al. 2000) and mildly elevates pulmonary arterial pressures (Balanos et al. 2002).
The known biochemistry of HIF regulation and its involvement in pulmonary physiology together suggest that clinical iron status may modify the effects of hypoxia on the pulmonary circulation. Such an interaction would have important implications for patients with hypoxia-induced pulmonary hypertension. This study of healthy iron-replete individuals aimed to determine the effects of increasing and decreasing iron availability on the pulmonary vasculature and its response to hypoxia. The primary outcome measures were pulmonary artery systolic pressure (PASP) and the change in PASP stimulated by exposure to acute hypoxia (ΔPASP), assessed by Doppler echocardiography.
Methods
There were two protocols to the study, which are illustrated schematically in Fig. 1. The Iron Protocol compared the effects of intravenous iron with those of a control infusion on the subsequent responses to 8 h of sustained hypoxia. This protocol tested the hypothesis that increasing iron availability would attenuate the increases in PASP and ΔPASP that normally accompany sustained exposure to hypoxia. The DFO Protocol investigated the effect of reducing iron availability on PASP and ΔPASP, by comparing the effects of an 8 h intravenous infusion of DFO with those of a control infusion. Sixteen healthy volunteers with normal iron status participated in the study (age 25 ± 3 year, mean ±s.d.; four men and four women in each protocol). Participants were taking no medications or supplements, and provided written informed consent. The Oxfordshire Clinical Research Ethics Committee approved the study, which was conducted in accordance with the Declaration of Helsinki.
Figure 1. Schematic representation of experimental protocols.

Each protocol was conducted over two separate days. For the Iron Protocol, a control infusion of normal saline was administered on the first experimental day and an infusion of iron sucrose was given on the second experimental day. For the DFO Protocol, a control infusion of normal saline was administered on one experimental day and an infusion of desferrioxamine (DFO) was given on the other experimental day. Pulmonary artery pressure was assessed echocardiographically during the acute hypoxic challenges. 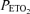 denotes end-tidal partial pressure of oxygen.
denotes end-tidal partial pressure of oxygen.
Iron Protocol
Each participant was studied twice with the studies separated by at least 1 week. The first protocol began with a control infusion of normal saline. Following the infusion, PASP and ΔPASP were measured during an acute hypoxic challenge. Subjects were then exposed to 8 h of sustained isocapnic hypoxia (end-tidal partial pressure of oxygen,  , of 55 mmHg) in a purpose built hypoxia chamber (Howard et al. 1995). After the 8 h exposure, PASP and ΔPASP were again determined during an acute hypoxic challenge. The second protocol began with an infusion of iron(iii)-hydroxide sucrose (200 mg over 105 min; Syner-Med (Pharmaceutical Products) Ltd, Purley, UK) and was otherwise identical to the first protocol.
, of 55 mmHg) in a purpose built hypoxia chamber (Howard et al. 1995). After the 8 h exposure, PASP and ΔPASP were again determined during an acute hypoxic challenge. The second protocol began with an infusion of iron(iii)-hydroxide sucrose (200 mg over 105 min; Syner-Med (Pharmaceutical Products) Ltd, Purley, UK) and was otherwise identical to the first protocol.
The order of the saline and iron infusions was not randomised between protocols as the duration of the potential downstream effects of iron administration is unknown. The plasma half-life of iron sucrose is 5–6 h (Danielson et al. 1996) and the immediately bioavailable labile iron fraction is approximately 5% (Van Wyck et al. 2004).
DFO Protocol
Each participant was studied on two days separated by at least 1 week. Participants received an 8 h infusion of desferrioxamine mesylate (4 g per 70 kg body weight; Ciba-Geigy, Cheshire, UK) on one experimental day and a control infusion of normal saline on the other. The infusion order was randomised. PASP and ΔPASP were measured during an acute hypoxic challenge before and after the 8 h infusions.
