

Abstract
Agrobacterium tumefaciens requires phosphatidylcholine (PC) in its membranes for plant infection. The phospholipid N-methyltransferase PmtA catalyzes all three transmethylation reactions of phosphatidylethanolamine (PE) to PC via the intermediates monomethylphosphatidylethanolamine (MMPE) and dimethylphosphatidylethanolamine (DMPE). The enzyme uses S-adenosylmethionine (SAM) as the methyl donor, converting it to S-adenosylhomocysteine (SAH). Little is known about the activity of bacterial Pmt enzymes, since PC biosynthesis in prokaryotes is rare. In this article, we present the purification and in vitro characterization of A. tumefaciens PmtA, which is a monomeric protein. It binds to PE, the intermediates MMPE and DMPE, the end product PC, and phosphatidylglycerol (PG) and phosphatidylinositol. Binding of the phospholipid substrates precedes binding of SAM. We used a coupled in vitro assay system to demonstrate the enzymatic activity of PmtA and to show that PmtA is inhibited by the end products PC and SAH and the antibiotic sinefungin. The presence of PG stimulates PmtA activity. Our study provides insights into the catalysis and control of a bacterial phospholipid N-methyltransferase.
Phospholipid N-methyltransferases (Pmt enzymes) catalyze the methylation of phosphatidylethanolamine (PE) to form either the N-methylated intermediates monomethylphosphatidylethanolamine (MMPE) and dimethylphosphatidylethanolamine (DMPE) or fully methylated phosphatidylcholine (PC). The methyl group derives from S-adenosylmethionine (SAM), which is converted to S-adenosylhomocysteine (SAH) (37) (Fig. 1).
FIG. 1.

Phospholipid N-methylation pathway of PC biosynthesis in bacteria. Phospholipid N-methyltransferase(s) (Pmt) catalyzes the three-step methylation of PE to PC via MMPE and DMPE. DAG, diacylglycerol.
Pmt enzymes occur in mammals, fungi, yeasts, and a restricted number of bacteria (37). The sequences of eukaryotic and prokaryotic enzymes differ substantially. Furthermore, the number of Pmt enzymes participating in the three-step methylation of PE to PC varies among organisms. In mammals, all three methylation reactions depend on a single gene coding for the two protein isoforms PEMT1 and PEMT2 (41). Hepatic phosphatidylethanolamine N-methyltransferase (PEMT) enzymes are transmembrane proteins localized to the endoplasmic reticulum (ER) and mitochondrion-associated membranes (9, 39, 40). The SAM binding site of PEMT is located at the cytosolic surface of the ER (35), but residues critical for the binding of PE to PEMT have not yet been identified. PEMT follows an ordered Bi-Bi mechanism in which phospholipid substrates and products are the first to bind to and the last to dissociate from the active site (32).
In Saccharomyces cerevisiae, two different genes encoding class I and class II Pmt enzymes are involved in phospholipid methylation. Class II Pmt enzymes catalyze the first methylation step from PE to MMPE, whereas class I Pmts catalyze the following steps from MMPE to PC via DMPE (21, 22, 25). A similar two-enzyme methylation pathway in the plant-symbiotic bacterium Bradyrhizobium japonicum has recently been described (17).
With the notable exception of Zymomonas mobilis Pmt (38), all bacterial Pmt enzymes known thus far are cytosolic enzymes. Two families can be distinguished, the Rhodobacter PmtA family (1) and the Sinorhizobium PmtA family (10). Members of the two families are only distantly related and are often more similar to methyltransferases with other substrate specificities than to each other (26, 37). The Sinorhizobium type Pmt enzymes show homology to rRNA methylases, whereas the Rhodobacter-like Pmt enzymes are similar to UbiE (ubiquinone/menaquinone biosynthesis methyltransferases) (37). Similarities between the Rhodobacter PmtA, the Sinorhizobium PmtA, and other SAM-dependent methyltransferases from prokaryotes and eukaryotes are restricted to the motif VL(E/D)XGXGXG, which is indicative of SAM-dependent methyltransferases (18) and which may bind to the methyl donor SAM, as discussed previously (19, 28).
While Pmt enzymes from mammals and yeast are biochemically well characterized, the activity of the bacterial enzymes is largely unexplored. PC biosynthesis in bacteria is the exception rather than the rule. It has been estimated that only around 10% of all bacterial species are capable of synthesizing PC (37). Quite intriguingly, a number of these bacteria interact with eukaryotic organisms, and it has been reported that bacterium-derived PC plays an important role in the interactions of B. japonicum, Agrobacterium tumefaciens, Brucella abortus, and Legionella pneumophila with their respective hosts (6-8, 15, 27, 30, 42). B. japonicum is the only bacterium known to possess a yeast- and fungi-like pathway involving multiple Pmt enzymes with different substrate specificities (17). PmtA predominantly performs the first methylation step, whereas PmtX1 catalyzes the second and third methylation reactions (17, 30). Three further pmt genes, some of which are upregulated in the absence of PmtA, were identified in this bacterium (16).
It was discovered more than 40 years ago that the plant pathogen A. tumefaciens produces PC by two alternative pathways, the methylation pathway (20) and the phosphatidylcholine synthase pathway (23, 29, 34). PE methylation activity was demonstrated in partially purified A. tumefaciens extracts (20) but has not been studied since this seminal finding. We reported recently that A. tumefaciens possesses a single pmtA gene that encodes a phospholipid N-methyltransferase belonging to the Sinorhizobium PmtA type. Disruption of the A. tumefaciens pmtA gene resulted in delayed tumor formation on plant leaves (42).
In the present study, we describe the recombinant production of A. tumefaciens PmtA in Escherichia coli and the in vitro characterization of lipid binding, SAM binding, and enzymatic activity. Our study paves the way for understanding bacterial phospholipid N-methyltransferases.
MATERIALS AND METHODS
Materials.
Phospholipids (3-sn-phosphatidyl-ethanolamine, 1,2-dipalmitoyl-sn-glycero-3-phospho-N-methylethanolamine, 1,2-dipalmi-toyl-sn-glycero-3-phospho-N, N-dimethyl-ethanol-amine, l-α-phosphatidylcholine, l-α-phosphatidyl-dl-glycerol, and l-α-phosphatidylinositol), S-adenosyl-l-methionine, S-adenosyl-l-homocysteine, sinefungin, and molybdenum blue spray reagent were purchased from Sigma Aldrich. S-Adenosyl-l-[methyl-3H]methionine (81.9 Ci/mmol) was obtained from Hartmann Analytic. HAWP 02500 filters for adenosylmethionine binding assays were purchased from Millipore and Hybond-C extra membranes for lipid overlay assays from GE Healthcare. Superdex 75 10/30 columns were from GE Healthcare. The HPTLC silica gel 60 plates were from Merck. All other reagents were of the highest standard commercially available.
Protein expression and purification.
