

Abstract
Mucoidy, or overproduction of the exopolysaccharide known as alginate, in Pseudomonas aeruginosa is a poor prognosticator for lung infections in cystic fibrosis. Mutation of the anti-σ factor MucA is a well-accepted mechanism for mucoid conversion. However, certain clinical mucoid strains of P. aeruginosa have a wild-type (wt) mucA. Here, we describe a loss-of-function mutation in kinB that causes overproduction of alginate in the wt mucA strain PAO1. KinB is the cognate histidine kinase for the transcriptional activator AlgB. Increased alginate production due to inactivation of kinB was correlated with high expression at the alginate-related promoters PalgU and PalgD. Deletion of alternative σ factor RpoN (σ54) or the response regulator AlgB in kinB mutants decreased alginate production to wt nonmucoid levels. Mucoidy was restored in the kinB algB double mutant by expression of wt AlgB or phosphorylation-defective AlgB.D59N, indicating that phosphorylation of AlgB was not required for alginate overproduction when kinB was inactivated. The inactivation of the DegS-like protease AlgW in the kinB mutant caused loss of alginate production and an accumulation of the hemagglutinin (HA)-tagged MucA. Furthermore, we observed that the kinB mutation increased the rate of HA-MucA degradation. Our results also indicate that AlgW-mediated MucA degradation required algB and rpoN in the kinB mutant. Collectively, these studies indicate that KinB is a negative regulator of alginate production in wt mucA strain PAO1.
Cystic fibrosis (CF) patients are predisposed to bacterial respiratory infections due to the mucus buildup in their airways (17). Mutation of the chloride ion transporter called CFTR creates a hospitable environment for the opportunistic pathogen Pseudomonas aeruginosa (27). The emergence of mucoid, or alginate-overproducing, strains marks the beginning of chronic infection by P. aeruginosa (13). The presence of mucoid strains causes significant deterioration of lung function (40). Mucoid strains produce alginate by increasing transcription of the algD promoter of the alginate biosynthetic operon (Fig. 1) (11). The first molecular mechanism for the conversion to mucoidy elucidated was mutation of the mucA gene (32). MucA is the anti-σ factor that sequesters the alternative sigma factor AlgU (also called AlgT or σ22) (Fig. 1) (33, 46). When MucA is not functional due to mutation, increased transcription directed by AlgU at the algD promoter (PalgD) activates alginate biosynthesis (Fig. 1) (57).
FIG. 1.
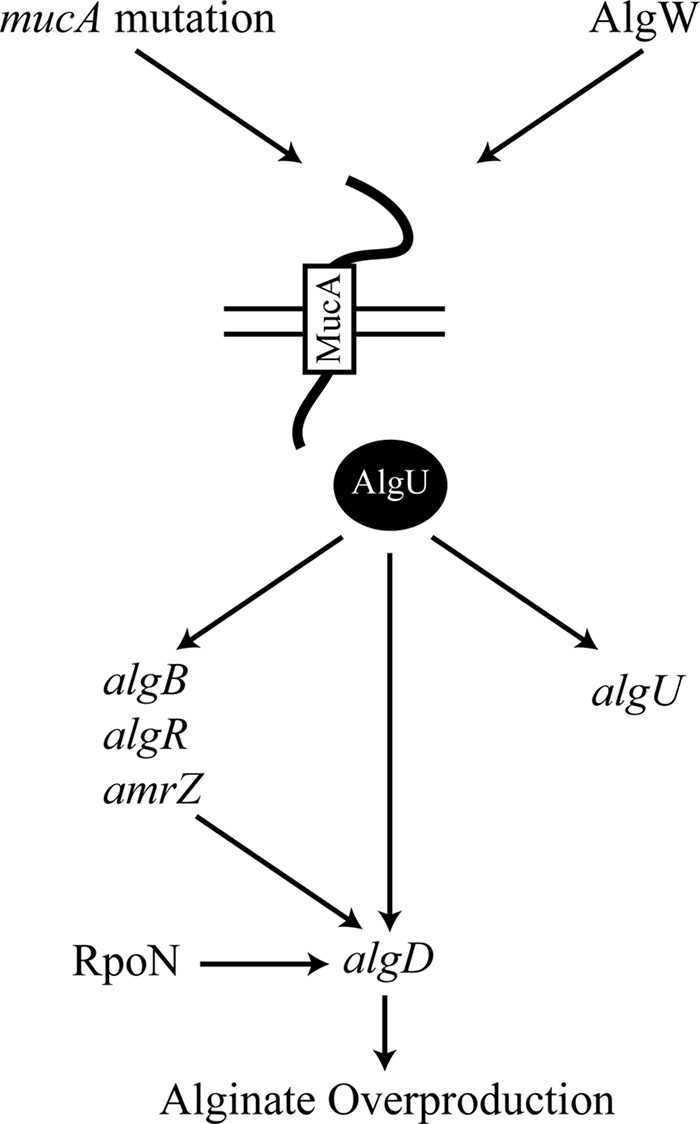
MucA-AlgU is the central regulatory pathway controlling the expression of the mucoid phenotype in P. aeruginosa. In mucA mutants, AlgU is not repressed (32) and activates transcription of downstream promoters. The algU gene is transcribed by five promoters, two of which (P1 and P3) are dependent on AlgU (46). AlgU activates transcription of algB, algR, and amrZ, whose gene products participate in transcriptional activation of PalgD (4, 38, 56). In a muc-23 mucoid mutant, RpoN has also been shown to bind to PalgD and activate or repress transcription under certain environmental conditions (7). AlgU also activates transcription of PalgD (57). Activation of transcription at PalgD results in alginate overproduction and a mucoid phenotype. MucA is the anti-σ-factor that sequesters AlgU(T) (46). The predicted protease AlgW can cleave MucA, which results in derepression of AlgU. Overexpression of the periplasmic peptide MucE results in mucoidy due to activation of AlgW (43), which leads to degradation of MucA. Cell wall inhibitors such as d-cycloserine have been shown to upregulate AlgW-dependent transcription at PalgD (54).
Activation of alginate production by AlgU is controlled at transcriptional and posttranslational levels (Fig. 1). Transcription of algU occurs from multiple promoters, two of which are AlgU dependent (12, 45), and therefore AlgU autoregulates its expression. Alginate production is also negatively controlled by MucB and MucD, which are encoded downstream of algU and mucA. MucB cooperates with MucA-AlgU sequestering, presumably by protecting the periplasmic portion of MucA from degradation and thus stabilizing the MucA-AlgU interaction (46). Inactivation of mucB in a wild-type (wt) mucA strain causes elevated alginate production (31). MucD is homologous to DegP of Escherichia coli, which degrades unfolded proteins in the periplasm (22) and also functions as a chaperone (49). In P. aeruginosa, mucD inactivation causes alginate overproduction and sensitivity to H2O2 and heat (6).
There is a high level of conservation between E. coli σE-RseA and P. aeruginosa AlgU-MucA. Activation of σE occurs after sequential proteolytic cleavage of the anti-σ factor RseA, first by activated DegS and finally by RseP proteases (3). DegS is a serine protease that is activated in response to unfolded outer membrane proteins via a conserved C-terminal sequence (51, 52). This conserved signal transduction pathway is referred to as regulated intramembrane proteolysis (1). In P. aeruginosa, AlgU is associated with the inner membrane and MucA in wt, nonmucoid strains (44). Recently, the P. aeruginosa DegS homologue, AlgW, has been shown to activate alginate production through regulated proteolysis of MucA in response to increased expression of mucE (Fig. 1) (43). Also, the cell wall inhibitor d-cycloserine can activate the AlgU stress response in P. aeruginosa, dependent upon AlgW (Fig. 1) (54).
When MucA does not repress AlgU, transcriptional activation at PalgD and alginate overproduction occurs. Significant research has focused on the multitude of regulators that bind and/or regulate transcriptional activity at PalgD. Most PalgD transcriptional regulators are AlgU dependent, such as AlgR, AmrZ, and AlgB (Fig. 1). The response regulator AlgR binds multiple sites within PalgD and is required for PalgD expression (21, 38). Additionally, the alginate and motility regulator Z (AmrZ) also promotes activity at PalgD (5, 50). The NtrC family response regulator AlgB has recently been shown to bind at PalgD and cause transcriptional activation (26). Beyond AlgU and the AlgU-dependent transcription factors, a second alternative sigma factor, RpoN, has been suggested to have dual roles as both a positive and a negative regulator at PalgD (Fig. 1) (7).