Acute hypoxic challenges: measurement of PASP
Plasma erythropoietin concentration was measured immediately prior to each acute hypoxic challenge. The apparatus and techniques used in the acute hypoxic challenges have been extensively validated and described in detail previously (Balanos et al. 2005; Smith et al. 2006). Challenges were conducted with the subject reclining in the left lateral position and breathing through a mouthpiece. Gas control was achieved by means of dynamic end-tidal forcing (Robbins et al. 1982). For each challenge an initial 5 min of euoxia (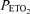 of 100 mmHg) preceded a period of 20 min of isocapnic hypoxia (
of 100 mmHg) preceded a period of 20 min of isocapnic hypoxia (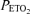 50 mmHg). This was followed by a final 10 min of euoxia. End-tidal partial pressure of carbon dioxide was maintained close to each subject's baseline value. A Sonos 5500 echocardiography machine (2–4 MHz transducer; Hewlett-Packard, Boston, MA, USA) was used to monitor PASP continuously. Using a standard Doppler technique, the maximum systolic pressure gradient across the tricuspid valve was determined and PASP was calculated using the modified Bernoulli equation and an estimated right atrial pressure of 5 mmHg (Swenson et al. 2002; Smith et al. 2006). Heart rate and ventilation were recorded, and cardiac output was determined echocardiographically every 2 min (Howson et al. 1986; Balanos et al. 2005).
50 mmHg). This was followed by a final 10 min of euoxia. End-tidal partial pressure of carbon dioxide was maintained close to each subject's baseline value. A Sonos 5500 echocardiography machine (2–4 MHz transducer; Hewlett-Packard, Boston, MA, USA) was used to monitor PASP continuously. Using a standard Doppler technique, the maximum systolic pressure gradient across the tricuspid valve was determined and PASP was calculated using the modified Bernoulli equation and an estimated right atrial pressure of 5 mmHg (Swenson et al. 2002; Smith et al. 2006). Heart rate and ventilation were recorded, and cardiac output was determined echocardiographically every 2 min (Howson et al. 1986; Balanos et al. 2005).
Statistical analyses
Differences between experiment days were assessed statistically using Student's paired t test (Microsoft Excel), and were considered significant at the P < 0.05 level. All values are expressed as means ±s.d. unless otherwise stated.
Results
All subjects were initially iron-replete (see Table 1). All infusions were well tolerated. Neither iron nor DFO significantly affected ventilation, heart rate or cardiac output, either during euoxia or in response to hypoxia.
Table 1.
Baseline venous blood analyses
| Analysis (normal range) | DFO Protocol | Iron Protocol |
|---|---|---|
| Haemoglobin (12–17 g dl−1) | 14.0 ± 1.0 | 14.0 ± 1.5 |
| Haematocrit (0.36–0.50 l l−1) | 0.42 ± 0.03 | 0.42 ± 0.04 |
| Serum iron (11–31 μmol l−1) | 21.9 ± 10.1 | 16.1 ± 5.6 |
| Serum ferritin (10–300 μg l−1) | 29.3 ± 14.6 | 72.7 ± 81.6 |
Mean ±s.d. values are shown. Where normal ranges vary with sex, the widest range is given.
Iron Protocol
Figure 2 shows that, of itself, infusion with iron did not affect PASP or ΔPASP. However, pretreatment with iron prevented the rise in PASP normally induced by 8 h of sustained hypoxia (increase in PASP of 23%± 13% with saline control versus 2%± 3% with iron, P < 0.001). Iron also substantially attenuated the increase in ΔPASP normally induced by sustained hypoxia (increase of 124%± 77% with saline control versus increase of 59%± 67% with iron, P = 0.002). The initial plasma erythropoietin concentration measured prior to the infusions was not significantly different on the two experimental days (13.2 ± 1.4 mIU ml−1 prior to control infusion and 13.3 ± 0.4 mIU ml−1 prior to iron infusion; P = 0.91). The elevation in plasma erythropoietin concentration generated by hypoxia was not significantly affected by iron (increase of 67%± 35% with saline control versus increase of 61%± 40% with iron, P = 0.79).
