

Abstract
Vibrio cholerae strains are capable of inhabiting multiple niches in the aquatic environment and in some cases cause disease in humans. However, the ecology and biodiversity of these bacteria in environmental settings remains poorly understood. We used the genomic fingerprinting technique enterobacterial repetitive intergenic consensus sequence PCR (ERIC-PCR) to profile 835 environmental isolates from waters and sediments obtained at nine sites along the central California coast. We identified 115 ERIC-PCR genotypes from 998 fingerprints, with a reproducibility of 98.5% and a discriminatory power of 0.971. When the temporal dynamics at a subset of sampling sites were explored, several genotypes provided evidence for cosmopolitan or geographically restricted distributions, and other genotypes displayed nonrandom patterns of cooccurrence. Partial Mantel tests confirmed that genotypic similarity of isolates across all sampling events was correlated with environmental similarity (0.04 ≤ r ≤ 0.05), temporal proximity (r = 0.09), and geographic distance (r = 0.09). A neutral community model for all sampling events explained 61% of the variation in genotype abundance. Cooccurrence indices (C-score, C-board, and Combo) were significantly different than expected by chance, suggesting that the V. cholerae population may have a competitive structure, especially at the regional scale. Even though stochastic processes are undoubtedly important in generating biogeographic patterns in diversity, deterministic factors appear to play a significant, albeit small, role in shaping the V. cholerae population structure in this system.
Recent advances in molecular methods have rapidly increased our ability to generate data sets that characterize microbial communities in the environment. We are now able to observe patterns in the spatial and temporal distribution of microorganisms and to evaluate these patterns in light of ecological theories developed for macroorganisms (26, 32). In accordance with MacArthur and Wilson's Theory of Island Biogeography (25), some microbial communities display a positive power law relationship between taxon richness and sample area in environments such as salt marshes (16), beech tree holes (2), and high mountain lakes (35). Communities of both macro- and microorganisms have also been shown to exhibit nonrandom patterns of taxon cooccurrence (17). Nonrandom cooccurrence patterns may indicate that community assembly is controlled by deterministic factors such as competitive interactions, habitat structuring, resource partitioning, commensal associations, and trophic level dynamics (11).
The field of microbial ecology is also subject to the controversies raised in macroecology. Several studies have raised the apparent conflict between explanations of community taxon abundance distributions using either niche assembly or neutral assembly models (18, 27). Sloan et al. (39, 40) derived a continuous form of a neutral community model (NCM) that explains microbial taxon abundance in terms of only stochastic dispersal, random speciation, and ecological drift. While the NCM provided a convincing fit to clone libraries for genes from a diverse collection of bacterial communities, there is some question as to whether these patterns could also be generated by deterministic mechanisms (27).
In this study, we describe patterns in the diversity of a coastal Vibrio cholerae population spanning 3 years and nine sampling sites along the central California coast, with the goal of testing whether population structure is controlled by deterministic and/or stochastic processes. The bacterial species V. cholerae was chosen for this analysis because of its importance as a human pathogen, its ubiquity in coastal waters across the globe (8, 9, 34), and its extensive genomic and phenotypic diversity (22, 28). Previous fingerprinting studies have repeatedly exposed a high degree of diversity among nontoxigenic V. cholerae isolates and especially non-O1/O139 serotype isolates compared to the primarily clonal relationships among both clinical and environmental isolates of the O1 and O139 serotypes (8, 21, 37). However, a thorough biogeographic analysis of environmental V. cholerae has not previously been performed.
Genomic fingerprinting based on conserved enterobacterial repetitive intergenic consensus sequence PCR (ERIC-PCR) is particularly amenable to characterizing genomic diversity within large collections of closely related isolates due to its low cost and technical ease (30, 44). This method provides a high level of discrimination between bacterial strains and correlates well with other measures of genome similarity, including pulsed-field gel electrophoresis, amplified fragment length polymorphism, multilocus enzyme electrophoresis, and DNA-DNA hybridization (33, 44). ERIC-PCR fingerprinting has been successfully implemented in prior studies of V. cholerae epidemiology (8), source tracking (36, 48), and genomic diversity (4, 21, 37).
Classifying our unique collection of environmental V. cholerae isolates from coastal waters and sediments into distinct ERIC-PCR genotypes allows us to assess patterns in the genomic diversity of isolates with respect to differences in geography, temporal proximity, and environmental conditions. Assembly of this microbial population in time and space is likely determined both by niche differentiation between genotypes or other deterministic mechanisms and by neutral processes, such as random dispersal, ecological drift, and chance (19). However, providing evidence for competitive structuring in a community beyond that which can be explained by stochastic processes is rarely possible (1, 27). Here, we use partial Mantel tests, NCMs, and cooccurrence indices to explore how stochastic and deterministic factors shape V. cholerae population structure.
MATERIALS AND METHODS
Sample collection and sediment processing.
During 11 sampling trips between January and December 2006, water column samples and hand-pushed sediment cores were collected at eight sampling sites along the central California coast (sites previously described by Keymer et al. [22]). A total of 601 novel vibrios were isolated from these samples, including 570 water column and 31 sediment isolates (see Tables S1 and S2 in the supplemental material). Water column isolates were obtained by membrane filtration of up to 100-ml samples and filter incubation on selective thiosulfate citrate bile sucrose (TCBS) agar plates without enrichment. The bacterial fraction was extracted from the sediment samples by a modified drill gun homogenization-centrifugation protocol adapted from the protocol of Lindahl and Bakken (24). Briefly, 1 g (wet weight) of sediment was homogenized in 5 ml sterile phosphate-buffered saline three times with a drill-mounted tissue macerator for 1 min. The suspension was centrifuged at 1,000 × g for 10 min, and the pooled supernatants were collected in a 15-ml sterile conical-bottom polypropylene tube. The tube was centrifuged at 10,000 × g for 20 min, and the supernatant was discarded. The cell pellet was resuspended in sterile phosphate-buffered saline and diluted serially. This cell fraction was then membrane filtered and incubated on TCBS agar like the water column samples. Suspected V. cholerae colonies on TCBS agar were screened and confirmed using biochemical and molecular methods as described previously (22). In brief, isolates were screened using l-ornithine decarboxylase broth and modified cellobiose-polymyxin B-collistin agar. Whole cells of presumptive positive isolates were used as the templates for PCR amplification of a 300-bp subset of the 16S-23S rRNA intergenic spacer with clinical O1 strain N16961 as a positive control and Escherichia coli as a negative control (7). Isolates yielding the correct band size in agarose gels were included in fingerprinting reactions described below.