The PalgD transcriptional regulator, AlgB (15, 26, 56), is a response regulator of a two-component signal transduction system. Typically, two-component signal transduction systems are comprised of a response regulator and a sensor kinase. Upon phosphorylation of the response regulator by the sensor kinase, the response regulator binds specific DNA sequences near a promoter and modulates transcription. The E. coli homologue of AlgB, known as NtrC, activates phosphorylation-dependent transcription at target promoters with the σ54-holoenzyme (24). σ54 (RpoN) is required for mucoidy in a P. aeruginosa prototype strain (muc-23) (7), but rpoN is not required for alginate synthesis in several different mucA mutant strains (7, 37, 38). AlgB is an NtrC family response regulator that mediates alginate biosynthesis in mucA mutants (16). The primary role of AlgB that has been elucidated thus far has been transcriptional activation of PalgD (26). KinB is the cognate sensor kinase of AlgB (29), and furthermore, KinB is capable of autophosphorylation and transfer of phosphate to AlgB (29). Interestingly, phosphorylation of AlgB is not required for PalgD activation (28). Unlike algB, kinB is not required for alginate production in a mucA22 mutant (28).
Previous extensive research has focused on regulation of alginate production in mucA mutant strains. However, recent data show that algD expression can occur independent of mucA mutations by regulated proteolysis of MucA (43, 54). Studies have shown that expression of algD is increased under anaerobic conditions (9, 19), which may occur in the CF lung (39). Given the data that P. aeruginosa can produce alginate irrespective of mucA mutation, we sought to further characterize mucoidy in wt mucA strain PAO1. In this report, we show that inactivation of kinB in nonmucoid P. aeruginosa strain PAO1 results in alginate overproduction that requires the predicted protease AlgW. We observed that algB and rpoN are also required in kinB mutants for alginate production and high PalgU and PalgD expression. We also show evidence of regulated MucA degradation in P. aeruginosa. A novel role for AlgB and RpoN in signal transduction of regulated proteolysis to release AlgU from sequestering by MucA in the kinB mutant background is proposed. Our results support a model in which KinB negatively regulates the AlgU signal transduction pathway in P. aeruginosa strain PAO1.
MATERIALS AND METHODS
Bacterial strains, plasmids, transposons, growth conditions, and oligonucleotides.
The bacterial strains, plasmids, and transposons used in this study are listed in Table 1. P. aeruginosa strains were grown at 37°C in Lennox broth (LB), on LB agar, or on Pseudomonas isolation agar (PIA) plates (Difco, Sparks, MD). PIA plates were prepared with 20 ml of glycerol per liter as recommended by the manufacturer. When necessary, PIA medium was supplemented with carbenicillin, tetracycline, or gentamicin at a concentration of 300 μg/ml. The sequences of the primers used in this study are available upon request.
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Phenotype, genotype, and/or descriptiona | Source or reference |
|---|---|---|
| P. aeruginosa strains | ||
| PAO1 | Alg−, prototroph | P. Phibbs |
| PAO1 ΔalgB | Alg−, PAO1 in-frame deletion of algB (PA5483) | This study |
| PAO1 ΔalgU | Alg−, PAO1 in-frame deletion of algU (PA0762) | This study |
| PAO1 ΔkinB | Alg+, PAO1 in-frame deletion of kinB (PA5484) | This study |
| PAO1 ΔrpoN | Alg−, PAO1 in-frame deletion of rpoN (PA4462) | This study |
| PAO1 kinB::aacC1 | Alg+, PAO1 kinB::Gmr | This study |
| PAO1 kinB::aacC1 ΔalgU | Alg−, PAO1 kinB::aacC1 in-frame deletion of algU (PA0762) | This study |
| PAO1 ΔalgB ΔkinB | Alg−, PAO1 in-frame deletion of algB (PA5483) and kinB (PA5484) | This study |
| PAO1 kinB::aacC1ΔalgW | Alg−, PAO1 kinB::aacC1 in-frame deletion of algW (PA4446) | This study |
| PAO1 kinB::aacC1 ΔrpoN | Alg−, PAO1 kinB::aacC1 in-frame deletion of rpoN (PA4462) | This study |
| E. coli strains | ||
| DH5α | F− φ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ−thi-1 gyrA96 relA1 | Lab strain |
| TOP10 | DH5α derivative | Invitrogen |
| SM10/λpir | thi recA thr leu tonA lacY supE RP4-2-Tc::Mu1::pir Kmr | Lab strain |
| Plasmids | ||
| pRK2013 | Kmr Tra Mob ColE1 | 14 |
| pFAC | Mini-himar1 mariner transposon in Pseudomonas suicide plasmid; Apr Gmr | 53 |
| pCR4-TOPO | TA cloning vector; 3.9 kb; Apr Kmr | Invitrogen |
| pUS56 | algB45 in pTrcHisA BamHI-EcoRI | 28 |
| pHERD20T | pUCP20T Plac replaced by 1.3-kb AflII-EcoRI fragment of araC-PBAD cassette | 41 |
| pHERD20T-algU | algU (PA0762) from PAO1 in pHERD20T EcoRI/HindIII | This study |
| pHERD20T-algB | algB from PAO1in pHERD20T EcoRI/HindIII | This study |
| pHERD20T-algB45 | algB45 from pUS56 in pHERD20T EcoRI/HindIII | This study |
| pHERD20T-algW | algW (PA4446) from PAO1in pHERD20T EcoRI/HindIII | This study |
| pHERD20T-MPDZalgW | algW with partial PDZ domain in pHERD20T EcoRI/HindIII | This study |
| pHERD20T-ΔPDZalgW | algW with complete deletion of PDZ domain in pHERD20T EcoRI/HindIII | This study |
| pHERD20T-kinB | kinB from PAO1 in pHERD20T KpnI/HindIII | This study |
| pHERD20T-HA-mucA | N-terminally tagged HA-mucA in pHERD20T EcoRI/HindIII | This study |
| pUCP20T-PBAD-rpoN | araC-PBAD-rpoN fusion in pUCP20 XbaI/HindIII | This study |
| MiniCTX-lacZ | Gene delivery vector for inserting genes at the CTX phage att site on the P. aeruginosa chromosome; Tcr | 20 |
| MiniCTX-PalgU-lacZ | Complete PalgU promoter (541 bp upstream of ATG) EcoRI/HindIII fused with lacZ for integration at the CTX phage att site in P. aeruginosa | This study |
| MiniCTX-PalgD-lacZ | Complete PalgD promoter (1,525 bp upstream of ATG) HindIII/BamHI fused with lacZ for integration at the CTX phage att site in P. aeruginosa | This study |
| pEX100T | Pseudomonas suicide vector, sacB oriT Cbr | 47 |
| pEX100T-NotI | Pseudomonas suicide vector with NotI restriction site fused into SmaI of pEX100T, sacB oriT Cbr | This study |
| pEX100T-ΔalgU | A 2.5-kb fragment flanking algU gene fused with pEX100T-NotI with in-frame deletion of algU with only 24 bp left coding for 8 amino acids of algU | This study |
| pEX100T-ΔalgB | A 1.5-kb fragment flanking algB gene fused with pEX100T-NotI with in-frame deletion of algB | This study |
| pEX100T-ΔalgBΔkinB | A 1.5-kb fragment flanking algB gene fused with pEX100T-NotI with in-frame deletion of algB and kinB | This study |
| pEX100T-ΔalgW | A 1.4-kb fragment flanking algW gene fused with pEX100T-NotI with in-frame deletion of algW | 43 |
| pEX100T-ΔkinB | A 2.5-kb fragment flanking kinB gene fused with pEX100T-NotI with in-frame deletion of kinB | This study |
| pEX100T-ΔrpoN | A 1.9-kb fragment flanking rpoN gene fused with pEX100T-NotI with in-frame deletion of rpoN | This study |
Alg−, nonmucoid phenotype; Alg+, mucoid phenotype.
Transposon mutagenesis.