Figure 2. Effects of 8 h of sustained hypoxia on pulmonary artery systolic pressure with and without prior infusion of iron.

Upper panels show end-tidal partial pressures of oxygen ( ) and carbon dioxide (
) and carbon dioxide (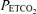 ) during acute hypoxic challenges. Lower panels show corresponding measurements of pulmonary artery systolic pressure (PASP). Values are means and error bars indicate s.e.m. The control measurements demonstrate the normal pulmonary vascular response to sustained hypoxia, consisting of an increase in PASP and a sensitized PASP response to subsequent acute hypoxia (ΔPASP). Intravenous infusion of iron prior to the 8 h hypoxic exposure prevented the increase in baseline PASP (P < 0.001) and markedly attenuated the degree to which ΔPASP was sensitized (P = 0.002).
) during acute hypoxic challenges. Lower panels show corresponding measurements of pulmonary artery systolic pressure (PASP). Values are means and error bars indicate s.e.m. The control measurements demonstrate the normal pulmonary vascular response to sustained hypoxia, consisting of an increase in PASP and a sensitized PASP response to subsequent acute hypoxia (ΔPASP). Intravenous infusion of iron prior to the 8 h hypoxic exposure prevented the increase in baseline PASP (P < 0.001) and markedly attenuated the degree to which ΔPASP was sensitized (P = 0.002).
DFO Protocol
Figure 3 shows that an 8 h infusion of DFO significantly increased basal PASP (increase of 0%± 3% with saline control versus increase of 10%± 4% with DFO, P < 0.001). DFO also increased pulmonary vascular reactivity to acute hypoxia (1%± 12% reduction in ΔPASP with saline control versus 36%± 26% increase with DFO, P = 0.01). The initial plasma erythropoietin concentration measured prior to the infusions was not significantly different on the two experimental days (10.4 ± 5.8 mIU ml−1 prior to control infusion and 8.3 ± 4.2 mIU ml−1 prior to DFO infusion; P = 0.14). DFO generated a significant rise in plasma erythropoietin (control increase of 2%± 65%versus increase of 794%± 566% with DFO, P = 0.03).
Figure 3. Effects of an 8 h infusion of desferrioxamine on pulmonary artery systolic pressure.

Upper panels show end-tidal partial pressures of oxygen (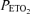 ) and carbon dioxide (
) and carbon dioxide (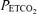 ) during acute hypoxic challenges. Lower panels show corresponding measurements of pulmonary artery systolic pressure (PASP). DFO denotes desferrioxamine. Values are means and error bars indicate s.e.m. Infusion of DFO increased PASP (P < 0.001) and sensitized the pulmonary vascular response to acute hypoxia (P = 0.01).
) during acute hypoxic challenges. Lower panels show corresponding measurements of pulmonary artery systolic pressure (PASP). DFO denotes desferrioxamine. Values are means and error bars indicate s.e.m. Infusion of DFO increased PASP (P < 0.001) and sensitized the pulmonary vascular response to acute hypoxia (P = 0.01).
Discussion
This study establishes the existence of a novel and substantial interaction between iron, hypoxia and the pulmonary circulation. We are not aware of any previous work describing such a link in animals or humans.
The results from the Iron Protocol demonstrate that, in iron-replete individuals, administration of iron abolishes the elevation in pulmonary arterial pressure that is normally induced following an 8 h exposure to hypoxia, and markedly inhibits the accompanying increase in acute hypoxic pulmonary vasoreactivity. This introduces the potential for benefit from iron therapy in disorders caused by pulmonary vasoconstrictive responses to hypoxia, such as COPD-related pulmonary hypertension and high altitude pulmonary oedema (HAPE).
The results from the DFO Protocol demonstrate that, for a given level of hypoxia, acute reduction of iron availability increases pulmonary arterial pressure. This raises the possibility that clinical iron deficiency might exacerbate hypoxically induced pulmonary hypertension, for example in cor pulmonale. Indeed, this might explain why the initial pulmonary antihypertensive benefits of venesection for secondary polycythaemia tend not to be sustained with continued venesection (Lewis et al. 1952; Hecht et al. 1955; Rakita et al. 1965; Segel & Bishop, 1966; Weisse et al. 1975).