In addition to the 601 isolates collected during 2006, we included the 41 environmental and 4 clinical isolates previously described by Keymer et al. (22), 1 water column isolate from August 2004, 162 water column isolates from June and July 2005, and 27 and 10 isolates obtained by alkaline peptone water (APW) enrichment from filters and crab shells, respectively (for a total of 846 isolates) (see Tables S1 and S2 in the supplemental material). For the filter enrichment isolates, water column samples were membrane filtered as described above, but the filters were vortexed in APW and incubated at 37°C for 6 h before aliquots of APW were spread plated on TCBS agar. One-square-centimeter pieces of crab shells recovered after 2 weeks of deployment at five sampling sites were also vortexed and incubated in APW prior to selection of isolates on TCBS agar. Some of these additional isolates were isolated from Kirby Park in Elkhorn Slough, which increased the number of coastal sampling sites to nine. Seven strains from the collection did not produce analyzable fingerprints and were classified as untyped, so the number of Vibrio isolates used in the study was 839. The genomic similarity of the 45 isolates described by Keymer et al. (22) has been well characterized using comparative genome hybridization (CGH), which provides a set of isolates that can be used to assess the accuracy of ERIC-PCR analysis methods. Below, these 45 isolates are referred to as the “calibration set”.
Environmental parameters.
Coincident with sample collection, water temperature, salinity, dissolved oxygen, pH, and turbidity were measured in situ using a calibrated Hydrolab Quanta water quality probe (HACH Environmental, Loveland, CO). Approximately 30-ml water samples were 0.2-μm syringe filtered into acid-washed containers and stored at −20°C prior to analysis for dissolved nutrients. Nutrient samples were sent to the UCSB Marine Science Institute Analytical Lab for analysis of ammonium, soluble reactive phosphate, nitrate plus nitrite, nitrite, and silicate using flow injection methods. Since the first four parameters were correlated over all sampling events, only ammonium concentration is included in the analysis of genotype distribution patterns. Log transformations of salinity, ammonium, and turbidity data were performed to ensure that the data were normally distributed. Raw environmental data are shown in Table S3 in the supplemental material.
ERIC-PCR.
Whole cells grown in LB broth with 1% NaCl were washed and resuspended in sterile water. One microliter of cell suspension was added to 24 μl of a master mixture containing 1.25 U high-fidelity Hot Star Taq polymerase (Qiagen), 1× HotStar HiFidelity PCR buffer, 1 mM additional MgSO4, and 2 μM each of 6-carboxyfluorescein-labeled primer ERIC2 and unlabeled primer ERIC1R (36). The thermal cycling conditions were the conditions described by Zo et al. (48), except for an initial 15-min hot start at 94°C. Three microliters of the PCR product was mixed with 22.5 μl of Hi-Di formamide (Applied Biosystems) and 0.5 μl of a custom MapMarker (Bioventures Inc., Murfreesboro, TN) consisting of 6-carboxyl-X-rhodamine-labeled size fragments (100 to 2,000 bp). Fragment analysis of the PCR-amplified genome fragments was performed with an ABI 3730XL DNA analyzer at the University of Wisconsin Biotechnology Center.
Processing of chromatograms.
The method described below for generating ERIC-PCR fingerprints from sample chromatograms was found to be superior to an automated band-calling method on the basis of precision, reproducibility, discriminatory power, and accuracy in classifying the calibration set (data not shown). Chromatogram files were uploaded to GelCompar II 5.0 (Applied Maths, Austin, TX) using the CrvConv filter. Curves were normalized to the internal standard and filtered using a rolling disk size of 16% to remove background noise and a least-squares cutoff of 0.03% to remove high-frequency noise. Filtered curves were compared using Pearson product-moment correlation (see Fig. S1 in the supplemental material), and the similarity matrix was exported to MATLAB 7.4 (MathWorks, Natick, MA).
Evaluation criteria for ERIC-PCR fingerprinting.
Four criteria were used to evaluate the ERIC-PCR fingerprinting method: precision, reproducibility, discriminatory power, and accuracy. Precision was computed as the mean of a lognormal distribution fit to the histogram of pairwise similarity between replicates. Replicates refer to multiple independent fingerprints (between 2 and 10 fingerprints; median, 2 fingerprints) obtained for the same isolate. For the other criteria, fingerprints were clustered into groups with intracluster similarity greater than a specified identity cutoff. Each cluster corresponds to one ERIC-PCR genotype and is designated by a unique identifier from 1 to 115. Reproducibility was calculated for all replicate fingerprints as the average percentage of replicates present in the cluster containing the most replicates of a given isolate. Discriminatory power (D) was computed for all fingerprints using equation 1 (20):
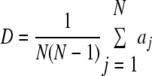 |
(1) |
where aj is the number of isolates represented by fingerprints in clusters besides the one containing isolate j and N is the total number of isolates examined. The value of D is equivalent to the average fraction of fingerprints distinguishable from a given fingerprint and ranges from 0 to 1, with 0 meaning that all fingerprints are indistinguishable and 1 indicating that each fingerprint is distinguishable from every other fingerprint. Accuracy was represented by the false-positive rate and false-negative rate for the calibration set, previously characterized using an independent CGH genotyping scheme (22). False positives were defined as two fingerprints from divergent isolates (identified by CGH) that fell into the same genotype cluster. In contrast, false negatives were defined as fingerprints from isolates with the same CGH profile that fell into different genotype clusters. Rates were computed using the following equations, where FP is false positives, FN is false negatives, TN is true negatives, and TP is true positives.
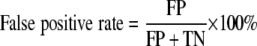 |
(2) |
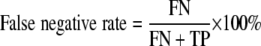 |
(3) |
Biogeography analysis.
Following the division of all fingerprints into ERIC-PCR genotypes, DNA sequences for the housekeeping gene dnaE were generated for representative isolates for all 115 genotypes (see Fig. S4 in the supplemental material). Six additional housekeeping genes were sequenced for isolates for 77 genotypes, and in all cases classification of isolates as species other than V. cholerae on the basis of dnaE sequence homology was verified by the additional locus sequences (D. P. Keymer and A. B. Boehm, unpublished data). Genotypes with <95% sequence similarity to V. cholerae (23) were classified as non-V. cholerae vibrios and excluded from further analysis.