The mariner transposon-containing plasmid pFAC (53) was introduced into PAO1 by biparental conjugations. The locations of the transposon insertion in the mucoid mutants were determined by inverse PCR (42, 43). The chromosomal DNAs of these strains were digested with SalI and ligated to generate circular closed DNA molecules (Fast-Link DNA ligation kit; Epicentre, Madison, WI). The ligated DNA was then used as the template for inverse PCR with primers (Gm3OUT and Gm5OUT) as previously described (42), which anneal to the gentamicin resistance (Gmr) gene. The resulting amplicons were sequenced by the Marshall University Genomics Core Facility.
Mutant strain construction.
For in-frame deletion of specific genes (algU, algB, algB-kinB, algW, kinB, and rpoN), the upstream and downstream sequence fragments (500 to 1,000 bp) flanking the target gene were PCR amplified and fused by using the crossover PCR method. The PCR products with the in-frame deletion of target gene were digested and ligated into pEX100T-NotI vector. A two-step allelic exchange procedure was employed with the pEX100T constructs for in-frame deletion. The single-crossover merodiploid exconjugants were selected based on carbenicillin resistance and sensitivity on PIA supplemented with 10% (wt/vol) sucrose (sacB). After overnight incubation of the merodiploids in LB broth at 37°C, the double-crossover recombinants were isolated on PIA with 10% sucrose. The in-frame deletion of the target gene was confirmed by antibiotic resistance assays and then PCR amplification of the flanking region of target gene with multiple sets of primers and amplicon sequencing.
Plasmid construction and complementation analyses.
Alleles were cloned into the shuttle vector pHERD20T (41) for complementation with gene expression driven by the PBAD arabinose-inducible promoter. For lacZ reporter analysis, algU and algD promoters were fused with lacZ in miniCTX-lacZ (20) (see Fig. 3A and C, respectively). All plasmid constructs containing PCR products were sequenced, and this confirmed that no mutations occurred.
FIG. 3.

Loss of kinB causes upregulation of both PalgU and PalgD. β-Galactosidase activity from PalgU-lacZ and PalgD-lacZ reporters on the chromosomes of PAO1, PAO1 kinB::aacC1, and PAO1 kinB::aacC1 isogenic mutants was determined. PalgU-lacZ and PalgD-lacZ reporter constructs were integrated into the chromosomes of the indicated strains. Genes indicated were expressed in trans from the PBAD promoter of pHERD20T. β-Galactosidase activities were determined after 24 h of growth on PIA with 0.1% arabinose. Values were normalized to PAO1 pHERD20T (empty vector) reporter expression and indicated as means ± standard deviations from three independent experiments. Student's t test was performed for comparison of activity of the strain with vector only or with the complementing gene in trans. Asterisks indicate significant differences (***, P < 0.0001). Strain PAO1 kinB::aacC1 is indicated as PAO1 kinB. Note that expression of algU in PAO1 is a positive control for the analysis due to the AlgU-dependent nature of both PalgU and PalgD. (A) A schematic of the entire PalgU promoter region with the relative positions of the five promoters that were utilized for the lacZ promoter fusion. (B) PalgU activity in PAO1, PAO1 kinB::aacC1, and strains isogenic to PAO1 kinB::aacC1. High PalgU activity in PAO1 kinB::aacC1 and PAO1 ΔkinB mutants requires algU, algB, rpoN, and algW. Note that kinB expression significantly lowers PalgU activity. (C) A schematic of the entire PalgD promoter region that was used for the lacZ promoter fusion. The relative binding sites of the PalgD transcriptional activators are indicated. (D) PalgD activity in PAO1, PAO1 kinB::aacC1, and strains isogenic to PAO1 kinB::aacC1. High PalgD activity in PAO1 kinB::aacC1 and PAO1 ΔkinB requires algU, algB, rpoN, and algW. Note that kinB expression significantly lowers PalgD activity.
Alginate assay.
P. aeruginosa strains were grown at 37°C on PIA plates supplemented with carbenicillin and 0.1% (wt/vol) arabinose for 24 h. Bacterial growth was removed from plates with phosphate-buffered saline (PBS) and suspended in 50 ml of PBS per plate. The optical density at 600 nm (OD600) of the bacterial suspension in PBS was measured and adjusted. Cell suspensions containing bacterial alginates were used for assay of the amounts of the uronic acid using a standard curve made with d-mannuronic acid lactone (Sigma-Aldrich, St. Louis, MO) in the range of 0 to 100 μg/ml as described previously (23).
β-Galactosidase activity assay.
The promoter fusion constructs miniCTX-PalgU-lacZ, and miniCTX-PalgD-lacZ were integrated onto the P. aeruginosa chromosome at the CTX phage att site (20). The β-galactosidase activity assay was based on the method originally described by Miller (36), with the modification that the cells were grown on PIA plates in triplicate for 24 h at 37°C and harvested in PBS, and the β-galactosidase activity was assayed after toluene permeabilization of the cells. The reported values represent the averages in triplicate from three independent experiments. The values displayed are normalized to PAO1 pHERD20T for each respective promoter fusion.
Western blot analysis.
Cell lysates were prepared with Ready-Preps (Epicentre, Madison, WI) by the manufacturer's protocol. Cell lysates were quantified by DC assay (Bio-Rad, Hercules, CA). Forty micrograms of protein was boiled in sodium dodecyl sulfate loading buffer. The samples were electrophoresed on 12% polyacrylamide gels or 15% ProteaGel (Protea, Morgantown, WV) polyacrylamide and then electroblotted (Trans-Blott cell; Bio-Rad, Hercules, CA) onto 0.45-μm nitrocellulose. The membrane was blocked with 3% nonfat dry milk in PBS (pH 7.4). Primary antibodies were diluted 1:1,000 in 3% nonfat dry milk in PBS. The membranes were probed with mouse monoclonal antibodies against AlgU (46), RpoN (Neoclone, Madison, WI), alpha RNA subunit polymerase subunit (Neoclone), rabbit polyclonal antibody against AlgB (28), or rat monoclonal antibody against hemagglutinin (HA) (Roche, Mannheim, Germany) overnight at 4°C with shaking. Horseradish peroxidase (HRP)-labeled goat anti-mouse immunoglobulin G (IgG) or HRP-labeled anti-rabbit IgG was diluted 1:5,000 in 3% nonfat dry milk in PBS and used as the secondary antibodies. Advanced ECL or ECL chemiluminescence (Amersham Biosciences, Piscataway, NJ) was used for detecting HRP-labeled goat anti-mouse IgG or anti-rabbit IgG (Roche) by the manufacturer's procedure. The signals were detected with an EC3 imaging system (UVP, Upland, CA) by capturing with a BioChemi HR camera. For reprobing, membranes were stripped with 62.5 mM Tris-HCl (pH 6.8)-2% sodium dodecyl sulfate-100 mM β-mercaptoethanol for 15 min at 50°C and then washed in PBS.
HA-MucA steady-state and kinetic concentration Western blot analysis.
To assay the HA-MucA degradation profile after 48 h of growth at 37°C, HA-MucA was expressed in trans in mucoid and nonmucoid strains from pHERD20T with 0.1% arabinose on PIA-carbenicillin plates. Cells were scraped from the plate, suspended in PBS (pH 7.4), and pelleted by centrifugation. Proteins were isolated and prepared as described for Western blot analysis.
To observe the rate of degradation of HA-MucA in PAO1 and PAO1 kinB::aacC1, we analyzed cell lysates over a time course. We utilized the conditional expression of HA-MucA from pHERD20T to compare the rates of degradation. Cultures of PAO1 and PAO1 kinB::aacC1 were incubated overnight at 37°C in LB-carbenicillin (100 μg/ml). The cultures were OD matched, and equal numbers of cells were inoculated into 500 ml of LB-carbenicillin (100 μg/ml) supplemented with 1% (wt/vol) arabinose to induce expression of HA-mucA. At an OD600 of 0.2, the cells were harvested by centrifugation at 7,000 × g for 10 min. The cells were then resuspended in 500 ml of M9 broth with 0.4% glucose supplemented with 100 μg/ml carbenicillin. Samples were taken at 10-min intervals by harvesting 50 ml of culture at 4,000 × g and 4°C for 10 min. Pellets were immediately stored at −80°C until proteins were prepared with Ready-Preps (Epicentre, Madison, WI) by the manufacturer's protocol and subjected to Western blot analysis with anti-HA and anti-alpha RNA polymerase subunit antibodies.