The A. tumefaciens C58 pmtA gene was PCR amplified with chromosomal DNA as a template and the appropriate primers (5′-GGAATTCCATATGGCACTCAACCTGAAGCAAC-3′ and 5′-CCGGTCGACTCACTC GAGGGCCCGCGTATAGGTCCACAA-3′; engineered NdeI and SalI restriction sites are underlined). The PCR product was digested with NdeI and SalI and cloned into vector plasmid pET28b+ (Novagen), resulting in hybrid plasmid pET28b_PmtA coding for an N-terminally His-tagged PmtA protein. E. coli BL21(DE3) cells carrying the pET28b_PmtA plasmid were grown in 1 liter of Luria-Bertani (LB) broth containing kanamycin (50 μg/ml) at 37°C. Protein production was induced at an optical density at 600 nm of 0.6 by the addition of isopropyl-ß-d-thiogalactopyranoside to a final concentration of 0.4 mM before the culture was incubated for 16 h at 18°C. After being harvested (6,000 × g; 10 min; 4°C) and washed with wash buffer (50 mM NaH2PO4-300 mM NaCl [pH 8.0]), cells were resuspended in 30 ml of wash buffer containing 10 mM imidazole, 0.05% (wt/vol) Triton X-100, 1 mM phenylmethylsulfonyl fluoride, and 1 mM DNase I. Cells were lysed by French press (five passages at 20,000 lb/in2), and insoluble material was removed by centrifugation (20,000 × g; 40 min; 4°C). Soluble fractions were applied to nickel-iminodiacetic acid (Ni-IDA) columns (2-ml-bed volume; Macherey-Nagel). The columns were washed with 8-column volumes (cv) of wash buffer containing 10 mM imidazole, 8 cv of wash buffer containing 40 mM imidazole, and 3 cv of wash buffer containing 70 mM imidazole. His-tagged proteins were eluted with 1 ml of wash buffer containing 250 mM imidazole. Protein purity was assessed by Coomassie blue staining of sodium dodecyl sulfate (SDS)-polyacrylamide gels (12.5%). Proteins from an unstained gel were transferred to a polyvinylidene difluoride membrane (Bio-Rad), and His-tagged proteins were detected by using anti-Penta-His horseradish peroxidase (HRP)-coupled antibody (Qiagen) and a chemiluminescence (ECL) Western blotting detection system (GE Healthcare) according to the manufacturer's instructions.
Size exclusion chromatography.
As a second purification step, size exclusion chromatography was performed, using a Superdex 75 10/30 column run with 50 mM NaH2PO4-50 mM NaCl (pH 8.0) at a flow rate of 0.5 ml/min. The column was calibrated with the globular proteins RNase (13.7 kDa), chymotrypsinogen A (25 kDa), ovalbumine (43 kDa), and albumin (67 kDa), obtaining an R2 value of 0.99 for the calibration curve. A 500-μl aliquot of protein at a concentration of 4 mg/ml was injected. Eluates (0.5 ml) were pooled and concentrated in Amicon Ultra concentrators (molecular weight cutoff, 10,000; Millipore). Protein concentrations were determined from A280 nm values with a calculated extinction coefficient of 14,440 M−1 cm−1.
TLC.
Fifty micrograms of PE, MMPE, DMPE, or E. coli membrane lipid mix in 10 μl of chloroform-methanol (1:1) was mixed with 3.6 μl of 1% (wt/vol) Triton X-100 and dried under vacuum before other components were added. Membrane lipid extracts from 2 ml of an E. coli DH5α culture isolated according to the method of Bligh and Dyer (2) were used for one assay. The reaction mixture (total volume, 180 μl) contained 0.4 mM (56 mol%) substrate lipids, 0.02% Triton X-100, 50 μg of recombinant PmtA protein, and 1.7 mM SAM in 100 mM Tris HCl (pH 9.5). Mixtures were incubated for 1 h in a 30°C water bath, and reactions were stopped by the addition of 180 μl of ice-cold 20% (wt/vol) trichloroacetic acid before precipitations were completed by 10 min of incubation on ice. Precipitates were pelleted by centrifugation (13,000 × g; 5 min), dissolved in 60 μl of 1 M Tris base, and neutralized with 40 μl of 20% (wt/vol) trichloroacetic acid. Methylated phospholipid reaction products resulting from reactions with PmtA were extracted according to the method of Bligh and Dyer (2). Chloroform phases were dried and dissolved in 20 μl of methanol-chloroform (1:1), and reaction products were analyzed by one-dimensional thin-layer chromatography (TLC) as described previously (42).
Photometric methyltransferase assay.
PmtA activity was photometrically determined by using a SAM 265 methyltransferase assay from G-Biosciences. The principle of this assay is as follows. SAH, the transmethylation product of SAM-dependent methyltransferases, is hydrolyzed to S-ribosylhomocysteine and adenine by adenosylhomocysteine nucleosidase (EC 3.2.2.9). This rapid conversion prevents the accumulation of SAH and its feedback inhibition on the methylation reaction. Finally, adenine is converted to hypoxanthine by adenine deaminase (EC 3.5.4.2). The deamination is associated with a decrease in absorbance at 265 nm (cf. Figure 7A) (11).
FIG. 7.

PmtA activity in enzyme-coupled SAM 265 assay. (A) Principle of the assay used to detect SAM-dependent methyltransferase activity according to Dorgan et al. (11). A detailed description is given in the Materials and Methods section. Nu, nucleophile. (B) Analysis of PmtA activity with natural substrate lipids isolated from A. tumefaciens ΔpmtA or ΔpmtA Δpcs mutant. Membrane lipids were isolated and separated by one-dimensional TLC. Phospholipids were specifically stained with molybdenum blue (inset). Reaction mixtures (200 μl) contained 0.01 μM adenine deaminase, 0.1 μM SAH nucleosidase, 110 μM SAM, 0.02% (wt/vol) Triton X-100, and lipids from a 2-ml culture of the A. tumefaciens ΔpmtA or ΔpmtA Δpcs mutant as the substrate. Reactions were started by the addition of 2 μM PmtA. Absorbance changes were measured at 265 nm with a 96-well plate reader (μQuant; BioTek). (C) Analysis of PmtA activity with PE micelles as substrate. Assays were performed as described for panel B with 0 to 800 μM (0 to 71 mol%) commercially available PE as the substrate. (D and E) Influence of PC and PG on PmtA activity. Assays were performed as described for panel B with 150 μM micellar PE (32 mol%) (D) or 150 μM liposomal PE (E) either in the absence (PE alone) or presence (PE+PG, PE+PC) of 60 μM or 150 μM PC or 60 μM PG. PmtA (3 μM) was used to start the assay.
The assay was carried out in 96-well UV microtiter-well plates (370-μl volume; Corning) according to the manufacturer's (G-Biosciences) instructions. Two-hundred-microliter reaction mixtures contained 0.01 μM adenine deaminase, 0.1 μM SAH nucleosidase, 110 μM SAM, and 50 to 800 μM (14 to 71 mol%) micellar PE with Triton X-100 (0.02% [wt/vol]) in 100 mM Tris HCl (pH 8). The mol% of PE in Triton-mixed micelles was calculated according to Carman et al. (4). Alternatively, PE liposomes (150 μM) were used as the substrate.