The number of ERIC-PCR genotypes detected was plotted as a function of the number of isolates collected for each sampling event. Data points were fitted to both linear and power responses using least-squares regression. The power curve had the form y = axb + c. Goodness of fit for the two models was evaluated using Fisher's F test.
Simple and partial Mantel tests were performed with the open-source program zt (5) to test for correlation between genotypic dissimilarity of samples and geographic distance, while accounting for differences in environmental conditions. Downloads of zt are free through the Journal of Statistical Software website (http://www.jstatsoft.org/v07/i10). Matrices for Mantel tests were assembled in MATLAB 7.4. The genetic dissimilarity matrix used the Dice coefficient dissimilarity between the incidences of ERIC-PCR genotypes within each sampling event. The geographical distance matrix was based on the spherical law of cosine distance from latitude-longitude coordinates at each site. Simple Mantel tests were used to determine which environmental parameters were correlated with the genotypic dissimilarity matrix. Measured parameters were added stepwise to the environmental dissimilarity matrix to generate a matrix that had maximum correlation with the genotypic dissimilarity matrix and included the fewest parameters. The environmental distance matrix consisted of standardized Euclidean distances between values for water temperature, log salinity, and log ammonium concentration measured during each sampling event. The temporal distance matrix was comprised of the number of days between sampling events normalized to the study period duration. A false-discovery rate correction was used to determine the significance of testing multiple hypotheses (3). Values and ranges of correlation coefficients presented below reflect the individual correlation of geographic, environmental, or temporal distance with genotypic dissimilarity when both of the remaining predictor variables were controlled for.
Distance-decay relationships were estimated for log-log plots of genotypic similarity versus geographic and temporal distance. Bootstrapped linear regressions were performed with 10,000 replicate resamples to verify that taxon-area and taxon-time exponents were nonzero, as described previously (16).
NCMs were fitted with least squares to mean relative abundance and frequency data for each ERIC-PCR genotype across all sampling events based on the equations derived by Sloan et al. (40). NCMs were also fitted to sampling events for individual sites to assess V. cholerae community assembly on a local-versus-regional spatial scale. Coefficients of determination and root mean square errors (RMSEs) (normalized to range in dependent variable) were computed to assess goodness of fit for each model. NCMs were not fitted for individual sites that contained fewer than 16 genotypes (Kirby Park, Lagunitas Creek, Moss Landing Harbor, and San Pedro Creek).
Cooccurrence indices (C-board, Combo, and C-score) for incidence matrices of samples within individual sites or all sites were computed as described by Horner-Devine et al. (17), using EcoSim 7.72 (14). Singleton genotypes were removed from the matrices before analysis. Indices were not computed for individual sites that contained fewer than three nonsingleton genotypes (Kirby Park, Lagunitas Creek, Moss Landing Harbor, and San Pedro Creek). Standardized effect scores, equivalent to statistical z scores, are included below to allow comparison between data sets of different sizes.
All statistical comparisons except the Mantel tests were performed in MATLAB 7.4 (MathWorks).
Nucleotide sequence accession numbers.
The GenBank accession numbers for the DNA sequences determined in this study are FJ609424 to FJ609633.
RESULTS
Evaluation of fingerprint comparisons.
Several criteria were used to optimize and evaluate the assignment of genotypes from the ERIC-PCR fingerprints. Precision was computed using 244 replicates from 110 individual V. cholerae isolates and was determined to be 90.4% (95% confidence interval, 89.5 to 91.3%). An optimal identity cutoff of 80% was chosen to produce the best trade-off between reproducibility and discriminatory power (see Fig. S2 in the supplemental material). At the 80% cutoff value, the method had a reproducibility of 98.5% and an index of discriminatory power of 0.971.
The high-resolution CGH approach used by Keymer et al. (22) identified several groups of apparently identical V. cholerae isolates. Using the same isolates characterized in that study, we compared clusters generated from the ERIC-PCR fingerprints with different identity cutoffs to the clusters defined using CGH. False positives (divergent isolates clustering together) and false negatives (identical isolates clustering separately) were tallied over a range of similarity cutoffs (see Fig. S3 in the supplemental material). When evaluated at the optimal identity cutoff, 80%, our genotype classification method had a false-positive rate of 0.57% and a false-negative rate of 15.0%.
Fingerprint analysis of entire strain collection.
The entire collection of ERIC-PCR fingerprints was analyzed using an 80% identity cutoff to discern distinct genotypes. A total of 998 fingerprints, including replicates, from 839 isolates were divided into 115 ERIC-PCR genotypes, each represented by between 1 and 72 isolates (see Fig. S1 in the supplemental material). Nucleotide sequence analysis using the DNA polymerase I gene (dnaE) (see Fig. S4 in the supplemental material) divided the genotypes into confirmed V. cholerae isolates (99 genotypes, 799 isolates) and other putative vibrios (16 genotypes, 40 isolates). Genotypes classified as non-V. cholerae vibrios (genotypes 38, 50, 62, 65 to 67, 70, 72 to 75, 103, 107, 112, and 114 to 115) were excluded from further analysis. Our calibration set of 45 V. cholerae isolates is ≥99% similar at the 16S rRNA gene level (data not shown); however, ERIC-PCR fingerprinting classified the 45 isolates into 21 different genotypes.
The genotype richness of isolates collected during most sampling events did not level off with increasing sampling effort, even though we collected up to 50 isolates per event. For the 97 sampling events, we observed a power law relationship in the number of genotypes detected with the number of isolates collected (Fig. 1) (r2 = 0.71). The power law curve provided a better fit than the linear model (r2 = 0.68, F = 9.49, P < 0.05). Samples below the regression line were relatively closer to saturation of diversity, while samples above the line were relatively further from saturation. The isolates obtained by enrichment had relatively low diversity and fell below the regression line, along with the isolates collected at Waddell Creek in June 2006. Conversely, the samples from San Lorenzo River collected in June, July, and August 2006 had relatively high diversity and fell above the regression line.
FIG. 1.

Relationship between sample size and ERIC-PCR genotype diversity. The number of ERIC-PCR genotypes detected in a sample increases according to a power law relationship with the total number of isolates collected (y = axb + c, where a is 1.48 ± 1.47, b is 0.60 ± 0.24, and c is −0.51 ± 1.97; r2 = 0.71). The power law model fit better than a linear model (r2 = 0.68) based on Fisher's F test (F = 9.49, P < 0.05). Samples discussed in the text are labeled for clarity.