RESULTS
Inactivation of kinB in P. aeruginosa strain PAO1 results in alginate overproduction.
To discover novel negative regulators of alginate biosynthesis, the standard genetic strain PAO1 was subjected to mariner transposon mutagenesis (53). Stable mucoid gentamicin-resistant mutants were isolated. Mucoid mutants were verified for single transposon insertions by Southern hybridization (data not shown), and the pFAC transposon insertions were mapped by inverse PCR and sequencing as previously described (42, 43). Numerous mucoid mutants with insertions into the well-characterized negative regulator genes mucA, mucB, and mucD were identified. Interestingly, an insertion into kinB converted PAO1 to the mucoid phenotype (GenBank accession number for the kinB insertion in PAO1, FJ209363) (Fig. 2A). To show that mucoidy due to kinB inactivation was not caused by polar effects on nearby genes, we constructed an in-frame deletion of kinB in PAO1. Alginate overproduction resulted when kinB was deleted (Fig. 2A). However, PAO1 ΔkinB produced less alginate, i.e., 62 ± 4 μg/ml/OD600 unit, versus 103 ± 10 μg/ml/OD600 unit for PAO1 kinB::aacC1 (Fig. 2A). The mucoid phenotypes of PAO1 kinB::aacC1 and PAO1 ΔkinB were complemented by conditional expression of kinB (Fig. 2A). Expression of kinB in trans in PAO1 kinB::aacC1 and PAO1 ΔkinB decreased alginate production to wt PAO1 levels, as expected (Fig. 2A). Furthermore, sequencing analysis confirmed that the mucA gene of PAO1 kinB::aacC1 did not harbor mutations (GenBank accession number, FJ209362). Thus, inactivation or deletion of kinB in a wt mucA background causes alginate overproduction. This suggests that KinB is a negative regulator of alginate in P. aeruginosa strain PAO1.
FIG. 2.

Mutation of kinB in PAO1 results in a mucoid phenotype dependent upon algB, rpoN, and algW. (A) Colony morphologies of P. aeruginosa PAO1 and mucoid kinB mutants with or without kinB expressed in trans. For complementation, kinB was expressed from the PBAD promoter of pHERD20T. Strains were grown on a PIA-carbenicillin plate supplemented with 0.1% arabinose at 37°C for 24 h and at room temperature for 24 h. Alginate production was assayed by the carbazole assay (23) after 24 h at 37°C. The amount of alginate is indicated as μg/ml/OD600 unit. Values are expressed as means ± standard deviations from three independent experiments. (B) kinB mutants require algB, rpoN, and algW for alginate overproduction. Each mutant strain was assayed for alginate production with a vector control (pHERD20T) or with the gene indicated in trans expressed from the PBAD promoter of pHERD20T. The strains were grown for 24 h at 37°C on PIA supplemented with carbenicillin and 0.1% arabinose.
Alginate production in kinB mutants requires algB and rpoN.
Alginate overproduction in mucA mutants requires AlgB, an NtrC-type transcriptional activator (56). The algB gene is located immediately upstream of kinB in the genome. The kinB gene encodes the cognate kinase that has been shown to phosphorylate AlgB (29). Deletion of both algB and kinB together results in wt nonmucoid alginate production (Fig. 2B, bars 1). Alginate production was restored in the PAO1 ΔalgB ΔkinB double mutant by expression of algB in trans (Fig. 2B, bars 1). Since rpoN has been shown to be required for alginate production in a mucoid strain with an undefined muc-23 mutation (7), we examined whether rpoN was required in PAO1 kinB::aacC1. Deletion of rpoN from PAO1 kinB::aacC1 resulted in loss of mucoidy and could be complemented with rpoN expressed in trans (Fig. 2B, bars 2).
Alginate production in kinB mutants requires algW.
Since the mucA gene is not mutated in PAO1 kinB::aacC1, one possible explanation for the mucoid phenotype is that MucA is being degraded. AlgW has been shown to be required for activation of the alginate biosynthetic operon by d-cycloserine (54), and AlgW mediates regulated proteolysis of MucA during overexpression of mucE (43). We next tested whether mucoidy due to loss of kinB was dependent upon AlgW-regulated proteolysis. Deletion of algW from PAO1 kinB::aacC1 resulted in a nonmucoid phenotype and lowered alginate production (Fig. 2B, bars 3). Expression of algW in trans restored alginate production (Fig. 2B, bars 3). The PDZ domain of AlgW is required for MucE-mediated signal transduction (43). Therefore, to show that PAO1 kinB::aacC1 utilizes activated AlgW for derepression of MucA, we introduced an algW allele with the PDZ domain truncated and an algW allele with the PDZ domain completely deleted. When these mutant algW alleles were expressed in trans in the double mutant PAO1 kinB::aacC1 ΔalgW, alginate overproduction was not restored (Fig. 2B, bars 3). These data suggest that activation of AlgW is required for alginate overproduction in PAO1 kinB::aacC1.
In the absence of kinB, phosphorylation of AlgB at D59 is not required for alginate production.
KinB has been shown to effectively phosphorylate AlgB in vitro (29). However, AlgB derivatives such as AlgB.D59N, which cannot be phosphorylated by KinB, still promote alginate production in mucA mutants (28). The algB45 allele encodes AlgB.D59N, where the phosphorylation site (D59) has been mutated to asparagine (N) (28) We presumed that AlgB was not phosphorylated in the absence of the cognate histidine kinase KinB. To confirm that phosphorylation of AlgB at position 59 was not required for alginate production in the absence of KinB, we cloned the algB45 allele into pHERD20T for conditional expression. The algB45 gene was PCR amplified from pUS56 (28) and directionally cloned. The construct was sequenced to observe the expected D59N mutation and to ensure that no other mutations resulted. Expression of algB45 from the PBAD promoter in the presence of arabinose complemented the PAO581 algB::aacC1 (mucA25 algB::Gmr) mutant (Table 2) (42), which is consistent with the previous finding that the algB45 allele can still promote alginate production in a mucA22 mutant (28). Since the construct was functional, we introduced algB45 into PAO1 ΔalgB ΔkinB. Alginate overproduction occurred when algB45 was expressed in PAO1 ΔalgB ΔkinB (Table 2). These data suggests that in the absence of KinB, phosphorylation of AlgB at position 59 was not required for mucoidy. Interestingly, when we overexpressed algB or algB45 in wt PAO1 and PAO1 ΔalgB, we did not observe an increase in alginate production even when culture was on 1% arabinose (data not shown). It seems that deletion of kinB affects alginate production independent of the phosphorylation status of AlgB. Similar to the case for mucA mutants, phosphorylation of AlgB is not required for alginate overproduction in the kinB mutant with wt mucA.
TABLE 2.
Complementation of alginate production by algB mutants with wt algB and phosphorylation-defective algB45
| Strain | Plasmid | Arabinose (%, wt/vol)a | Phenotypeb | Alginate, μg/ml/OD600 unit (mean ± SD) |
|---|---|---|---|---|
| PAO581 algB::aacCC1 | pHERD20T- | 0 | NM | 49.0 ± 7.3 |
| (mucA25 algB::Gmr) | algB | 1 | M | 285.7 ± 12.9 |
| pHERD20T- | 0 | NM | 51.9 ± 3.0 | |
| algB45 | 1 | M | 228.2 ± 21.0 | |
| PAO1 Δ algBΔkinB | pHERD20T- | 0 | NM | 64.7 ± 10.9 |
| algB | 1 | M | 215.7 ± 13.5 | |
| pHERD20T- | 0 | NM | 49.5 ± 2.9 | |
| algB45 | 1 | M | 263.8 ± 2.5 |
Strains were cultured for 24 h at 37°C on PIA supplemented with carbenicillin and arabinose.
NM and M, nonmucoid and mucoid phenotypes, respectively.
PalgU and PalgD activities in kinB null mutants are dependent on algU, algB, rpoN, and algW.