For PE micelles, phospholipids dissolved in methanol-chloroform (1:1) were incubated with Triton X-100 and dried under vacuum prior to being resuspended in reaction buffer and the addition of the other components. Reactions were started with 2 to 3 μM recombinant PmtA. Absorbance measurements (taken at 1-min intervals for 40 min) were performed with a 96-well plate reader (μQuant; BioTek) and analyzed with BioTek Gen5 data analysis software.
Liposome preparation.
PE or PE-PC and PE-phosphatidylglycerol (PG) (molar ratio, 1:0.4 or 1:1) mixtures dissolved in chloroform were mixed in a glass vial and dried under nitrogen flow. Dried lipids were rehydrated in buffer (100 mM Tris HCl [pH 8]) at a final phospholipid concentration of 600 μM at room temperature for 1 h. The lipid suspension was then sonicated for 10 min at 30°C in a bath sonicator (Sonorex Super RK 102H; Bandelin). The sonicated lipid suspension was extruded 10 times, using an Avanti miniextruder according to the manufacturer's instructions. Polycarbonate membranes with a pore size of 100 nm were used. The particle size of the extruded liposome vesicles was measured by a high-performance particle sizer (Malvern HPPS; Malvern Instruments). The extruded liposomes were stored in a glass vial at 4°C.
Protein lipid overlay assay.
Protein lipid overlay assays were carried out as described previously (12). One-microliter lipid solutions (containing 0.44 nmol, 0.88 nmol, 1.75 nmol, 3.5 nmol, 7 nmol, or 14 nmol of phospholipids) in a mixture of chloroform-methanol-water (1:2:0.8) were spotted onto Hybond-C extra membrane strips and air dried for 1 h at room temperature. Membranes were blocked in TBST blocking buffer (50 mM Tris HCl [pH 7.5], 150 mM NaCl, 0.1% [vol/vol] Tween 20, 2% [wt/vol] fatty acid-free bovine serum albumin) for 1 h at room temperature and incubated with 4 nmol of recombinant PmtA protein in 5 ml blocking buffer overnight at 4°C. Membranes were washed six times for 5 min in TBST buffer (50 mM Tris HCl [pH 7.5], 150 mM NaCl, 0.1% [vol/vol] Tween 20), and His-tagged proteins bound to the lipids were detected with an anti-Penta-His HRP-coupled antibody (Qiagen) and a chemiluminescence (ECL) Western blotting detection system according to the manufacturer's (GE Healthcare) instructions.
Radioactive SAM binding studies.
SAM binding assays were carried out as described previously (44) with slight modifications. Briefly, 0.4 nmol of recombinant PmtA protein was incubated with 2.5 μCi of S-adenosyl-l-[methyl-3H]methionine (81.9 Ci/mmol) in binding buffer (100 mM Tris HCl [pH 8.0]) for 10 min at 30°C (total assay volume, 50 μl). Binding assay mixtures were passed over HAWP 02500 filters (Millipore) on a filtration funnel, and unbound S-adenosyl-l-[methyl-3H]methionine was removed by washing four times with 300 μl of binding buffer. Bound S-adenosyl-l-[methyl-3H]methionine was quantified by liquid scintillation spectrometry (LS 6000 TA counter; Beckman Coulter). If required, phospholipids were added to the reaction mixture to a final concentration of 300 μM.
RESULTS
Purification of A. tumefaciens PmtA.
N-terminally His6-tagged PmtA was produced in E. coli BL21(DE3) cells and purified by nickel-chelate chromatography. Although the majority of recombinant PmtA was insoluble, a typical preparation from 1 liter of bacterial culture yielded about 8 mg of soluble PmtA protein with an estimated 85% purity (Fig. 2A). Consistent with the calculated mass of the His-tagged protein (24.3 kDa), recombinant PmtA migrated as a band with an apparent molecular mass of 24 kDa. To yield highly pure protein and to analyze the oligomeric state of PmtA, size exclusion chromatography was performed. As judged by SDS- polyacrylamide gel electrophoresis (PAGE), PmtA protein of >95% purity was obtained after size exclusion chromatography (Fig. 2B, inset). The PmtA peak corresponded to a molecular mass of about 23 kDa (Fig. 2B), indicating that PmtA forms monomers.
FIG. 2.

Purification of recombinant PmtA. (A) SDS-PAGE of Ni-IDA purification of N-terminal His6-tagged PmtA (HisPmtA). The fractions loaded were P, pellet; C, crude extract; W, wash fraction; E1 and E2, elution fractions 1 and 2; M, BenchMark protein standard (Invitrogen). The two right lanes (marked as “blot”) show Western blot analyses of E1 and E2, using anti-Penta-His HRP-coupled antibody (Qiagen). (B) Size exclusion chromatography of Ni-IDA-purified PmtA. The positions of two standard proteins (chymotrypsinogen, 25 kDa; ovalbumine, 43 kDa) and the void volume (vv) are indicated. The peak fractions 1 and 2 were analyzed by SDS-PAGE as depicted on the right. mAU, milli-absorbance units.
PmtA activity and substrate preferences.
Recombinant A. tumefaciens PmtA is able to convert E. coli PE to PC in the heterologous host (24). Therefore, the activity of purified PmtA was initially tested by using isolated E. coli lipids as the substrate. Reaction products were analyzed by one-dimensional TLC. E. coli lipids contain neither MMPE, DMPE, nor PC (Fig. 3, lane 1). In the presence of SAM, purified PmtA converted PE (the most abundant phospholipid in E. coli) to MMPE, DMPE, and PC, demonstrating that the A. tumefaciens enzyme catalyzes all three methylation steps (Fig. 3, lane 2).
FIG. 3.
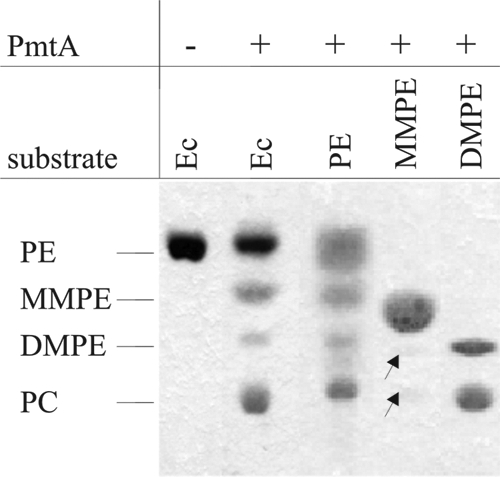
Initial characterization of PmtA activity. The assay was carried out in the absence (−) or presence (+) of recombinant PmtA. PmtA activity was analyzed with different phospholipid substrates (E. coli lipids [Ec], PE, MMPE, or DMPE). The products were extracted and separated by one-dimensional TLC. Phospholipid products were specifically stained with molybdenum blue. Barely detectable lipids are marked with arrows.