Striking patterns of temporal and spatial occurrence were observed for some genotypes. Forty-seven different genotypes were detected during three or more sampling months, and nine genotypes were found in more than 5 months. Thus, several genotypes appear to be temporally persistent in the coastal environment. Analogously, 26 genotypes displayed relatively wide geographic occurrence, since they were present at between three and six of our nine sites. These 26 genotypes appear to be cosmopolitan in nature and able to tolerate a range of environmental conditions (see Table S2 in the supplemental material). Interestingly, geographically restricted genotypes (the genotypes found during multiple months at just one site) were relatively rare. Only five genotypes were collected in three or more months and were restricted to a single site. These five potentially endemic genotypes were isolated from Pescadero Creek, Old Salinas River, San Lorenzo River, and Waddell Creek. Latitude-longitude coordinates for each site are shown in Table S4 in the supplemental material, along with pairwise distances (in kilometers) between sites.
The distribution of persistent genotypes at Pescadero Creek, Old Salinas River, San Lorenzo River, and Waddell Creek is shown in Fig. 2 to illustrate interesting spatial and temporal patterns in genotype occurrence. For instance, genotypes 27, 45, 86, and 89 are cosmopolitan and were repeatedly found across the sites. In contrast, genotypes 1, 98, 99, and 113 are restricted to a single site. Temporal shifts in the genotypic composition of particular sites are also evident in Fig. 2. Genotype 90 appears to dominate the V. cholerae population in Waddell Creek in June 2006, but less than 30 days later most of its contribution to the sample diversity has been replaced by a number of other genotypes. Genotype 54 is numerically dominant in the Old Salinas River during many summer months, but its contribution declines throughout the rest of the year. Cooccurrence of some genotypes also appears to be common. For example, genotypes 1 and 111, 16 and 33, and 21, 42, 56, and 82 repeatedly occur together during various sampling events.
FIG. 2.

Proportions of selected genotypes collected from four sites over the entire sampling period. The numbers above the bars indicate the numbers of water column and sediment isolates collected and typed for the sampling event. The letters at the bottom indicate the months in which samples were collected. Each sample bar is divided into areas representing different genotypes (indicated by different colors and patterns) detected in the sample. To improve visual appearance, rare genotypes that appeared only once in the entire data set are indicated by white areas, while other genotypes that were not persistent in time or space are indicated by light gray areas. Genotypes classified as non-V. cholerae vibrios (non-VC) are indicated by black areas.
Biogeographic patterns in sample diversity.
We examined how the distribution of isolates collected throughout our study is controlled by geographical distance, temporal separation, and environmental dissimilarity. First, a log-log relationship was estimated for genotypic similarity with geographic distance and temporal distance (29). The taxon-area exponent (z) for a least-squares fit is significantly negative (P < 0.0001) and equal to −0.043 ± 0.013 (see Fig. S5A in the supplemental material). The time-taxon exponent is similar in magnitude (−0.047 ± 0.024, P = 0.0001) (see Fig. S5B in the supplemental material). It should be noted that the correlations of diversity with distance or time do not distinguish between effects of contemporary environmental heterogeneity and effects of dispersal and history.
Simple Mantel tests revealed significant intercorrelations (P < 0.05) between our deterministic predictor variables (environmental, geographic, and temporal distances). Ammonium concentration was the most highly correlated individual environmental predictor of genotypic dissimilarity, followed by salinity and then water temperature. The partial Mantel tests revealed that the environmental conditions, sampling date, and geographic locations of the sampling events had significant, although small, independent effects on the genetic dissimilarity between samples (Table 1). Recall that the partial Mantel tests control for intercorrelation between predictor variables. Environmental differences had a relatively smaller effect on genotype similarity among samples (0.04 ≤ r ≤ 0.05, P < 0.008), and days between samples (r = 0.09, P = 0.0001) and kilometers between sites (r = 0.09, P < 0.0001) were equally significant predictors. While the geographic, temporal, and environmental associations were significantly correlated with the genetic similarity between samples, greater than 95% of the variability in the data remains unexplained by the variability in our predictor matrices.
TABLE 1.
Results of simple and partial Mantel tests between distance matrices for genotypic dissimilarity, environmental dissimilarity, geographic distance, and temporal separation of isolates from each sampling eventa
| Dependent variable | Independent variable | Controlling for | r | P value |
|---|---|---|---|---|
| Environmental dissimilarity | Temporal separation | 0.081 | 0.0034 | |
| Geographic distance | Environmental dissimilarity | 0.141 | 0.0019 | |
| Genotypic dissimilarity | Water temperature | 0.023 | 0.1094 | |
| Genotypic dissimilarity | Salinity | 0.033 | 0.0366 | |
| Genotypic dissimilarity | Turbidity | −0.010 | 0.3094 | |
| Genotypic dissimilarity | Ammonium | 0.049 | 0.0078 | |
| Genotypic dissimilarity | Environmental dissimilarity | 0.061 | 0.0004 | |
| Genotypic dissimilarity | Environmental dissimilarity | Geographic distance | 0.047 | 0.0074 |
| Genotypic dissimilarity | Environmental dissimilarity | Temporal separation | 0.053 | 0.0028 |
| Genotypic dissimilarity | Geographic distance | 0.102 | 0.0001 | |
| Genotypic dissimilarity | Geographic distance | Environmental dissimilarity | 0.094 | 0.0001 |
| Genotypic dissimilarity | Geographic distance | Temporal separation | 0.097 | 0.0001 |
| Genotypic dissimilarity | Temporal separation | 0.101 | 0.0001 | |
| Genotypic dissimilarity | Temporal separation | Geographic distance | 0.095 | 0.0001 |
| Genotypic dissimilarity | Temporal separation | Environmental dissimilarity | 0.096 | 0.0001 |
Simple and partial Mantel test data are shown in rows containing two and three comparison variables, respectively. All correlations were statistically significant (P < 0.05 with false discovery rate correction), except for the simple Mantel tests between genotypic dissimilarity and water temperature and between genotypic dissimilarity and turbidity. Ranges of r in the text represent the individual correlations of geographic, environmental, or temporal distance with genotypic dissimilarity when both remaining factors were accounted for.