To examine the effect of the kinB mutation on the alginate-related promoters PalgU and PalgD, we integrated a single copy of the entire algU or algD promoter region (Fig. 3A and C, respectively) fused with lacZ onto the chromosomes of PAO1 and PAO1 kinB::aacC1 as well as kinB algU, kinB algB, kinB rpoN, and kinB algW double mutants. The effect of each deletion or inactivated gene on the expression of the promoter fusions in the PAO1 and kinB backgrounds was assessed by complementation. The β-galactosidase activity was measured with vector alone (pHERD20T) and compared to that when the mutation was complemented with expression of the gene from the PBAD promoter of pHERD20T (41) in the presence of 0.1% arabinose. As a control for these experiments, PalgU and PalgD expression was measured when algU was overexpressed (Fig. 3B, bars 1, and D, bars 1, respectively).
Previous studies have shown that only small changes in PalgU expression are required for mucoidy (33). Inactivation of kinB in PAO1 kinB::aacC1 caused significantly increased PalgU expression compared to that in parent strain PAO1 (Fig. 3B, bars 2). The high PalgU expression in PAO1 kinB::aacC1 can be reduced with kinB expressed in trans (Fig. 3B, bars 2). Deletion of algU eliminated detectable PalgU expression in PAO1 kinB::aacC1 (Fig. 3B, bars 3). Since algB was observed to be required for alginate production in kinB mutants, we next examined whether algB was required for high levels of expression of PalgU. The high level of PalgU expression in the absence of kinB required algB (Fig. 3B, bars 4). AlgB has been established as a transcriptional activator at PalgD in the mucA22 mutant FRD-1 (56). Here we show a possible new role for AlgB in addition to the role at PalgD. We also observed that rpoN has a role in influencing high expression of PalgU (Fig. 3B, bars 5) that can be restored with rpoN expressed in trans. This information shows a possible role of rpoN outside of characterized interactions at PalgD (7). As expected, PalgU expression is also influenced by the serine protease AlgW in PAO1 kinB::aacC1 (Fig. 3B, bars 6). However, the level of expression of PalgU with algW in trans exceeded the PalgU expression level in PAO1 kinB::aacC1. A possible explanation for this is that algW expression from the arabinose promoter in the presence of 0.1% on a multicopy vector may exceed endogenous expression levels of algW in vivo.
PalgD expression was measured with the same strategy utilized for PalgU. Unlike PalgU activity, PalgD activity was minimally detectable in PAO1 (Fig. 3D, bars 1). The elevated level of PalgD expression in PAO1 kinB::aacC1 was significantly reduced when kinB was expressed in trans (Fig. 3D, bars 2). The elevated level of PalgD in kinB mutants required algU, algB, rpoN, and algW, which correlates with the observations of PalgU expression. The kinB mutants with deletions of algU, algB, rpoN, and algW had minimally detectable PalgD (Fig. 3D, bars 3 to 6). When algU, algB, algW, and rpoN were expressed in trans to complement their respective gene deletions in kinB mutants, elevated PalgD expression was returned. Collectively these promoter fusions in the PAO1 and kinB backgrounds show that algU, algB, algW, and rpoN influence the PalgU and PalgD activity, which correlates with alginate production (Fig. 2B).
AlgU and AlgB expression is increased in PAO1 kinB::aacC1.
Next we measured the expression of AlgU and AlgB in whole-cell lysates of PAO1 kinB::aacC1 (Fig. 4A). To control for cross-reactivity of anti-AlgU and anti-AlgB, total lysates of PAO1 ΔalgU and PAO1 ΔalgB were blotted, and very low cross-reactivity was noted (Fig. 4A, lanes 1 and 5, respectively). Western blot analysis revealed that AlgU was upregulated 2.6- ± 0.8-fold in PAO1 kinB::aacC1 compared to PAO1 (Fig. 4A, lanes 2 and 3). AlgB expression was also increased in PAO1 kinB::aacC1, which is consistent with a previous observation that algB transcription requires algT/U (57). Interestingly, AlgB was detected in PAO1 ΔalgU cell lysate, which suggests that AlgB expression may also be controlled by another σ factor in addition to AlgU (Fig. 4A, lane 1).
FIG. 4.

PAO1 kinB::aacC1 exhibits elevated levels of AlgB and AlgU, and HA-MucA degradation in PAO1 kinB::aacC1 requires algW, algB, and rpoN. Shown are representative panels of blots from three independent experiments with 40 μg of total lysate. (A) A Western blot of total cell lysate of PAO1 kinB::aacC1 shows elevated levels of AlgB and AlgU. Western blots of cell lysates were prepared from cells after 24 h growth on PIA. The membranes were probed with anti-AlgU, anti-AlgB, and anti-alpha subunit of RNA polymerase (loading control). Levels of each protein were adjusted for loading and then normalized to PAO1 levels and expressed as means ± standard deviations. Note that deletion of algU did not abolish AlgB expression. (B) Western blot analysis of N-terminally HA-tagged MucA in PAO1 and PAO1 kinB::aacC1 isogenic backgrounds. Cell lysates were prepared after 48 h of growth on PIA-carbenicillin plates supplemented with 0.1% arabinose. The membranes were immunoblotted with rat anti-HA diluted 1:1,000 (Roche). Lane 2, PAO1 pHERD20T-mucA is a negative control for background and cross-reactivity. Lanes 1 and 3 to 7, HA-mucA expressed in trans from pHERD20T. Levels of each protein were adjusted for loading and then normalized to PAO1 pHERD20T-HA-mucA levels and expressed as means ± standard deviations. Apparent molecular masses are depicted. NM and M indicate nonmucoid and mucoid phenotypes, respectively.
MucA proteolytic degradation facilitates alginate overproduction in PAO1 kinB::aacC1.
Since mucA is wt in PAO1 kinB::aacC1, MucA repression of AlgU must be relieved for activation of AlgU and alginate production. Based on the fact that alginate overproduction by PAO1 kinB::aacC1 requires AlgW, our hypothesis is that alginate production in the kinB mutant occurs by regulated proteolysis of MucA. To test this model, we needed to observe MucA degradation. N-terminal HA-tagged MucA was expressed from pHERD20T-HA-mucA under induction of arabinose into nonmucoid and mucoid PAO1 derivative strains. The wt mucA gene without HA was expressed in trans as the negative control. Western blotting of PAO1 without HA-tagged mucA showed no background or cross-reactivity with other proteins (Fig. 4B, lane 2). In PAO1, full-length HA-MucA existed as well as other truncated degradation products (Fig. 4B lane 1). HA-MucA degradation in PAO1 is consistent with degradation of RseA in E. coli, which occurs in the absence of stress signals (2). Also, PIA contains triclosan, which has been shown to activate PalgD activity (54), suggesting that regulated proteolysis occurs in the presence of cell wall-inhibitory antibiotics. In PAO1 ΔalgW, full-length HA-MucA is 2.4- ± 0.3-fold increased relative to PAO1 HA-MucA (Fig. 4B, lanes 1 and 3). This implies that HA-MucA is not as rapidly degraded in PAO1 ΔalgW as in PAO1. However, PAO1 ΔalgW also exhibited a truncated HA-MucA with an apparent molecular mass of 19 kDa (Fig. 4B, lane 3). The absence of this band in PAO1 suggests that deletion of algW inhibited efficient proteolysis of HA-MucA, resulting in accumulation of two major fragments of HA-MucA. Mucoid PAO1 kinB::aacC1 lacks the secondary truncated peptide of HA-MucA (Fig. 4B, lane 4), and the concentration of full-length HA-MucA is lower than that in PAO1 kinB::aacC1ΔalgW (Fig. 4B, lane 5). Interestingly, smaller degradation intermediates of HA-MucA (∼15 kDa) were still observed when algW was deleted in PAO1 and PAO1 kinB::aacC1 (Fig. 4B, lanes 3 and 5). These data suggest that regulated proteolysis of MucA may occur independent of AlgW, as has been shown when PAO1 algW::Tcr converts to mucoidy in the presence of the reactive oxygen-producing paraquat (6). The presence of the truncated HA-MucA was apparent upon deletion of algW, algB, and rpoN in PAO1 kinB mutants (Fig. 4B, lanes 5, 6, and 7). Taken together, these observations suggest that algB, algW, and rpoN influence HA-MucA degradation and derepression of AlgU in PAO1 kinB::aacC1.