To examine whether each individual intermediate can be bound and methylated by PmtA, the assay was also carried out in the presence of commercially available phospholipids. Significant amounts of MMPE, DMPE, and PC were produced with PE as the substrate (Fig. 3, lane 3). When MMPE was used as the substrate, it caused the formation of very small amounts of DMPE and PC (Fig. 3, lane 4), whereas DMPE was efficiently methylated to form PC (Fig. 3, lane 5). Thus, A. tumefaciens PmtA uses not only PE but also DMPE and (to a lesser extent) MMPE as substrates.
Lipid binding of PmtA.
Phospholipid binding of PmtA was analyzed by protein lipid overlay assay, using serial dilutions of phospholipids spotted onto nitrocellulose filters. As expected, PmtA bound to PE, MMPE, DMPE, and PC (Fig. 4A). Binding to MMPE was more efficient than to other phospholipids. Lipid binding was not dependent on SAM (data not shown). Additionally, we tested whether PmtA binds to nonsubstrate phospholipids. It did not bind to triglyceride and cholesterol (data not shown). However, PmtA bound strongly to PG and phosphatidylinositol (PI) (Fig. 4B). Even 30-fold-lower concentrations (0.44 nmol) of PG or PI were sufficient to achieve equal binding, as observed with 14 nmol PE, MMPE, DMPE, or PC (Fig. 4A and B).
FIG. 4.
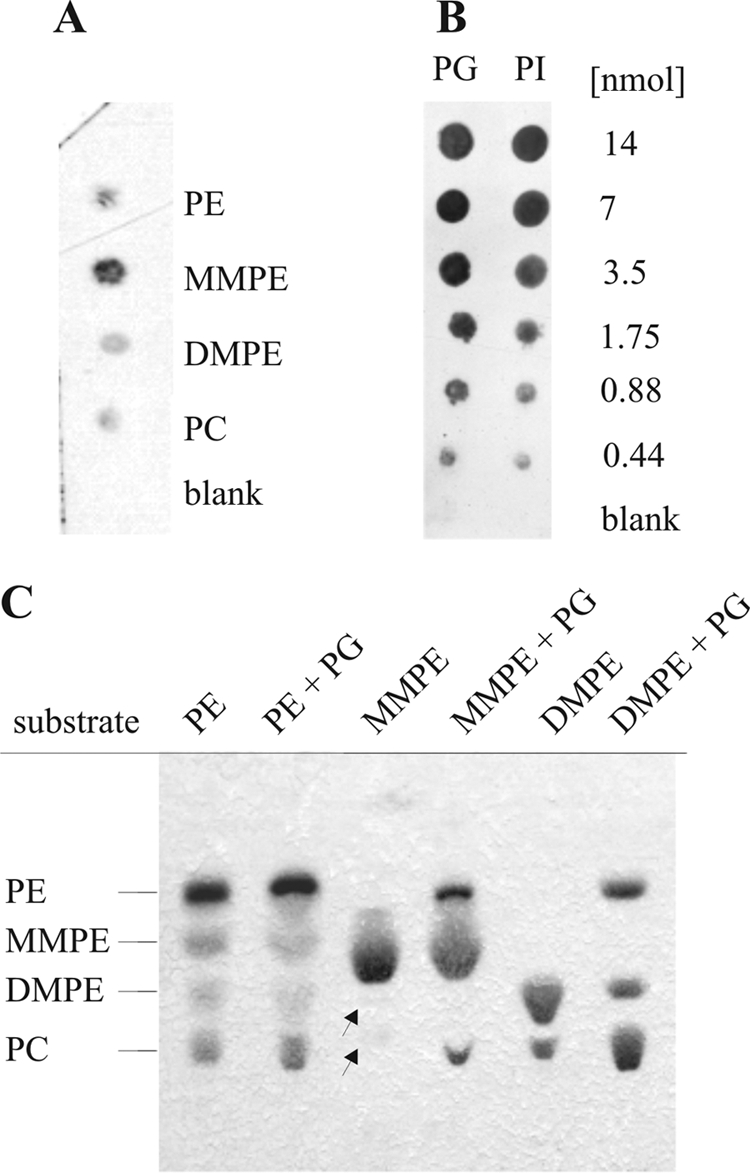
Protein lipid overlay assay. His6-tagged PmtA (4 nmol) was incubated with nitrocellulose strips carrying equal amounts (14 nmol) of various phospholipids (PE, MMPE, DMPE, and PC) (A) or serial dilutions of PG and PI (0 to 14 nmol) (B). Bound protein was detected with anti-Penta-His HRP-coupled antibody (Qiagen). “Blank” is a chloroform-methanol-water (1:2:0.8) solvent. (C) PmtA activity in the presence (+) or absence of PG (12.5 μg). Assays contained 50 μg of PE, MMPE, or DMPE and 166 μM SAM. The enzymatic products were analyzed by one-dimensional TLC. Lipid products were visualized with molybdenum blue staining. Barely detectable lipids are marked with arrows.
This finding prompted us to ask whether PG or PI affects PmtA activity. PG or PI (12.5 μg) was added to PmtA-catalyzed methylation reactions. PmtA activity was not influenced in the presence of PI (data not shown). However, regardless of whether PE, MMPE, or DMPE was used as the substrate, increased amounts of the corresponding enzyme products were formed in the presence of PG (Fig. 4C). These results clearly indicate that PG stimulates PmtA activity.
SAM binding depends on phospholipids.
To determine SAM binding of PmtA, a filter binding assay for S-adenosyl-l-[methyl-3H]methionine was developed. In the absence of phospholipids, only negligible binding was detected (Fig. 5A, column 1). S-Adenosyl-l-[methyl-3H]methionine binding by PmtA was observed only in the presence of PE, MMPE, DMPE, or PC (Fig. 5A). A 500-fold excess of unlabeled SAM efficiently competed with S-adenosyl-l-[methyl-3H]methionine binding to PmtA (Fig. 5A, column 3). In a more detailed competition experiment, a series of concentrations of unlabeled SAM was used (Fig. 5B). In the presence of a 10-fold molar excess of unlabeled SAM (10 μM), about 50% of the radioligand was exchanged. A 100-fold molar excess displaced the radioligand to less than 20%. Although PG binds to PmtA and enhances PmtA activity (Fig. 4B and C), it did not promote SAM binding by PmtA (Fig. 5A, column 7).
FIG. 5.