To assess whether the diversity not explained by the Mantel tests could be due to a neutral community structure, we used Sloan et al.'s NCM to fit mean relative abundance and frequency data for each ERIC-PCR genotype (40). It has been argued that good agreement between observed taxon abundance patterns and the NCM fit is evidence for a prominent role of neutral, stochastic processes in assembly of the observed communities (39, 40, 47). When abundance and frequency of genotypes over all 97 sampling events are used to fit the NCM, an r2 value of 0.61 is obtained. Using the total number of isolates as a minimum population size (NT), a maximum possible immigration probability (m) of 0.0003 is calculated (Table 2). In spite of the curve capturing the general trend in the data, there is a fair amount of disagreement between the observed data and the model (RMSE = 0.143) (Fig. 3). When fitted to abundance data from individual sites, the NCMs produced r2 values ranging from 0.37 to 0.85, with 0.0009 ≤ m ≤ 0.0045 and equally prominent scatter in the observed abundance data (Table 2 and Fig. 3).
TABLE 2.
Fitting parameters (NT and m) and goodness of fit (r2 and RMSE) for NCMsa
| Group | NTm | 95% confidence interval | NT | r2 | RMSE | m |
|---|---|---|---|---|---|---|
| All samples | 0.26 | 0.21-0.30 | 795 | 0.61 | 0.143 | 0.0003 |
| Old Salinas River | 0.28 | 0.20-0.35 | 199 | 0.56 | 0.107 | 0.0014 |
| Pescadero Creek | 0.50 | 0.44-0.57 | 157 | 0.85 | 0.075 | 0.0032 |
| San Lorenzo River | 0.32 | 0.23-0.41 | 196 | 0.37 | 0.152 | 0.0016 |
| Soquel Creek | 0.19 | 0.14-0.23 | 41 | 0.74 | 0.066 | 0.0045 |
| Waddell Creek | 0.15 | 0.10-0.20 | 169 | 0.49 | 0.171 | 0.0009 |
NT is the number of individuals in the population (minimum population size), and m is the maximum probability at which a dead individual is replaced by an immigrant from the source community. RMSE is normalized to the range of the dependent variable, a measure of scatter from the predicted curve.
FIG. 3.

Fit of NCM to observed isolation frequency and mean relative abundance for ERIC-PCR genotypes from all environmental samples and from a subset of individual sampling sites. The open circles represent individual genotypes, and the dashed lines indicate the least-squares best fit. Fitting parameters are shown in Table 2.
Nonrandom patterns in cooccurrence of ERIC-PCR genotypes may indicate the importance of deterministic factors such as competition and niche differences in shaping the V. cholerae population assembly. Three cooccurrence indices (C-board, Combo, and C-score) were computed for incidence matrices from all sampling events or individual sites and compared to indices computed for null matrices (Table 3). C-board is the number of genotype pairs never occurring in the same sample, Combo is the number of unique genotype cooccurrences between sites, and C-score is the average number of “checkerboard” units, as described by Stone and Roberts (41), between all genotype pairs. Based on Diamond's community assembly rules, a competitively structured community should yield higher C-board, higher C-score, and lower Combo values than expected by chance (11, 15). Indeed, across all sampling events, C-board and C-score are significantly higher than expected by chance (P = 0.001 and P = 0.019, respectively), and Combo is significantly lower (P < 0.001), pointing to a competitively structured community. Similar results were obtained for cooccurrence at individual sites (Table 3). C-score was higher than expected by chance for Old Salinas River (P = 0.044), Pescadero Creek (P = 0.024), and Waddell Creek (P = 0.002). C-board was also statistically higher than the null model value for two sites: San Lorenzo River (P = 0.012) and Waddell Creek (P = 0.028).
TABLE 3.
Cooccurrence indices computed for incidence matrices from all sampling events or individual sites relative to null matrices using EcoSim 7.72a
| Samples | C-score
|
C-board
|
Combo
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Observed | Mean null | Variance null | P (observed ≤ null) | P (observed ≥ null) | SES | Observed | Mean null | Variance null | P (observed ≤ null) | P (observed ≥ null) | SES | Observed | Mean null | Variance null | P (observed ≤ null) | P (observed ≥ null) | SES | |
| All | 19.4 | 19.1 | 0.015 | 0.981 | 0.019b | 2.45 | 1,192 | 1,137 | 250 | 0.999 | 0.001b | 3.47 | 69.0 | 75.2 | 4.92 | 0.000b | 1.000 | −2.80 |
| Old Salinas River | 4.04 | 3.81 | 0.014 | 0.960 | 0.044b | 1.94 | 66.0 | 64.3 | 24.1 | 0.676 | 0.404 | 0.35 | 15.0 | 14.8 | 0.72 | 0.773 | 0.657 | 0.21 |
| Pescadero Creek | 9.35 | 9.07 | 0.016 | 0.098 | 0.024b | 2.16 | 79.0 | 73.0 | 15.1 | 0.947 | 0.085 | 1.54 | 20.0 | 20.4 | 0.44 | 0.544 | 0.903 | −0.53 |
| San Lorenzo River | 3.22 | 3.23 | 0.007 | 0.497 | 0.524 | −0.14 | 87.0 | 71.0 | 45.3 | 0.993 | 0.012b | 2.38 | 14.0 | 15.0 | 0.66 | 0.263 | 0.966 | −1.21 |
| Waddell Creek | 2.40 | 1.77 | 0.026 | 0.998 | 0.002b | 3.92 | 16.0 | 7.71 | 7.55 | 0.996 | 0.028b | 3.02 | 8.00 | 9.19 | 0.47 | 0.145 | 0.994 | −1.74 |
See reference 14. C-board is the number of genotype pairs never occurring in the same sample, Combo is the number of unique genotype cooccurrences between sites, and C-score is the average number of “checkerboard” units between samples. SES, standardized effect score. Kirby Park, Lagunitas Creek, Moss Landing Harbor, San Pedro Creek, and Soquel Creek were excluded due to small numbers of nonsingleton genotypes (<3).
There was significant deviation in the indices (P < 0.05) from values expected by chance (null distribution).