In order to better measure the differences in HA-MucA degradation between PAO1 and PAO1 kinB::aacC1, a time course was conducted to show in vivo depletion of HA-MucA in PAO1 and PAO1 kinB::aacC1. To quantify the degradation of HA-MucA, cells were grown in LB broth containing arabinose to express HA-MucA and then transferred to M9 minimal medium supplemented with 0.4% glucose lacking arabinose. The OD600 remained stable during the time course. Therefore, depletion of HA-MucA was due to in vivo proteolytic degradation and not to division of the cells. Samples were taken every 10 min for the cell lysis and Western blotting. We also performed the time course with 30-min intervals but found that degradation of HA-MucA even in PAO1 was rapid, indicating that shorter time points need to be used (data not shown). The HA-MucA levels were assayed by Western blotting of 40 μg of total cell lysate (Fig. 5B). The rate of HA-MucA degradation in PAO1 kinB::aacC1 was 2.6-fold greater than that of PAO1 from 0 to 10 min (Fig. 5C). However, after 10 min the amounts of HA-MucA leveled off and were not significantly different between PAO1 and PAO1 kinB::aacC1 (Fig. 5C). Assuming that the mobility of HA-MucA peptide 1 was not aberrant, it appears that there is a cleavage site between the transmembrane domain and the C terminus of MucA (Fig. 5A). The smaller major truncated MucA peptide recognized with an HA epitope (HA-MucA peptide 2) was likely created by cleavage near the transmembrane domain. This study indicates that HA-MucA degradation is rapid even in PAO1; however, inactivation of kinB causes an increased rate of degradation. The increased HA-MucA degradation likely contributes to the mucoid phenotype of kinB mutants.
FIG. 5.

Kinetic comparison of HA-MucA degradation in PAO1 and PAO1 kinB::aacC1. (A) Schematic diagram of HA-MucA. Indicated are the N-terminal HA tag, the transmembrane domain, and the relative cleavage sites resulting in the truncated HA-MucA peptides observed by Western blotting. (B) Western blotting analysis of a time course of HA-MucA degradation. PAO1 and PAO1 kinB::aacC1 expressing HA-mucA from pHERD20T were grown at 37°C with shaking in LB supplemented with carbenicillin and arabinose until the OD600 reached 0.2. Cells were harvested and resuspended in M9 broth supplemented with 0.4% glucose. Glucose enhances repression of the PBAD promoter. During the time course the OD600 was monitored and remained stable throughout. Equal numbers of cells were extracted at 10-min intervals and harvested at 4°C, and pellets were frozen at −80°C until cell lysates were prepared. Shown is a representative panel of blots with 40 μg of total lysate transferred and blotted with anti-HA from three independent experiments. Apparent molecular masses are indicated. (C) Quantitative measurement of wt HA-MucA in PAO1 and PAO1 kinB::aacC1 from 0 min to 30 min. Levels of each protein were adjusted for loading and then normalized to PAO1 pHERD20T-HA-mucA or PAO1 kinB::aacC1 pHERD20T-HA-mucA levels and expressed as means ± standard deviations from three independent experiment (*, P < 0.01).
DISCUSSION
We discovered that mutation of kinB in PAO1 results in overproduction of alginate (Fig. 2A). Alginate regulation in mucA mutant strains was the first characterized mode of conversion to mucoidy and is the best elucidated (32). However, recent studies have shown that regulated proteolysis mediated by AlgW is a mechanism for alginate production in P. aeruginosa (43, 54). Here we have presented data showing that inactivation of kinB causes mucoidy and is dependent upon algB, algW, and rpoN (Fig. 2B). We also observed through complementation analysis that phosphorylation of AlgB at the confirmed phosphorylation site is not required for alginate production in the kinB mutant. Our data suggest that the kinB mutation increases the rate of degradation of MucA by regulated proteolysis, which causes the mucoid phenotype of kinB mutants.
KinB is the cognate kinase of the alginate regulator AlgB (29), and alginate biosynthesis occurs independent of phosphorylation of AlgB (28). However, the role of kinB in alginate production has been examined only in mucoid mucA22 mutant strains such as FRD-1 (28). In mucA mutants, the requirement for regulated proteolysis to activate AlgU would likely be bypassed due to the mucA mutation. We observed that in kinB mutants, algB and rpoN are both required for alginate production (Fig. 2B) and increased PalgU and PalgD promoter activity (Fig. 3B and D, respectively). Previously both rpoN (7) and algB (26) have been shown to affect transcription at PalgD. Conversely, our data show that these regulators, AlgB and RpoN, also affect PalgU transcription. Only relatively small changes in PalgU expression are required for mucoidy (33); however, PAO1 kinB::aacC1 exhibits significantly elevated expression of both PalgU and PalgD (Fig. 3B and D, respectively). We also noted that deletion of algU from PAO1 kinB::aacC1 resulted in complete loss of detectable PalgU-lacZ activity as measured by β-galactosidase assay (Fig. 3B, bars 3). This has also been observed when algU is deleted from PAO1 (data not shown). Two of the algU promoters are AlgU dependent (12, 45); however, it is not clear which σ factors the other promoters depend upon. Therefore, it is possible that in vivo AlgU contributes the bulk of transcriptional activation of the AlgU promoters that is detectable by our reporter assay, but further analysis is required to fully understand the algU promoters.
Based on our data, we propose two alternative models for activation of alginate production through regulated proteolysis in kinB mutants (Fig. 6). In both models, regulated proteolysis of MucA by AlgW occurs, but the cause of the increased concentration of activating signals differs. The first model suggests that mutation of kinB affects expression of a protease or chaperone responsible for removal of misfolded proteins (Fig. 6). Mutation of an aminopeptidase gene, phpA, has been shown to cause increased PalgD activity and mucoidy (55). The second proposed model is that algB and rpoN directly control expression of peptide signals in the absence of kinB that activate AlgW and therefore increase proteolytic degradation of MucA (Fig. 6). Deletion of algB and rpoN in kinB mutants caused an accumulation of the major HA-MucA truncation product that was also observed when algW was deleted in PAO1 or PAO1 kinB::aacC1 (Fig. 4B). We have also observed that algB and rpoN are not required for algW expression (data not shown), which suggested that loss of algB or rpoN may affect the proteolytic activity of AlgW. From this information, we hypothesize that algB and rpoN may be required for expression of signals that activate AlgW and regulated proteolysis. Our data suggest that increased regulated proteolysis occurs in kinB mutants of PAO1 (Fig. 4B and 5B and C). In E. coli, many outer membrane and periplasmic proteins have been shown to activate DegS protease activity through interaction with the PDZ domain (18). Interestingly, in E. coli, inactivation of the two-component histidine kinase EnvZ causes upregulation of the porin OmpC (48). Porins such as OmpC can activate regulated proteolysis (18). Analysis of the P. aeruginosa genome shows no significant homologues to the DegS-activating peptides such as OmpC of E. coli. This is conceivable because P. aeruginosa and E. coli reside in different habitats, and therefore it is likely that activation of AlgU and activation of σE require different types of signals. However, proteins with probable activating sequences are encoded throughout the P. aeruginosa genome (43). Thus, P. aeruginosa likely has novel proteins that could potentially activate AlgW degradation of MucA. It is possible that RpoN, in tandem with response regulators such as AlgB, controls numerous genes with various functions which may be involved in signal transduction of the AlgU stress response.
FIG. 6.

Proposed models of negative regulation of alginate production by KinB in P. aeruginosa. Mutation of kinB in wt mucA strain PAO1 causes alginate overproduction. Alginate overproduction by kinB mutants requires algB and rpoN. In model 1, we propose that mutation of kinB causes loss of expression of a periplasmic protease(s), which leads to accumulation of AlgW-activating factors. In model 2, we propose that algB and rpoN control expression of factors which can activate the AlgW protease to release repression of MucA by proteolytic degradation and activate AlgU. In either case, derepression of MucA by regulated proteolysis causes AlgU activation, which facilitates algU expression, resulting in mucoidy by upregulating the alginate biosynthetic operon. OM and IM, outer membrane and inner membrane, respectively.