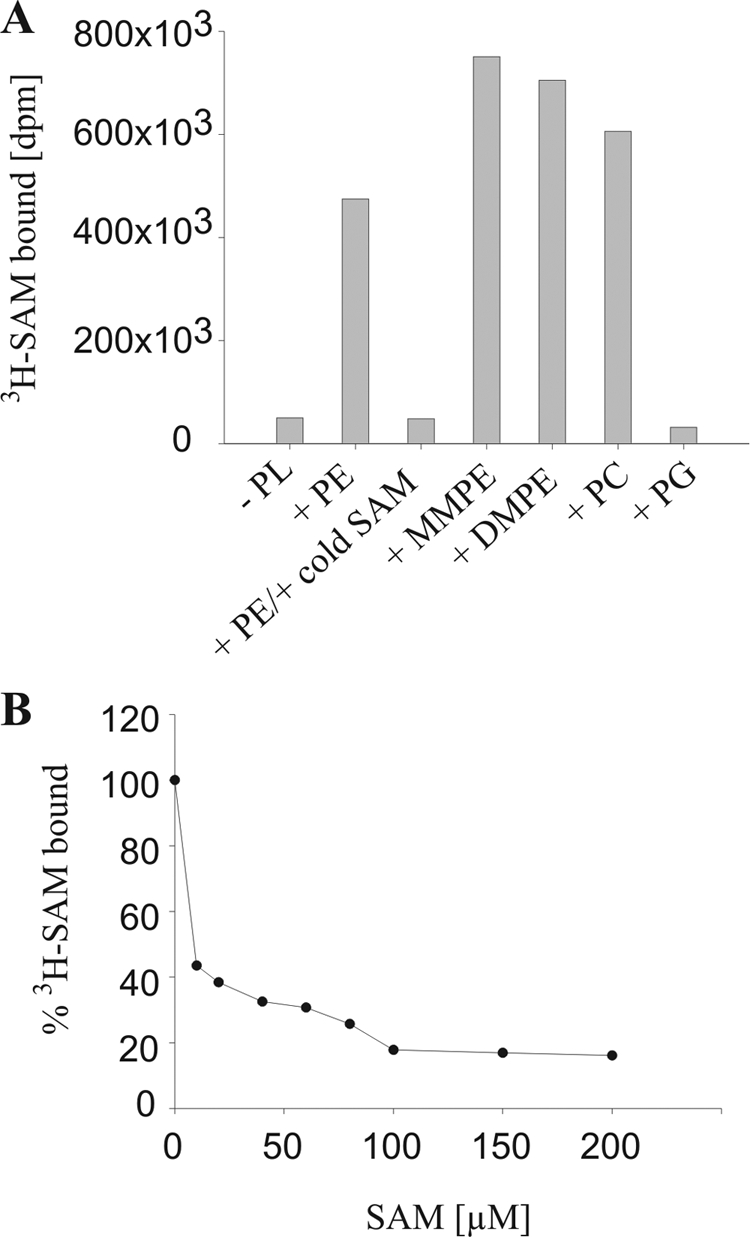
Radioactive SAM binding assay. (A) SAM binding activity was analyzed with 0.4 nmol of recombinant PmtA and 2.5 μCi (0.6 μM SAM) of S-adenosyl-l-[methyl-3H]methionine (3H-SAM; 81.9 Ci/mmol) in the presence (+) or absence (−) of 300 μM of the phospholipid (PL) PE, MMPE, DMPE, PC, or PG. The displacement assay contained, in addition, 500-fold unlabeled (“cold”) SAM (third column). (B) Competitive displacement of radiolabeled SAM by unlabeled SAM (0 to 200 μM). The assay mixture contained 0.4 nmol of recombinant PmtA, 300 μM PE, and 2.5 μCi (0.6 μM SAM) of S-adenosyl-l-[methyl-3H]methionine (81.9 Ci/mmol). One hundred percent S-adenosyl-l-[methyl-3H]methionine corresponds to 477 × 103 dpm.
SAH and sinefungin compete with SAM binding and inhibit PmtA activity.
Several methyltransferases are known to be inhibited by their structurally related transmethylation product SAH or by sinefungin (adenosyl-ornithine) (Fig. 6A), an antibiotic originally isolated from Streptomyces griseolus (3). Therefore, we asked whether SAH and sinefungin would interfere with SAM binding and the enzyme activity of PmtA. To this end, binding of radioactively labeled SAM was analyzed in the presence of various SAH or sinefungin concentrations (0 to 200 μM). Evidently, both SAH and sinefungin interfered severely with SAM binding (Fig. 6B). A twofold molar excess of SAH or sinefungin (∼1.3 μM) displaced nearly 50% of S-adenosyl-l-[methyl-3H]methionine. As a consequence, the presence of SAH or sinefungin inhibited PmtA activity, as shown in a TLC-based enzyme assay. PmtA activity was analyzed in the presence of a constant SAM concentration (166 μM) with either equimolar (166 μM) or double molar (332 μM) amounts of SAH or sinefungin. Both SAH and sinefungin significantly reduced PmtA activity, as indicated by reduced PC and elevated PE concentrations (Fig. 6C).
FIG. 6.

Displacement of radiolabeled SAM by SAH and sinefungin. (A) Chemical structures of SAM, SAH, and sinefungin. (B) Recombinant PmtA (0.4 nmol) was assayed for SAM binding activity in the presence of S-adenosyl-l-[methyl-3H]methionine (3H-SAM; 2,5 μCi) and PE (300 μM) and various concentrations (0 to 200 μM) of unlabeled SAH or sinefungin. One hundred percent S-adenosyl-l-[methyl-3H]methionine corresponds to 221 × 103 dpm in the SAH experiment and 252 × 103 dpm in the sinefungin experiment. (C) Inhibition of in vitro PmtA activity by SAH and sinefungin. PmtA activity was analyzed as previously described with E. coli lipids and 166 μM SAM (control). An inhibition assay was performed in the presence of 166 μM (1) and 332 μM (2) of SAH and sinefungin, respectively. Enzymatic products were analyzed by one-dimensional TLC. Lipid products were visualized with molybdenum blue staining.
The phospholipid composition affects PmtA activity.
Since there is only a little spectral difference between SAM and its demethylated product SAH, most quantitative phospholipid N-methyltransferase activity assays described in the literature are based on radiolabeled SAM. However, radioactive assays require a subsequent separation of product and substrate. Here, we used a more convenient and reliable novel approach (Fig. 7A) to quantify phospholipid N-methyltransferase activity by the continuous enzyme-coupled spectrophotometric SAM 265 assay (G-Biosciences).
We first analyzed the PmtA activity in the presence of its natural lipid substrates. For this purpose, lipids of 2-ml A. tumefaciens ΔpmtA and ΔpmtA Δpcs cultures were isolated and used as substrates. The double mutant A. tumefaciens ΔpmtA Δpcs is unable to produce MMPE, DMPE, or PC (42). In contrast, lipids of the ΔpmtA strain contain both PE and PC (Fig. 7B, inset). PmtA activity was reduced when PC was present (Fig. 7B), suggesting that PC inhibits PmtA activity.
To analyze this effect in more detail, PmtA activity was measured in the presence of commercially available phospholipids. The optimal phospholipid concentration was determined in the presence of 0 to 800 μM (0 to 71 mol%) PE micelles (Fig. 7C). PmtA activity increased with increasing PE concentrations, reaching maximal activity at 800 μM (71 mol%) PE micelles. Higher PE concentrations could not be used, as they resulted in a turbid assay solution. In the presence of 71 mol% PE, an initial rate of 28.47 ± 3.6 μmol SAH mg−1 min−1 was determined.