DISCUSSION
Similarity in the composition of V. cholerae assemblages in central California coastal waters decreased as a function of both time and space between sampling events. We observed a significant negative log-log relationship between genotypic diversity and geographic distance. The taxon-area exponent (z = −0.043 ± 0.013) is very small but significantly nonzero. This value is within the range of values computed for salt marsh bacteria and other microbial communities (−0.02 ≤ z ≤ −0.07) (12, 16) but lower than values computed for high-mountain-lake bacteria (z = 0.16) (35) and beech tree hole bacterial communities (z = 0.26) (2). The small sample sizes used here and in other studies allow only systematic shifts in abundant taxa to be detected using the taxon-area relationship, so the actual absolute value of the exponent could be much larger if rare taxa behave similarly (46). The small exponent observed for the V. cholerae taxon-area relationship probably indicates some niche overlap between intraspecific genotypes (29). A negative log-log relationship was also observed between genotypic similarity and temporal distance of sampling events (z = −0.047 ± 0.024). The taxon-time exponent is much smaller than the exponents estimated for bacteria treating industrial wastewater (−0.512 ≤ z ≤ −0.162) (43). Our smaller exponent compared to the industrial wastewater reactor exponents suggests that there is higher temporal stability in the V. cholerae population, but it could stem from differences in the taxon-level resolution as well.
The distance-decay relationships described above provide an elementary tool to describe distribution patterns in microbial communities, but they cannot distinguish the effects of multiple interrelated factors. Therefore, we used partial Mantel tests to quantify independent effects of our geographic, temporal, and environmental predictor matrices on genotype occurrence across sampling events. All three of our predictor variables had significant, but minor effects on the genotype dissimilarity between samples. The correlation coefficients for the partial Mantel tests are somewhat lower than the values that have been seen for environmental effects in salt marsh bacteria (0.26 ≤ r ≤ 0.37) (16), for spatial effects in high-mountain-lake bacteria (r = 0.29) (35), and for pH effects on soil bacterial communities (r = 0.75) (12). In contrast to previous studies, we found evidence for independent effects on genotypic similarity from both environmental heterogeneity and spatial distance, as well as temporal proximity. The fact that we are able to detect geographic effects in our data set means that dispersal rates are not high enough to mask a distance-decay relationship (26). Correlation between environmental similarity and genotypic similarity implies that local deterministic factors and niche differences play a role in shaping V. cholerae population structure. However, the relatively strong temporal influence on genotypic similarity across sites up to 100 km apart suggests that regional factors acting simultaneously at all sites are also important. These factors could include biological interactions with other seasonally abundant taxa or fluctuations in climate and near-shore oceanographic phenomena.
The amount of variability in the genotype dissimilarity matrix explained by deterministic factors (location, time, and environmental conditions) according to the partial Mantel tests was less than 5% of the total variability, so much of the variability remains unexplained. There are several possible explanations for this result. We may not have measured some environmental parameters that are important in controlling V. cholerae population structure. V. cholerae may inhabit microniches where environmental parameters vary immensely from those measured and assigned to the sampling event (31, 45). Niche selection may occur at the level of gene content or gene regulation, which is finer than the resolution that we observe with genome fingerprinting. If this is the case, genotypes with similar genome fingerprints may respond very differently to changes in their environment. Alternatively, it may be possible that isolates that are classified as having different ERIC-PCR genotypes but are similar in some portion of their gene content experience selection with the same environmental cues (22). Finally, most of the diversity observed among isolates in this environment may be neutral and shaped by stochastic processes (42). To further explore the latter explanation, the role of stochastic and deterministic processes in controlling genotype distribution patterns was examined using NCMs and cooccurrence indices.
NCMs appear to describe much of the distribution of genotypes, suggesting that stochastic processes play an important role in shaping population structure. The NCMs fit our taxon abundance data better than the human fecal community example described by Sloan et al. (40), but our r2 values are relatively low and the curve fits are less convincing than those for other bacterial communities (39, 47). However, the simple NCMs can very rarely be rejected based on statistical considerations, and simulations show that under most circumstances niche and neutral models are indistinguishable, even under strong selective pressure (1, 27). Therefore, caution should be taken in interpretation of these results; they do not imply that deterministic processes are unimportant. To the contrary, we interpret the relatively weak NCM fit as evidence that stochastic processes, while important, are not sufficient to fully explain the observed patterns in genotype diversity.
The nonrandom cooccurrence indices suggest that competitive structuring of the V. cholerae population is important. The C-board and C-score indices were significantly larger than the indices generated for the null matrices (P = 0.001 and P = 0.019, respectively), and Combo was significantly smaller (P < 0.001), when all samples were compared across sites. Cooccurrence indices for samples within individual sites gave similar results, with three sites and two sites of four sites tested having significantly larger C-score and C-board indices than expected by chance, respectively. Competitive exclusion within the same niche is one mechanism that could produce higher-than-expected C-board and C-score values, but environmental differences between sites could also lead to habitat exclusion for some genotypes at the regional scale (1). These results highlight the potential importance of deterministic processes in shaping population structure. However, the work described here did not identify which deterministic factors ultimately control the observed patterns in V. cholerae biogeography, so further work is needed to verify effects of individual factors.
The results presented here ultimately depend on the tool that we used to discriminate genotypes. ERIC-PCR has been used by other researchers to explore V. cholerae diversity (21, 36, 37), and our independent evaluation of the ERIC-PCR analysis method confirmed that this genomic fingerprinting tool could be used to accurately define V. cholerae genotypes. However, other genomic fingerprinting methods (e.g., repetitive extragenic palindromic PCR with BOX or GTG5 primers), as well as multilocus sequence analysis, may provide greater discrimination between Vibrio isolates (13, 33, 38). It will be interesting to see how the results of biogeographic analysis of V. cholerae using these tools to define genotypes compare to our results.
The vast difference between practical sample size and actual population size in a site is an inherent problem in environmental microbial ecology studies (10). Due to constraints on time and resources, we could collect only up to 50 V. cholerae isolates for each sampling event. Based on our analysis of the relationship between sample size and genotype diversity, we found that there was a power law increase in the number of genotypes detected as the number of isolates collected increased. Therefore, the patterns that we observed take into account only the most abundant culturable genotypes, even though rare genotypes should be primarily responsible for driving spatial or other patterns in genotype distribution (46). We cannot currently assess how the results observed here would be affected by the use of culture-independent methods or larger sample sizes, but future work in these and other ecosystems should address this issue. Although future work must assess how well the patterns observed for our collection of isolates can be extrapolated to the larger coastal V. cholerae population, the nonrandom genotype distributions observed here provide evidence that deterministic factors have a small, but meaningful role in shaping the coastal V. cholerae population.