AlgB and or RpoN could drive both algU and algD transcription. This is an alternative hypothesis to the models already described. Both AlgB and RpoN have been shown to bind at PalgD and are required for algD expression (7, 26). It has been suggested that AlgB may interact with other σ factors than RpoN (26). We have attempted to show AlgB binding with PalgU using a gel shift assay; however, interaction has not been observed (data not shown). Recent studies have employed special conditions to detect AlgB DNA binding at PalgD (26). Since exhaustive studies have not been performed, we cannot dismiss the possibility that AlgB and/or RpoN may initiate transcription at PalgU. Based on our data, both the PalgU and the PalgD promoters are highly upregulated in kinB mutants (Fig. 3B and D). Therefore, it is possible that in the absence of kinB, AlgB could activate transcription of both the PalgU and PalgD promoters.
Do P. aeruginosa CF isolates have kinB mutations? Most clinical observations have focused on surveying mucA, mucB, and mucD (8, 10, 32). Therefore, large-scale surveys looking for kinB mutants have not been performed. However, one recently sequenced epidemic CF isolate, C3719, does have a mutation that truncates the KinB protein to 526 amino acids instead of the wt 595 amino acids of PAO1 KinB (http://www.broad.mit.edu). Therefore, a CF isolate has been shown to have a kinB mutation, but C3719 is apparently nonmucoid (34). This suggests that either the mutation is not completely detrimental to KinB regulation or C3719 may have additional suppressor mutations in either known or novel alginate regulators. PAO579 is another strain that requires rpoN for mucoidy (7); however, the mucoid phenotype cannot be suppressed by complementation with kinB (data not shown). We are currently surveying for wt mucA CF isolates for kinB mutations. Many two-component signal systems can be activated by environmental conditions. The PhoP-PhoQ (30) and PmrA-PmrB (35) systems of P. aeruginosa are activated by low Mg2+ concentrations, whereas the conserved PhoB-PhoR system is activated by low phosphate concentrations (25). Therefore, elucidation of the environmental signals that relieve the negative regulation of KinB on alginate overproduction will be as interesting as finding kinB mutant CF isolates.
In this report we have characterized KinB as a negative regulator of alginate production and have proposed novel regulation of AlgW-dependent MucA derepression that is mediated by AlgB and RpoN. These data bring us a step closer toward understanding the molecular events leading to alginate production which preclude the classically described mucA mutations in P. aeruginosa. It will be interesting to further elucidate the unknown genes that may be under the negative control of the sensor kinase KinB and to determine the environmental stimulus that affects KinB regulation in P. aeruginosa.
Acknowledgments
This work was supported by a research grant (NNA04CC74G) from the National Aeronautics and Space Administration (NASA) and research grants from the NASA West Virginia Space Grant Consortium. F.H.D. was supported by a training grant (NNX06AH20H) from the NASA Graduate Student Researchers Program (GSRP).
We thank D. Wozniak for the gift of the AlgB antibodies and plasmid pUS56, N. Head for the initial cloning and sequencing work, V. Eisinger and M. Bartley for technical assistance, and J. Denvir for statistical assistance. We also thank M. J. Schurr and the anonymous reviewers for helpful comments on the manuscript.
Footnotes
Published ahead of print on 23 January 2009.
REFERENCES
- 1.Ades, S. E. 2004. Control of the alternative sigma factor sigmaE in Escherichia coli. Curr. Opin. Microbiol. 7157-162. [DOI] [PubMed] [Google Scholar]
- 2.Ades, S. E., I. L. Grigorova, and C. A. Gross. 2003. Regulation of the alternative sigma factor sigma(E) during initiation, adaptation, and shutoff of the extracytoplasmic heat shock response in Escherichia coli. J. Bacteriol. 1852512-2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aiba, B. M., J. A. Leeds, C. Onufryk, C. Z. Lu, and C. A. Gross. 2002. DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigma(E)-dependent extracytoplasmic stress response. Genes Dev. 162156-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baynham, P. J., A. L. Brown, L. L. Hall, and D. J. Wozniak. 1999. Pseudomonas aeruginosa AlgZ, a ribbon-helix-helix DNA-binding protein, is essential for alginate synthesis and algD transcriptional activation. Mol. Microbiol. 331069-1080. [DOI] [PubMed] [Google Scholar]
- 5.Baynham, P. J., D. M. Ramsey, B. V. Gvozdyev, E. M. Cordonnier, and D. J. Wozniak. 2006. The Pseudomonas aeruginosa ribbon-helix-helix DNA-binding protein AlgZ (AmrZ) controls twitching motility and biogenesis of type IV pili. J. Bacteriol. 188132-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boucher, J. C., J. Martinezsalazar, M. J. Schurr, M. H. Mudd, H. Yu, and V. Deretic. 1996. Two distinct loci affecting conversion to mucoidy in Pseudomonas aeruginosa in cystic fibrosis encode homologs of the serine protease htrA. J. Bacteriol. 178511-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boucher, J. C., M. J. Schurr, and V. Deretic. 2000. Dual regulation of mucoidy in Pseudomonas aeruginosa and sigma factor antagonism. Mol. Microbiol. 36341-351. [DOI] [PubMed] [Google Scholar]
- 8.Bragonzi, A., L. Wiehlmann, J. Klockgether, N. Cramer, D. Worlitzsch, G. Doring, and B. Tummler. 2006. Sequence diversity of the mucABD locus in Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 1523261-3269. [DOI] [PubMed] [Google Scholar]
- 9.Bragonzi, A., D. Worlitzsch, G. B. Pier, P. Timpert, M. Ulrich, M. Hentzer, J. B. Andersen, M. Givskov, M. Conese, and G. Doring. 2005. Nonmucoid Pseudomonas aeruginosa expresses alginate in the lungs of patients with cystic fibrosis and in a mouse model. J. Infect. Dis. 192410-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciofu, O., B. Lee, M. Johannesson, N. O. Hermansen, P. Meyer, and N. Hoiby. 2008. Investigation of the algT operon sequence in mucoid and non-mucoid Pseudomonas aeruginosa isolates from 115 Scandinavian patients with cystic fibrosis and in 88 in vitro non-mucoid revertants. Microbiology 154103-113. [DOI] [PubMed] [Google Scholar]
- 11.Deretic, V., J. F. Gill, and A. M. Chakrabarty. 1987. Gene algD coding for GDPmannose dehydrogenase is transcriptionally activated in mucoid Pseudomonas aeruginosa. J. Bacteriol. 169351-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeVries, C. A., and D. E. Ohman. 1994. Mucoid-to-nonmucoid conversion in alginate-producing Pseudomonas aeruginosa often results from spontaneous mutations in algT, encoding a putative alternate sigma factor, and shows evidence for autoregulation. J. Bacteriol. 1766677-6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doggett, R. G., G. M. Harrison, R. N. Stillwell, and E. S. Wallis. 1966. An atypical Pseudomonas aeruginosa associated with cystic fibrosis of the pancreas. J. Pediatr. 68215-221. [Google Scholar]
- 14.Figurski, D. H., and D. R. Helinski. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. USA 761648-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldberg, J. B., and T. Dahnke. 1992. Pseudomonas aeruginosa AlgB, which modulates the expression of alginate, is a member of the NtrC subclass of prokaryotic regulators. Mol. Microbiol. 659-66. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg, J. B., and D. E. Ohman. 1984. Cloning and expression in Pseudomonas aeruginosa of a gene involved in the production of alginate. J. Bacteriol. 1581115-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Govan, J. R., and V. Deretic. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60539-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasselblatt, H., R. Kurzbauer, C. Wilken, T. Krojer, J. Sawa, J. Kurt, R. Kirk, S. Hasenbein, M. Ehrmann, and T. Clausen. 2007. Regulation of the sigmaE stress response by DegS: how the PDZ domain keeps the protease inactive in the resting state and allows integration of different OMP-derived stress signals upon folding stress. Genes Dev. 212659-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hassett, D. J. 1996. Anaerobic production of alginate by Pseudomonas aeruginosa: alginate restricts diffusion of oxygen. J. Bacteriol. 1787322-7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoang, T. T., A. J. Kutchma, A. Becher, and H. P. Schweizer. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 4359-72. [DOI] [PubMed] [Google Scholar]