In addition, the photometric assay was used to study the influence of phospholipids on PmtA activity. To exclude that PC or PG per se influences the assay system, we analyzed thiopurine-S-methyltransferase activity in the presence of PC or PG. Neither phospholipid affected thiopurine-S-methyltransferase activity (data not shown). Next, the assay was performed with 150 μM (32 mol%) PE micelles and 110 μM SAM in the absence or presence of 60 μM PC. Reactions were started by the addition of 3 μM PmtA. PmtA activity decreased about 60% when PC was present (initial enzyme rate without PC, 28.95 μmol SAH mg−1 min−1; rate with PC, 10.85 μmol SAH mg−1 min−1) (Fig. 7D). Consistent with the TLC assay (Fig. 4C), PG had a stimulating effect on PmtA activity (Fig. 7D).
To validate the effect of PC and PG on PmtA activity, we assayed PmtA activity with liposomes. Although PmtA activity was lower than with PE-Triton micelles, the inhibitory and stimulatory effects of PC and PG, respectively, were clearly confirmed with liposomes (Fig. 7E).
DISCUSSION
Bacterial PC biosynthesis pathways and the enzymes involved differ substantially from PC formation in eukaryotes, in which PC is the most abundant phospholipid. Although PC biosynthesis is restricted to a minority of bacteria, it plays an important physiological role in these microorganisms (15, 26). In A. tumefaciens, PC is required for plant infection, stress management, motility, and normal biofilm formation (24, 42).
The major PC biosynthesis pathway in A. tumefaciens depends on PmtA (42), whose activity was initially described in 1964 (20). In this study, we present a detailed characterization of the properties of this enzyme. By the use of highly purified recombinant protein we examined each subreaction, including lipid binding, SAM binding, and the conversion of PE to PC in vitro. We show that PmtA is a monomeric, single-subunit enzyme (Fig. 2) that catalyzes all three-step methylations of PE to form PC via the intermediates MMPE and DMPE (Fig. 3). This finding correlates well with the in vivo situation described earlier (42). Membrane lipids of wild-type A. tumefaciens contain PE, MMPE, DMPE, and PC. The mono- and dimethylated intermediates are lacking in the pmtA mutant. PC, however, is still formed, owing to the presence of a phosphatidylcholine synthase, which condenses externally provided choline directly to CDP-diacylglycerol to form PC.
Taking into account that PmtA catalyzes a fairly complex series of reactions involving binding and metabolic conversion of a phospholipid and the methyl donor SAM, the A. tumefaciens enzyme is a surprisingly small monomeric enzyme (∼21.7 kDa). Likewise, rat liver PEMT (18.3 kDa; monomeric) catalyzes all three methylations (33). PmtA and most other bacterial Pmts are cytosolic enzymes. One exception is the membrane-bound Z. mobilis Pmt enzyme that was isolated from Z. mobilis membrane fractions (38). Although it catalyzes all three steps of PE methylation, the enzyme is quite different from other known bacterial Pmts. Since Z. mobilis Pmt is much larger than sinorhizobial and rhodobacterial Pmt enzymes (42 kDa versus ∼23 kDa) and due to its membrane localization, it has been proposed that Z. mobilis Pmt represents a third type of bacterial phospholipid N-methyltransferase (37). Like Zymomonas Pmt, eukaryotic Pmts are integral membrane proteins. Liver PEMT, for example, is localized in membranes of the ER (9). Recently, a C-terminal ER-targeting motif in mice liver PEMT was identified. This motif contains a lysine residue (lys-197) that is essential for targeting PEMT to the ER (36). An obvious membrane-targeting motif in A. tumefaciens PmtA is missing. However, it is worth noting that the extreme C-terminal part of A. tumefaciens PmtA and other bacterial Pmts contain a conserved stretch of aromatic amino acids that play a critical role in the binding of peripheral proteins to the membranes, composed mainly of zwitterionic phospholipids, as previously discussed (5). Thus, we speculate that PmtA, an enzyme that converts the membrane lipid PE, might in fact be a peripheral membrane protein which (transiently) associates with the cytoplasmic membrane.
Although PmtA binds all possible substrates (PE, MMPE, DMPE) (Fig. 4A), it is likely that in vivo, only binding and the successive conversion of PE are relevant, since free MMPE and DMPE occur only in marginal amounts in A. tumefaciens membranes. PmtA also bound the fully methylated end product PC (Fig. 4A), which inhibited the reaction (Fig. 7D). This is the first report of PC-mediated product inhibition of phospholipid N-methyltransferases. It is possible that this in vitro finding is relevant in vivo, since it might be an elegant way to adjust the proper balance between PE and PC in the bacterial membrane. The PC content in A. tumefaciens membranes is around 23% (24). The relative level of PC does not exceed 40 to 50% of total membrane lipids in B. japonicum membranes (17, 30) or 25% in E. coli cells overexpressing Pmt enzymes (17). In contrast to A. tumefaciens PmtA, rat liver PEMT is not inhibited, but activated, by PC (32). Rat liver PEMT possesses only a single substrate phospholipid binding site, and therefore, PE, MMPE, and DMPE compete for the same binding site. The end product PC binds at a second phospholipid binding site, as discussed previously (32). We assume that A. tumefaciens PmtA contains a single phospholipid binding site for PE, MMPE, DMPE, and PC. PE and PC would thus compete for one binding site, resulting in inhibition of PmtA activity by PC.
Interestingly, PmtA was also shown to bind the nonsubstrate phospholipid PG (Fig. 4B), which stimulated PmtA activity (Fig. 4C). Since SAM binding of PmtA was not influenced by PG (Fig. 5A), we speculate that PmtA requires a defined phospholipid environment, specifically the presence of PG, for full enzymatic activity. A. tumefaciens membranes are known to possess PG (23, 34). Since PG stimulates PE, MMPE, or DMPE methylation, it seems likely that PG binds to PmtA at a site different from the substrate binding site.
Like all SAM-dependent methyltransferases, PmtA contains a highly conserved N-terminal SAM binding motif (E/DXGXGXG) (28). Thus, we speculate that SAM binding occurs in the N-terminal part of PmtA protein. We showed that SAM binding by PmtA takes place only if the substrates PE, MMPE, and DMPE or the end product PC was present (Fig. 5A). Thus, lipid binding is proposed to alter the structure of PmtA, allowing the cofactor to bind. This is reminiscent of the Pmt activity in rat liver microsomes, which follows an ordered Bi-Bi mechanism in which the phospholipids first bind to the enzyme, followed by SAM binding (32).
It has been postulated that drugs directed against bacterial PC biosynthesis enzymes might have an antibiotic effect (26). Our study directly demonstrates that PmtA activity can be inhibited not only by the end products PC and SAH but also by the streptomycelial compound sinefungin. We also tested whether sinefungin had an effect on A. tumefaciens growth. Even at high concentrations (up to 1,000 μg/ml), sinefungin did not inhibit growth (data not shown). The PC content was reduced only slightly, if at all, in the pcs mutant possessing only the PmtA pathway (data not shown). Therefore, sinefungin may not be effectively transported into the cell.