Overall this study demonstrates the importance of iron in pulmonary vascular responses to hypoxia and suggests that even modest alterations in iron balance might be clinically significant in this respect. Although prolonged administration might eventually be limited by iron overload, it is nonetheless remarkable for a pharmacological agent as familiar, safe and inexpensive as iron to offer promise for an entirely new indication, and further studies are warranted to determine whether careful adjustment of iron balance has any place in the management of hypoxic pulmonary hypertensive disease. Perhaps most immediately, potential protective effects of iron loading should be investigated for HAPE, which afflicts non-acclimatized individuals ascending rapidly to high altitude. HAPE is associated with an excessive hypoxic pulmonary vasoconstrictive response that is characteristic of HAPE-susceptible individuals (Hackett & Roach, 2001) and that might be attenuated by parenteral iron, as was observed in this study.
Our hypothesis – that alterations in iron availability affect the pulmonary vascular response to hypoxia – was predicated on the known effects of iron manipulation on HIF hydroxylation, and on evidence implicating HIF in pulmonary vascular responses in vivo. The positive results we obtained would therefore imply that iron availability indeed modifies HIF hydroxylation in the pulmonary circulation, which appears to be limited by normal physiological levels of iron. This is difficult to prove formally as there is no means of directly quantifying HIF in the human lung, and peripheral concentrations of HIF-regulated gene products such as erythropoietin do not necessarily reflect HIF activity in the pulmonary vasculature. However, it is of interest that iron chelation and iron supplementation emulated the pulmonary vascular manifestations of up- and down-regulation of HIF that are seen in Chuvash polycythaemia (Smith et al. 2006) and HIF+/− mice (Yu et al. 1999; Brusselmans et al. 2003), respectively.
An alternative explanation for our findings is that the responses were due to some novel iron-dependent oxygen sensor, possibly another member of the iron-dependent dioxygenase enzyme family. Up to 40 such enzymes may sense changes in iron and oxygen availability and might mediate HIF-independent responses to these stimuli (Elkins et al. 2003; Elvidge et al. 2006; Ratcliffe, 2006; Ozer & Bruick, 2007). A further theoretical explanation is that iron availability influenced pulmonary vascular tone through the formation of oxygen free radicals. However, the precise role of reactive oxygen species in hypoxic pulmonary vasoconstriction is controversial (Aaronson et al. 2006). Furthermore, it is likely that any such hypothetical effect would be rapid, yet PASP and ΔPASP were normal when measured shortly after the iron infusion. Nevertheless, involvement of reactive oxygen species cannot be excluded as the time course of the observed effects could reflect, for example, that of iron uptake into pulmonary arterial myocytes.
We conclude that iron availability modifies pulmonary arterial pressures and pulmonary vasoconstrictive responses to acute and sustained hypoxia, and suggest that this interaction may derive from respective inhibition and potentiation of HIF hydroxylation in the lung. Until further evidence is forthcoming it may be prudent to avoid iron deficiency in hypoxic patients with end-stage pulmonary hypertension, and in HAPE-susceptible individuals returning to high altitude. Further research should investigate the link between iron status and pulmonary arterial pressure, and explore the therapeutic potential of manipulating iron and HIF in hypoxic pulmonary hypertensive disease.
Acknowledgments
T.G.S. was supported by a Rhodes Scholarship. This work was funded by the Wellcome Trust. The authors thank Mr David O’Connor for expert technical assistance, and the volunteers who took part in this study.