Supplementary Material
Acknowledgments
We thank Chris A. Francis, Alyson E. Santoro, Jason M. Smith, George F. Wells, and two anonymous reviewers for their constructive comments on a previous version of the manuscript. Nick de Sieyes, Tim Julian, Blythe Layton, Alyson Santoro, and Kevan Yamahara provided valuable support in the laboratory.
This work was funded by NOAA Oceans and Human Health Initiative grant NA04OAR4600195 (D.P.K., L.H.L., and A.B.B.), NSF grant OCE-0742048 (D.P.K. and A.B.B.), and a Gerhard Casper Stanford Graduate Fellowship (D.P.K.).
Footnotes
Published ahead of print on 9 January 2009.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Bell, G. 2005. The co-distribution of species in relation to the neutral theory of community ecology. Ecology 86:1757-1770. [Google Scholar]
- 2.Bell, T., D. Ager, J. I. Song, J. A. Newman, I. P. Thompson, A. K. Lilley, and C. J. van der Gast. 2005. Larger islands house more bacterial taxa. Science 308:1884. [DOI] [PubMed] [Google Scholar]
- 3.Benjamini, Y., and Y. Hochberg. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57:289-300. [Google Scholar]
- 4.Bhanumathi, R., F. Sabeena, S. R. Isac, B. N. Shukla, and D. V. Singh. 2003. Molecular characterization of Vibrio cholerae O139 Bengal isolated from water and the aquatic plant Eichhornia crassipes in the River Ganga, Varanasi, India. Appl. Environ. Microbiol. 69:2389-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonnet, E., and Y. Van de Peer. 2002. zt: a software tool for simple and partial Mantel tests. J. Stat. Software. http://www.jstatsoft.org/v07/i10.
- 6.Reference deleted.
- 7.Chun, J., A. Huq, and R. R. Colwell. 1999. Analysis of 16S-23S rRNA intergenic spacer regions of Vibrio cholerae and Vibrio mimicus. Appl. Environ. Microbiol. 65:2202-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colombo, M. M., S. Mastrandrea, F. Leite, A. Santona, S. Uzzau, P. Rappelli, M. Pisano, S. Rubino, and P. Cappuccinelli. 1997. Tracking of clinical and environmental Vibrio cholerae O1 strains by combined analysis of the presence of toxin cassette, plasmid content and ERIC PCR. FEMS Immunol. Med. Microbiol. 19:33-45. [DOI] [PubMed] [Google Scholar]
- 9.Colwell, R. R., J. Kaper, and S. W. Joseph. 1977. Vibrio cholerae, Vibrio parahaemolyticus, and other vibrios: occurrence and distribution in Chesapeake Bay. Science 198:394-396. [PubMed] [Google Scholar]
- 10.Curtis, T. P., I. M. Head, M. Lunn, S. Woodcock, P. D. Schloss, and W. T. Sloan. 2006. What is the extent of prokaryotic diversity? Philos. Trans. R. Soc. Lond. B 361:2023-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diamond, J. M. 1975. Assembly of species communities, p. 342-444. In M. L. Cody and J. M. Diamond (ed.), Ecology and evolution of communities. Harvard University Press, Cambridge, MA.
- 12.Fierer, N., and R. B. Jackson. 2006. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 103:626-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez-Gil, B., S. Soto-Rodriguez, A. Garcia-Gasca, A. Roque, R. Vazquez-Juarez, F. L. Thompson, and J. Swings. 2004. Molecular identification of Vibrio harveyi-related isolates associated with diseased aquatic organisms. Microbiology 150:1769-1777. [DOI] [PubMed] [Google Scholar]
- 14.Gotelli, N. J., and G. L. Entsminger. 2007. EcoSim: null models software for ecology, 7.72 ed. Acquired Intelligence Inc. & Kesey-Bear, Jericho, VT.
- 15.Gotelli, N. J., and D. J. McCabe. 2002. Species co-occurrence: a meta-analysis of J. M. Diamond's assembly rules model. Ecology 83:2091-2096. [Google Scholar]
- 16.Horner-Devine, M. C., M. Lage, J. B. Hughes, and B. J. Bohannan. 2004. A taxa-area relationship for bacteria. Nature 432:750-753. [DOI] [PubMed] [Google Scholar]
- 17.Horner-Devine, M. C., J. M. Silver, M. A. Leibold, B. J. M. Bohannan, R. K. Colwell, J. A. Fuhrman, J. L. Green, C. R. Kuske, J. B. H. Martiny, G. Muyzer, L. Overas, A.-L. Reysenbach, and V. H. Smith. 2007. A comparison of taxon co-occurrence patterns for macro- and microorganisms. Ecology 88:1345-1353. [DOI] [PubMed] [Google Scholar]
- 18.Hubbell, S. P. 2006. Neutral theory and the evolution of ecological equivalence. Ecology 87:1387-1398. [DOI] [PubMed] [Google Scholar]
- 19.Hubbell, S. P. 2001. The unified theory of biodiversity and biogeography. Princeton University Press, Princeton, NJ.
- 20.Hunter, P. R. 1990. Reproducibility and indices of discriminatory power of microbial typing methods. J. Clin. Microbiol. 28:1903-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang, S., W. Chu, and W. Fu. 2003. Prevalence of cholera toxin genes (ctxA and zot) among non-O1/O139 Vibrio cholerae strains from Newport Bay, California. Appl. Environ. Microbiol. 69:7541-7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keymer, D. P., M. C. Miller, G. K. Schoolnik, and A. B. Boehm. 2007. Genomic and phenotypic diversity of coastal Vibrio cholerae strains is linked to environmental factors. Appl. Environ. Microbiol. 73:3705-3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konstantinidis, K. T., A. Ramette, and J. M. Tiedje. 2006. The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. B 361:1929-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindahl, V., and L. R. Bakken. 1995. Evaluation of methods for extraction of bacteria from soil. FEMS Microbiol. Ecol. 16:135-142. [Google Scholar]
- 25.MacArthur, R. H., and E. O. Wilson. 1967. The theory of island biogeography. Princeton University Press, Princeton, NJ.