Inhibition of SAM-dependent transmethylation by SAH was first demonstrated for PEMT from rat liver (14) and later shown for both yeast phospholipid N-methyltransferases (13). The first hint that A. tumefaciens PmtA activity might be inhibited by SAH was derived from studies with partially purified phospholipid N-methyltransferase from A. tumefaciens cell extracts (20). Although several pathogenic bacteria predominantly use the alternative phosphatidylcholine synthase pathway for PC biosynthesis (6-8, 43), some of them also encode a functional PmtA enzyme. Given the fact that there are substantial differences between eukaryotic and prokaryotic PC biosynthesis enzymes in sequence, structure, and biochemical properties (32, 35-37), this might open new therapeutic avenues that use PC biosynthesis in human pathogens as a novel drug target. It is noteworthy in this context that PC biosynthesis in Plasmodium falciparum is considered to be a useful target of antimalarial treatment (31). P. falciparum uses a phosphoethanolamine methyltransferase to convert phosphoethanolamine by threefold, SAM-dependent methylation to phosphocholine, a precursor for the synthesis of PC. Activity of the methyltransferase is inhibited by the phosphocholine analog miltefosine, and this compound was shown to inhibit parasite proliferation in human erythrocytes. Interfering with membrane lipid homeostasis might thus emerge as a promising strategy for future drug design against harmful microbes.
Acknowledgments
We are very grateful to Heike Holländer-Czytko for introduction into the filter binding assay and for many thoughtful comments. We thank Jan Gleichenhagen for technical assistance; Silke Johanning and Eckhard Hofmann for help with liposome preparation; and Bernd Masepohl, Nicole Frankenberg-Dinkel, and Stephanie Hacker for critical readings of the manuscript.
The study was funded in part by a grant from the German Research Foundation (DFG, SFB 480) to F.N. and a fellowship from the Promotionskolleg der Ruhr-Universität Bochum to M.A.
Footnotes
Published ahead of print on 30 January 2009.
REFERENCES
- 1.Arondel, V., C. Benning, and C. R. Somerville. 1993. Isolation and functional expression in Escherichia coli of a gene encoding phosphatidylethanolamine methyltransferase (EC 2.1.1.17) from Rhodobacter sphaeroides. J. Biol. Chem. 26816002-16008. [PubMed] [Google Scholar]
- 2.Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37911-917. [DOI] [PubMed] [Google Scholar]
- 3.Boeck, L. D., G. M. Clem, M. M. Wilson, and J. E. Westhead. 1973. A9145, a new adenine-containing antifungal antibiotic: fermentation. Antimicrob. Agents Chemother. 349-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carman, G. M., R. A. Deems, and E. A. Dennis. 1995. Lipid signaling enzymes and surface dilution kinetics. J. Biol. Chem. 27018711-18714. [DOI] [PubMed] [Google Scholar]
- 5.Cho, W., and R. V. Stahelin. 2005. Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 34119-151. [DOI] [PubMed] [Google Scholar]
- 6.Comerci, D. J., S. Altabe, D. de Mendoza, and R. A. Ugalde. 2006. Brucella abortus synthesizes phosphatidylcholine from choline provided by the host. J. Bacteriol. 1881929-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conde-Alvarez, R., M. J. Grilló, S. P. Salcedo, M. J. de Miguel, E. Fugier, J. P. Gorvel, I. Moriyón, and M. Iriarte. 2006. Synthesis of phosphatidylcholine, a typical eukaryotic phospholipid, is necessary for full virulence of the intracellular bacterial parasite Brucella abortus. Cell. Microbiol. 81322-1335. [DOI] [PubMed] [Google Scholar]
- 8.Conover, G. M., F. Martínez-Morales, M. I. Heidtman, Z. Q. Luo, M. Tang, C. Chen, O. Geiger, and R. R. Isberg. 2008. Phosphatidylcholine synthesis is required for optimal function of Legionella pneumophila virulence determinants. Cell. Microbiol. 10514-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui, Z., J. E. Vance, M. H. Chen, D. R. Voelker, and D. E. Vance. 1993. Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. J. Biol. Chem. 26816655-16663. [PubMed] [Google Scholar]
- 10.de Rudder, K. E., I. M. López-Lara, and O. Geiger. 2000. Inactivation of the gene for phospholipid N-methyltransferase in Sinorhizobium meliloti: phosphatidylcholine is required for normal growth. Mol. Microbiol. 37763-772. [DOI] [PubMed] [Google Scholar]
- 11.Dorgan, K. M., W. L. Wooderchak, D. P. Wynn, E. L. Karschner, J. F. Alfaro, Y. Cui, Z. S. Zhou, and J. M. Hevel. 2006. An enzyme-coupled continuous spectrophotometric assay for S-adenosylmethionine-dependent methyltransferases. Anal. Biochem. 350249-255. [DOI] [PubMed] [Google Scholar]
- 12.Dowler, S., G. Kular, and D. R. Alessi. 2002. Protein lipid overlay assay. Sci. STKE 2002(129)PL6. [DOI] [PubMed] [Google Scholar]
- 13.Gaynor, P. M., and G. M. Carman. 1990. Phosphatidylethanolamine methyltransferase and phospholipid methyltransferase activities from Saccharomyces cerevisiae. Enzymological and kinetic properties. Biochim. Biophys. Acta 1045156-163. [DOI] [PubMed] [Google Scholar]
- 14.Gibson, K. D., J. D. Wilson, and S. Udenfriend. 1961. The enzymatic conversion of phospholipid ethanolamine to phospholipid choline in rat liver. J. Biol. Chem. 236673-679. [PubMed] [Google Scholar]
- 15.Goldfine, H. 1984. Bacterial membranes and lipid packing theory. J. Lipid Res. 251501-1507. [PubMed] [Google Scholar]
- 16.Hacker, S., J. Gödeke, A. Lindemann, S. Mesa, G. Pessi, and F. Narberhaus. 2008. Global consequences of phosphatidylcholine reduction in Bradyrhizobium japonicum. Mol. Genet. Genomics 28059-72. [DOI] [PubMed] [Google Scholar]
- 17.Hacker, S., C. Sohlenkamp, M. Aktas, O. Geiger, and F. Narberhaus. 2008. Multiple phospholipid N-methyltransferases with distinct substrate specificities are encoded in Bradyrhizobium japonicum. J. Bacteriol. 190571-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haydock, S. F., J. A. Dowson, N. Dhillon, G. A. Roberts, J. Cortes, and P. F. Leadlay. 1991. Cloning and sequence analysis of genes involved in erythromycin biosynthesis in Saccharopolyspora erythraea: sequence similarities between EryG and a family of S-adenosylmethionine-dependent methyltransferases. Mol. Gen. Genet. 230120-128. [DOI] [PubMed] [Google Scholar]