References
- Aaronson PI, Robertson TP, Knock GA, Becker S, Lewis TH, Snetkov V, Ward JP. Hypoxic pulmonary vasoconstriction: mechanisms and controversies. J Physiol. 2006;570:53–58. doi: 10.1113/jphysiol.2005.098855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balanos GM, Dorrington KL, Robbins PA. Desferrioxamine elevates pulmonary vascular resistance in humans: potential for involvement of HIF-1. J Appl Physiol. 2002;92:2501–2507. doi: 10.1152/japplphysiol.00965.2001. [DOI] [PubMed] [Google Scholar]
- Balanos GM, Talbot NP, Robbins PA, Dorrington KL. Separating the direct effect of hypoxia from the indirect effect of changes in cardiac output on the maximum pressure difference across the tricuspid valve in healthy humans. Pflugers Arch. 2005;450:372–380. doi: 10.1007/s00424-005-1422-6. [DOI] [PubMed] [Google Scholar]
- Barbera JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J. 2003;21:892–905. doi: 10.1183/09031936.03.00115402. [DOI] [PubMed] [Google Scholar]
- Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. 2003;111:1519–1527. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushuev VI, Miasnikova GY, Sergueeva AI, Polyakova LA, Okhotin D, Gaskin PR, Debebe Z, Nekhai S, Castro OL, Prchal JT, Gordeuk VR. Endothelin-1, vascular endothelial growth factor and systolic pulmonary artery pressure in patients with Chuvash polycythemia. Haematologica. 2006;91:744–749. [PubMed] [Google Scholar]
- Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducolone A, Ehrhart M, Kessler R, Weitzenblum E. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172:189–194. doi: 10.1164/rccm.200401-006OC. [DOI] [PubMed] [Google Scholar]
- Danielson BG, Salmonson T, Derendorf H, Geisser P. Pharmacokinetics of iron (III)-hydroxide sucrose complex after a single intravenous dose in healthy volunteers. Arzneimittelforschung. 1996;46:615–621. [PubMed] [Google Scholar]
- Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1α. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1α, HIF-2α, and other pathways. J Biol Chem. 2006;281:15215–15226. doi: 10.1074/jbc.M511408200. [DOI] [PubMed] [Google Scholar]
- Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2α mutation. Blood. 2008;112:919–921. doi: 10.1182/blood-2008-04-153718. [DOI] [PubMed] [Google Scholar]
- Hackett PH, Roach RC. High-altitude illness. N Engl J Med. 2001;345:107–114. doi: 10.1056/NEJM200107123450206. [DOI] [PubMed] [Google Scholar]
- Hecht HH, Gaylor W, Stein D. Vascular adjustments after phlebotomy in polycythemic subjects. J Clin Invest. 1955;34:939. [Google Scholar]
- Howard LSGE, Barson RA, Howse BPA, McGill TR, McIntyre ME, O’Connor DF, Robbins PA. Chamber for controlling the end-tidal gas tensions over sustained periods in humans. J Appl Physiol. 1995;78:1088–1091. doi: 10.1152/jappl.1995.78.3.1088. [DOI] [PubMed] [Google Scholar]
- Howson MG, Khamnei S, O’Connor DF, Robbins PA. The properties of a turbine device for measuring respiratory volumes in man. J Physiol. 1986;382:12P. [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian Y-M, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim Av, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Knowles HJ, Raval RR, Harris AL, Ratcliffe PJ. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 2003;63:1764–1768. [PubMed] [Google Scholar]
- Lewis CS, Samuels AJ, Daines MC, Hecht HH. Chronic lung disease, polycythemia and congestive heart failure; cardiorespiratory, vascular and renal adjustments in cor pulmonale. Circulation. 1952;6:874–887. doi: 10.1161/01.cir.6.6.874. [DOI] [PubMed] [Google Scholar]
- Mal H. Prevalence and diagnosis of severe pulmonary hypertension in patients with chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2007;13:114–119. doi: 10.1097/MCP.0b013e32801d217f. [DOI] [PubMed] [Google Scholar]
- McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–1431. doi: 10.1161/CIRCULATIONAHA.104.503540. [DOI] [PubMed] [Google Scholar]