- 21.Kato, J., and A. M. Chakrabarty. 1991. Purification of the regulatory protein AlgR1 and its binding in the far upstream region of the algD promoter in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 881760-1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, K. I., S. C. Park, S. H. Kang, G. W. Cheong, and C. H. Chung. 1999. Selective degradation of unfolded proteins by the self-compartmentalizing HtrA protease, a periplasmic heat shock protein in Escherichia coli. J. Mol. Biol. 2941363-1374. [DOI] [PubMed] [Google Scholar]
- 23.Knutson, C. A., and A. Jeanes. 1968. A new modification of the carbazole reaction: application to heteropolysaccharides. Anal. Biochem. 24470-481. [DOI] [PubMed] [Google Scholar]
- 24.Kustu, S., A. K. North, and D. S. Weiss. 1991. Prokaryotic transcriptional enhancers and enhancer-binding proteins. Trends Biochem. Sci. 16397-402. [DOI] [PubMed] [Google Scholar]
- 25.Lamarche, M. G., B. L. Wanner, S. Crepin, and J. Harel. 2008. The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol. Rev. 32461-473. [DOI] [PubMed] [Google Scholar]
- 26.Leech, A. J., A. Sprinkle, L. Wood, D. J. Wozniak, and D. E. Ohman. 2008. The NtrC family regulator AlgB, which controls alginate biosynthesis in mucoid Pseudomonas aeruginosa, binds directly to the algD promoter. J. Bacteriol. 190581-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyczak, J. B., C. L. Cannon, and G. B. Pier. 2002. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 15194-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma, S., U. Selvaraj, D. E. Ohman, R. Quarless, D. J. Hassett, and D. J. Wozniak. 1998. Phosphorylation-independent activity of the response regulators AlgB and AlgR in promoting alginate biosynthesis in mucoid Pseudomonas aeruginosa. J. Bacteriol. 180956-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma, S., D. J. Wozniak, and D. E. Ohman. 1997. Identification of the histidine protein kinase KinB in Pseudomonas aeruginosa and its phosphorylation of the alginate regulator algB. J. Biol. Chem. 27217952-17960. [DOI] [PubMed] [Google Scholar]
- 30.Macfarlane, E. L., A. Kwasnicka, M. M. Ochs, and R. E. Hancock. 1999. PhoP-PhoQ homologues in Pseudomonas aeruginosa regulate expression of the outer-membrane protein OprH and polymyxin B resistance. Mol. Microbiol. 34305-316. [DOI] [PubMed] [Google Scholar]
- 31.Martin, D. W., M. J. Schurr, M. H. Mudd, and V. Deretic. 1993. Differentiation of Pseudomonas aeruginosa into the alginate-producing form: inactivation of mucB causes conversion to mucoidy. Mol. Microbiol. 9497-506. [DOI] [PubMed] [Google Scholar]
- 32.Martin, D. W., M. J. Schurr, M. H. Mudd, J. R. Govan, B. W. Holloway, and V. Deretic. 1993. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 908377-8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathee, K., C. J. McPherson, and D. E. Ohman. 1997. Posttranslational control of the algT (algU)-encoded sigma22 for expression of the alginate regulon in Pseudomonas aeruginosa and localization of its antagonist proteins MucA and MucB (AlgN). J. Bacteriol. 1793711-3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathee, K., G. Narasimhan, C. Valdes, X. Qiu, J. M. Matewish, M. Koehrsen, A. Rokas, C. N. Yandava, R. Engels, E. Zeng, R. Olavarietta, M. Doud, R. S. Smith, P. Montgomery, J. R. White, P. A. Godfrey, C. Kodira, B. Birren, J. E. Galagan, and S. Lory. 2008. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. USA 1053100-3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McPhee, J. B., S. Lewenza, and R. E. Hancock. 2003. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 50205-217. [DOI] [PubMed] [Google Scholar]
- 36.Miller, J. H. 1972. Beta-galactosidase assay, p. 352-355. In J. H. Miller (ed.), Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 37.Mohr, C. D., and V. Deretic. 1990. Gene-scrambling mutagenesis: generation and analysis of insertional mutations in the alginate regulatory region of Pseudomonas aeruginosa. J. Bacteriol. 1726252-6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohr, C. D., N. S. Hibler, and V. Deretic. 1991. AlgR, a response regulator controlling mucoidy in Pseudomonas aeruginosa, binds to the FUS sites of the algD promoter located unusually far upstream from the mRNA start site. J. Bacteriol. 1735136-5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer, K. L., S. A. Brown, and M. Whiteley. 2007. Membrane-bound nitrate reductase is required for anaerobic growth in cystic fibrosis sputum. J. Bacteriol. 1894449-4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedersen, S. S., N. Hoiby, F. Espersen, and C. Koch. 1992. Role of alginate in infection with mucoid Pseudomonas aeruginosa in cystic fibrosis. Thorax 476-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu, D., F. H. Damron, T. Mima, H. P. Schweizer, and H. D. Yu. 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl. Environ. Microbiol. 747422-7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiu, D., V. M. Eisinger, N. E. Head, G. B. Pier, and H. D. Yu. 2008. ClpXP proteases positively regulate alginate overexpression and mucoid conversion in Pseudomonas aeruginosa. Microbiology 1542119-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu, D., V. M. Eisinger, D. W. Rowen, and H. D. Yu. 2007. Regulated proteolysis controls mucoid conversion in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1048107-8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowen, D. W., and V. Deretic. 2000. Membrane-to-cytosol redistribution of ECF sigma factor AlgU and conversion to mucoidy in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Mol. Microbiol. 36314-327. [DOI] [PubMed] [Google Scholar]
- 45.Schurr, M. J., H. Yu, J. C. Boucher, N. S. Hibler, and V. Deretic. 1995. Multiple promoters and induction by heat shock of the gene encoding the alternative sigma factor AlgU (σE) which controls mucoidy in cystic fibrosis isolates of Pseudomonas aeruginosa. J. Bacteriol. 1775670-5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schurr, M. J., H. Yu, J. M. Martinez-Salazar, J. C. Boucher, and V. Deretic. 1996. Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J. Bacteriol. 1784997-5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schweizer, H. P., and T. T. Hoang. 1995. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene 15815-22. [DOI] [PubMed] [Google Scholar]
- 48.Slauch, J. M., S. Garrett, D. E. Jackson, and T. J. Silhavy. 1988. EnvZ functions through OmpR to control porin gene expression in Escherichia coli K-12. J. Bacteriol. 170439-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spiess, C., A. Beil, and M. Ehrmann. 1999. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 97339-347. [DOI] [PubMed] [Google Scholar]
- 50.Tart, A. H., M. J. Blanks, and D. J. Wozniak. 2006. The AlgT-dependent transcriptional regulator AmrZ (AlgZ) inhibits flagellum biosynthesis in mucoid, nonmotile Pseudomonas aeruginosa cystic fibrosis isolates. J. Bacteriol. 1886483-6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh, N. P., B. M. Alba, B. Bose, C. A. Gross, and R. T. Sauer. 2003. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 11361-71. [DOI] [PubMed] [Google Scholar]
- 52.Wilken, C., K. Kitzing, R. Kurzbauer, M. Ehrmann, and T. Clausen. 2004. Crystal structure of the DegS stress sensor: how a PDZ domain recognizes misfolded protein and activates a protease. Cell 117483-494. [DOI] [PubMed] [Google Scholar]
- 53.Wong, S. M., and J. J. Mekalanos. 2000. Genetic footprinting with mariner-based transposition in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 9710191-10196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wood, L. F., A. J. Leech, and D. E. Ohman. 2006. Cell wall-inhibitory antibiotics activate the alginate biosynthesis operon in Pseudomonas aeruginosa: roles of sigma (AlgT) and the AlgW and Prc proteases. Mol. Microbiol. 62412-426. [DOI] [PubMed] [Google Scholar]
- 55.Woolwine, S. C., A. B. Sprinkle, and D. J. Wozniak. 2001. Loss of Pseudomonas aeruginosa PhpA aminopeptidase activity results in increased algD transcription. J. Bacteriol. 1834674-4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wozniak, D. J., and D. E. Ohman. 1991. Pseudomonas aeruginosa AlgB, a two-component response regulator of the NtrC family, is required for algD transcription. J. Bacteriol. 1731406-1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wozniak, D. J., and D. E. Ohman. 1994. Transcriptional analysis of the Pseudomonas aeruginosa genes algR, algB, and algD reveals a hierarchy of alginate gene expression which is modulated by algT. J. Bacteriol. 1766007-6014. [DOI] [PMC free article] [PubMed] [Google Scholar]