- 26.Martiny, J. B. H., B. J. M. Bohannan, J. H. Brown, R. K. Colwell, J. A. Fuhrman, J. L. Green, M. C. Horner-Devine, M. Kane, J. A. Krumins, C. R. Kuske, P. J. Morin, S. Naeem, L. Ovreas, A.-L. Reysenbach, V. H. Smith, and J. T. Staley. 2006. Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4:102-112. [DOI] [PubMed] [Google Scholar]
- 27.McGill, B. J., B. A. Maurer, and M. D. Weiser. 2006. Empirical evaluation of neutral theory. Ecology 87:1411-1423. [DOI] [PubMed] [Google Scholar]
- 28.Miller, M. C., D. P. Keymer, A. Avelar, A. B. Boehm, and G. K. Schoolnik. 2007. Detection and transformation of genome segments that differ within a coastal population of Vibrio cholerae strains. Appl. Environ. Microbiol. 73:3695-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nekola, J. C., and P. S. White. 1999. The distance decay of similarity in biogeography and ecology. J. Biogeogr. 26:867-878. [Google Scholar]
- 30.Olive, D. M., and P. Bean. 1999. Principles and applications of methods for DNA-based typing of microbial organisms. J. Clin. Microbiol. 37:1661-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polz, M. F., D. E. Hunt, S. P. Preheim, and D. M. Weinreich. 2006. Patterns and mechanisms of genetic and phenotypic differentiation in marine microbes. Philos. Trans. R. Soc. Lond. B 361:2009-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prosser, J. I., B. J. M. Bohannan, T. P. Curtis, R. J. Ellis, M. K. Firestone, R. P. Freckleton, J. L. Green, L. E. Green, K. Killham, J. J. Lennon, A. M. Osborn, M. Solan, C. J. van der Gast, and J. P. W. Young. 2007. The role of ecological theory in microbial ecology. Nat. Rev. Microbiol. 5:384-392. [DOI] [PubMed] [Google Scholar]
- 33.Rademaker, J. L. W., B. Hoste, F. J. Louws, K. Kersters, J. Swings, L. Vauterin, P. Vauterin, and F. J. de Bruijn. 2000. Comparison of AFLP and rep-PCR genomic fingerprinting with DNA-DNA homology studies: Xanthomonas as a model system. Int. J. Syst. Evol. Microbiol. 50:665-677. [DOI] [PubMed] [Google Scholar]
- 34.Radu, S., Y. K. Ho, S. Lihan, Yuherman, G. Rusul, R. M. Yasin, J. Khair, and N. Elhadi. 1999. Molecular characterization of Vibrio cholerae O1 and non-O1 from human and environmental sources in Malaysia. Epidemiol. Infect. 123:225-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reche, I., E. Pulido-Villena, R. Morales-Baquero, and E. O. Casamayor. 2005. Does ecosystem size determine aquatic bacterial richness? Ecology 86:1715-1722. [Google Scholar]
- 36.Rivera, I. G., M. A. Chowdhury, A. Huq, D. Jacobs, M. T. Martins, and R. R. Colwell. 1995. Enterobacterial repetitive intergenic consensus sequences and the PCR to generate fingerprints of genomic DNAs from Vibrio cholerae O1, O139, and non-O1 strains. Appl. Environ. Microbiol. 61:2898-2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shangkuan, Y. H., H. C. Lin, and T. M. Wang. 1997. Diversity of DNA sequences among Vibrio cholerae O1 and non-O1 isolates detected by whole-cell repetitive element sequence-based polymerase chain reaction. J. Appl. Microbiol. 82:335-344. [DOI] [PubMed] [Google Scholar]
- 38.Singh, D. V., M. H. Matte, G. R. Matte, S. Jiang, F. Sabeena, B. N. Shukla, S. C. Sanyal, A. Huq, and R. R. Colwell. 2001. Molecular analysis of Vibrio cholerae O1, O139, non-O1, and non-O139 strains: clonal relationships between clinical and environmental isolates. Appl. Environ. Microbiol. 67:910-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sloan, W., S. Woodcock, M. Lunn, I. Head, and T. Curtis. 2007. Modeling taxa-abundance distributions in microbial communities using environmental sequence data. Microb. Ecol. 53:443-455. [DOI] [PubMed] [Google Scholar]
- 40.Sloan, W. T., M. Lunn, S. Woodcock, I. M. Head, S. Nee, and T. P. Curtis. 2006. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 8:732-740. [DOI] [PubMed] [Google Scholar]
- 41.Stone, L., and A. Roberts. 1990. The checkerboard score and species distributions. Oecologia 85:74-79. [DOI] [PubMed] [Google Scholar]
- 42.Thompson, J. R., S. Pacocha, C. Pharino, V. Klepac-Ceraj, D. E. Hunt, J. Benoit, R. Sarma-Rupavtarm, D. L. Distel, and M. F. Polz. 2005. Genotypic diversity within a natural coastal bacterioplankton population. Science 307:1311-1313. [DOI] [PubMed] [Google Scholar]
- 43.van der Gast, C. J., D. Ager, and A. K. Lilley. 2008. Temporal scaling of bacterial taxa is influenced by both stochastic and deterministic ecological factors. Environ. Microbiol. 10:1411-1418. [DOI] [PubMed] [Google Scholar]
- 44.Versalovic, J., F. J. de Bruijn, and J. R. Lupski. 1998. Repetitive sequence-based PCR (rep-PCR) DNA fingerprinting of bacterial genomes, p. 437-454. In F. J. de Bruijn, J. R. Lupski, and G. M. Weinstock (ed.), Bacterial genomes: physical structure and analysis. Chapman & Hall, New York, NY.
- 45.Whitaker, R. J., and J. F. Banfield. 2006. Population genomics in natural microbial communities. Trends Ecol. Evol. 21:508-516. [DOI] [PubMed] [Google Scholar]
- 46.Woodcock, S., T. P. Curtis, I. M. Head, M. Lunn, and W. T. Sloan. 2006. Taxa-area relationships for microbes: the unsampled and the unseen. Ecol. Lett. 9:805-812. [DOI] [PubMed] [Google Scholar]
- 47.Woodcock, S., C. J. van der Gast, T. Bell, M. Lunn, T. P. Curtis, I. M. Head, and W. T. Sloan. 2007. Neutral assembly of bacterial communities. FEMS Microbiol. Ecol. 62:171-180. [DOI] [PubMed] [Google Scholar]
- 48.Zo, Y. G., I. N. Rivera, E. Russek-Cohen, M. S. Islam, A. K. Siddique, M. Yunus, R. B. Sack, A. Huq, and R. R. Colwell. 2002. Genomic profiles of clinical and environmental isolates of Vibrio cholerae O1 in cholera-endemic areas of Bangladesh. Proc. Natl. Acad. Sci. USA 99:12409-12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.