- 19.Ingrosso, D., A. V. Fowler, J. Bleibaum, and S. Clarke. 1989. Sequence of the d-aspartyl/l-isoaspartyl protein methyltransferase from human erythrocytes. Common sequence motifs for protein, DNA, RNA, and small molecule S-adenosylmethionine-dependent methyltransferases. J. Biol. Chem. 26420131-20139. [PubMed] [Google Scholar]
- 20.Kaneshiro, T., and J. H. Law. 1964. Phosphatidylcholine synthesis in Agrobacterium tumefaciens. I. Purification and properties of a phosphatidylethanolamine N-methyltransferase. J. Biol. Chem. 2391705-1713. [PubMed] [Google Scholar]
- 21.Kanipes, M. I., and S. A. Henry. 1997. The phospholipid methyltransferases in yeast. Biochim. Biophys. Acta 1348134-141. [DOI] [PubMed] [Google Scholar]
- 22.Kanipes, M. I., J. E. Hill, and S. A. Henry. 1998. The Schizosaccharomyces pombe cho1+ gene encodes a phospholipid methyltransferase. Genetics 150553-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karnezis, T., H. C. Fisher, G. M. Neumann, B. A. Stone, and V. A. Stanisich. 2002. Cloning and characterization of the phosphatidylserine synthase gene of Agrobacterium sp. strain ATCC 31749 and effect of its inactivation on production of high-molecular-mass (1→3)-ß-d-glucan (curdlan). J. Bacteriol. 1844114-4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klüsener, S., M. Aktas, K. M. Thormann, M. Wessel, and F. Narberhaus. 2009. Expression and physiological relevance of Agrobacterium tumefaciens phosphatidylcholine biosynthesis genes. J. Bacteriol. 191365-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kodaki, T., and S. Yamashita. 1987. Yeast phosphatidylethanolamine methylation pathway. Cloning and characterization of two distinct methyltransferase genes. J. Biol. Chem. 26215428-15435. [PubMed] [Google Scholar]
- 26.López-Lara, I. M., and O. Geiger. 2001. Novel pathway for phosphatidylcholine biosynthesis in bacteria associated with eukaryotes. J. Biotechnol. 91211-221. [DOI] [PubMed] [Google Scholar]
- 27.López-Lara, I. M., C. Sohlenkamp, and O. Geiger. 2003. Membrane lipids in plant-associated bacteria: their biosyntheses and possible functions. Mol. Plant-Microbe Interact. 16567-579. [DOI] [PubMed] [Google Scholar]
- 28.Martin, J. L., and F. M. McMillan. 2002. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 12783-793. [DOI] [PubMed] [Google Scholar]
- 29.Martínez-Morales, F., M. Schobert, I. M. López-Lara, and O. Geiger. 2003. Pathways for phosphatidylcholine biosynthesis in bacteria. Microbiology 1493461-3471. [DOI] [PubMed] [Google Scholar]
- 30.Minder, A. C., K. E. de Rudder, F. Narberhaus, H. M. Fischer, H. Hennecke, and O. Geiger. 2001. Phosphatidylcholine levels in Bradyrhizobium japonicum membranes are critical for an efficient symbiosis with the soybean host plant. Mol. Microbiol. 391186-1198. [PubMed] [Google Scholar]
- 31.Pessi, G., G. Kociubinski, and C. B. Mamoun. 2004. A pathway for phosphatidylcholine biosynthesis in Plasmodium falciparum involving phosphoethanolamine methylation. Proc. Natl. Acad. Sci. USA 1016206-6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ridgway, N. D., and D. E. Vance. 1988. Kinetic mechanism of phosphatidylethanolamine N-methyltransferase. J. Biol. Chem. 26316864-16871. [PubMed] [Google Scholar]
- 33.Ridgway, N. D., and D. E. Vance. 1987. Purification of phosphatidylethanolamine N-methyltransferase from rat liver. J. Biol. Chem. 26217231-17239. [PubMed] [Google Scholar]
- 34.Sherr, S. I., and J. H. Law. 1965. Phosphatidylcholine synthesis in Agrobacterium tumefaciens. II. Uptake and utilization of choline. J. Biol. Chem. 2403760-3765. [PubMed] [Google Scholar]
- 35.Shields, D. J., J. Y. Altarejos, X. Wang, L. B. Agellon, and D. E. Vance. 2003. Molecular dissection of the S-adenosylmethionine-binding site of phosphatidylethanolamine N-methyltransferase. J. Biol. Chem. 27835826-35836. [DOI] [PubMed] [Google Scholar]
- 36.Shields, D. J., S. Lingrell, L. B. Agellon, J. T. Brosnan, and D. E. Vance. 2005. Localization-independent regulation of homocysteine secretion by phosphatidylethanolamine N-methyltransferase. J. Biol. Chem. 28027339-27344. [DOI] [PubMed] [Google Scholar]
- 37.Sohlenkamp, C., I. M. López-Lara, and O. Geiger. 2003. Biosynthesis of phosphatidylcholine in bacteria. Prog. Lipid Res. 42115-162. [DOI] [PubMed] [Google Scholar]
- 38.Tahara, Y., Y. Ogawa, T. Sakakibara, and Y. Yamada. 1987. Purification and characterization of phosphatidylethanolamine N-methyltransferase from Zymomonas mobilis. Agric. Biol. Chem. 511425-1430. [Google Scholar]
- 39.Vance, D. E., Z. Li, and R. L. Jacobs. 2007. Hepatic phosphatidylethanolamine N-methyltransferase, unexpected roles in animal biochemistry and physiology. J. Biol. Chem. 28233237-33241. [DOI] [PubMed] [Google Scholar]
- 40.Vance, J. E. 1990. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 2657248-7256. [PubMed] [Google Scholar]
- 41.Walkey, C. J., L. R. Donohue, R. Bronson, L. B. Agellon, and D. E. Vance. 1997. Disruption of the murine gene encoding phosphatidylethanolamine N-methyltransferase. Proc. Natl. Acad. Sci. USA 9412880-12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wessel, M., S. Klüsener, J. Gödeke, C. Fritz, S. Hacker, and F. Narberhaus. 2006. Virulence of Agrobacterium tumefaciens requires phosphatidylcholine in the bacterial membrane. Mol. Microbiol. 62906-9015. [DOI] [PubMed] [Google Scholar]
- 43.Wilderman, P. J., A. I. Vasil, W. E. Martin, R. C. Murphy, and M. L. Vasil. 2002. Pseudomonas aeruginosa synthesizes phosphatidylcholine by use of the phosphatidylcholine synthase pathway. J. Bacteriol. 1844792-4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu, Y., C. Qi, W. Q. Cao, A. V. Yeldandi, M. S. Rao, and J. K. Reddy. 2001. Cloning and characterization of PIMT, a protein with a methyltransferase domain, which interacts with and enhances nuclear receptor coactivator PRIP function. Proc. Natl. Acad. Sci. USA 9810380-10385. [DOI] [PMC free article] [PubMed] [Google Scholar]