- Oswald-Mammosser M, Weitzenblum E, Quoix E, Moser G, Chaouat A, Charpentier C, Kessler R. Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure. Chest. 1995;107:1193–1198. doi: 10.1378/chest.107.5.1193. [DOI] [PubMed] [Google Scholar]
- Ozer A, Bruick RK. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3:144–153. doi: 10.1038/nchembio863. [DOI] [PubMed] [Google Scholar]
- Penaloza D, Arias-Stella J. The heart and pulmonary circulation at high altitudes: healthy highlanders and chronic mountain sickness. Circulation. 2007;115:1132–1146. doi: 10.1161/CIRCULATIONAHA.106.624544. [DOI] [PubMed] [Google Scholar]
- Rakita L, Gillespie DG, Sancetta SM. The acute and chronic effects of phlebotomy on general hemodynamics and pulmonary functions of patients with secondary polycythemia associated with pulmonary emphysema. Am Heart J. 1965;70:466–475. doi: 10.1016/0002-8703(65)90358-3. [DOI] [PubMed] [Google Scholar]
- Ratcliffe PJ. Understanding hypoxia signalling in cells – a new therapeutic opportunity? Clin Med. 2006;6:573–578. doi: 10.7861/clinmedicine.6-6-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Dorrington KL, Maxwell PH, Robbins PA. Effects of desferrioxamine on serum erythropoietin and ventilatory sensitivity to hypoxia in humans. J App Physiol. 2000;89:680–686. doi: 10.1152/jappl.2000.89.2.680. [DOI] [PubMed] [Google Scholar]
- Robbins PA, Swanson GD, Howson MG. A prediction-correction scheme for forcing alveolar gases along certain time courses. J Appl Physiol. 1982;52:1353–1357. doi: 10.1152/jappl.1982.52.5.1353. [DOI] [PubMed] [Google Scholar]
- Scharf SM, Iqbal M, Keller C, Criner G, Lee S, Fessler HE. Hemodynamic characterization of patients with severe emphysema. Am J Respir Crit Care Med. 2002;166:314–322. doi: 10.1164/rccm.2107027. [DOI] [PubMed] [Google Scholar]
- Segel N, Bishop JM. The circulation in patients with chronic bronchitis and emphysema at rest and during exercise, with special reference to the influence of changes in blood viscosity and blood volume on the pulmonary circulation. J Clin Invest. 1966;45:1555–1568. doi: 10.1172/JCI105462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- Shimoda LA, Manalo DJ, Sham JSK, Semenza GL, Sylvester JT. Partial HIF-1α deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L202–L208. doi: 10.1152/ajplung.2001.281.1.L202. [DOI] [PubMed] [Google Scholar]
- Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, Liu C, Maxwell PH, McMullin MF, McNamara CJ, Percy MJ, Pugh CW, Ratcliffe PJ, Talbot NP, Treacy M, Robbins PA. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006;3:e290. doi: 10.1371/journal.pmed.0030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TG, Robbins PA, Ratcliffe PJ. The human side of hypoxia-inducible factor. Br J Haematol. 2008;141:325–334. doi: 10.1111/j.1365-2141.2008.07029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson ER, Maggiorini M, Mongovin S, Gibbs JS, Greve I, Mairbäurl H, Bärtsch P. Pathogenesis of high-altitude pulmonary edema: inflammation is not an etiologic factor. JAMA. 2002;287:2228–2235. doi: 10.1001/jama.287.17.2228. [DOI] [PubMed] [Google Scholar]
- Van Wyck D, Anderson J, Johnson K. Labile iron in parenteral iron formulations: a quantitative and comparative study. Nephrol Dial Transplant. 2004;19:561–565. doi: 10.1093/ndt/gfg579. [DOI] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood. 1993;82:3610–3615. [PubMed] [Google Scholar]
- Weisse AB, Moschos CB, Frank MJ, Levinson GE, Cannilla JE, Regan TJ. Hemodynamic effects of staged hematocrit reduction in patients with stable cor pulmonale and severely elevated hematocrit levels. Am J Med. 1975;58:92–98. doi: 10.1016/0002-9343(75)90538-0. [DOI] [PubMed] [Google Scholar]
- Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JSK, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]