

Abstract
Representatives of several picornavirus genera have been shown previously to significantly enhance noncontrollable bidirectional exchange of proteins between nuclei and cytoplasm. In enteroviruses and rhinoviruses, enhanced permeabilization of the nuclear pores appears to be primarily due to proteolytic degradation of some nucleoporins (protein components of the pore), whereas this effect in cardiovirus-infected cells is triggered by the leader (L) protein, devoid of any enzymatic activities. Here, we present evidence that expression of L alone was sufficient to cause permeabilization of the nuclear envelope in HeLa cells. In contrast to poliovirus, mengovirus infection of these cells did not elicit loss of nucleoporins Nup62 and Nup153 from the nuclear pore complex. Instead, nuclear envelope permeabilization was accompanied by hyperphosphorylation of Nup62 in cells infected with wild-type mengovirus, whereas both of these alterations were suppressed in L-deficient virus mutants. Since phosphorylation of Nup62 (although less prominent) did accompany permeabilization of the nuclear envelope prior to its mitotic disassembly in uninfected cells, we hypothesize that cardiovirus L alters the nucleocytoplasmic traffic by hijacking some components of the normal cell division machinery. The variability and biological significance of picornaviral interactions with the nucleocytoplasmic transport in the infected cells are discussed.
Productive viral interaction with host cells requires generation of an intracellular environment ensuring efficient synthesis and assembly of virally encoded macromolecules. A fundamental aspect of this process is overcoming a variety of host defensive tools, collectively named innate immunity. Some of the viral antidefensive measures are direct results of enzymatic activities of viral proteins, as exemplified by proteolytic degradation of host translation or transcription factors. However, the viral antidefensive armament in the case of small-genome viruses is usually relatively limited, and therefore such viruses have evolved the capacity to divert (“hijack”) cellular proteins, pathways, and structures to perform proviral functions. A group of viruses relatively well studied in this respect, is represented by picornaviruses—small, nonenveloped animal viruses with a 7.2- to 8.5-kb RNA genome of positive polarity (46).
The Picornaviridae family is composed of at least nine genera (48), which share general genome organization but differ from each other in some important aspects such as, for example, the capsid structure (and consequently the usage of cellular receptors), the structure of replicative and translational cis elements, and the presence or absence of structurally and functionally unrelated accessory (usually nonessential) proteins (2).
In recent years, we have studied, among other activities exhibited by picornaviruses, their ability to trigger bidirectional permeabilization of the nuclear envelope, facilitating noncontrollable exchange of macromolecules between the nucleus and cytoplasm (5, 6, 25). It is thought that such exchange could stimulate virus-specific protein and/or RNA synthesis in the cytoplasm by making available stimulatory nuclear proteins, and it could, on the other hand, represent a viral antidefensive tool, permitting entry into the nucleus of viral inhibitors of host transcription.
In uninfected cells, the nucleocytoplasmic transport is a tightly regulated and energy-dependent process, at least as far as proteins with molecular masses of more than 40 to 60 kDa and RNA molecules are concerned (15, 50). The proteins destined into or out of the nuclei should possess specific amino acid motifs, nuclear localization signals (NLS) and nuclear export signals, respectively. These signaling motifs are recognized by soluble protein receptors, importins (38). The direction of traffic is controlled by Ran, a small GTPase, which exists in either GTP-bound (nuclear) or GDP-bound (cytoplasmic) forms. The traffic occurs through specialized “gates” in the nuclear envelope, so-called nuclear pore complexes (NPC), large (∼125-MDa) barrel-like assemblies build from about 30 protein species called nucleoporins (Nups). The core part of the pore is comprised by eight granules arranged around the permeable channel and anchored to the nuclear envelope. Attached to this core are peripheral components, the cytoplasmic and nuclear rings and filaments (26).
The recognition of transportins is carried out by the Nup's amino acid motifs, so-called phenylalanine-glycine (FG)-containing repeats (FXFG, GLFG, and FG) (44). The FG repeats containing domains of Nups are thought to exist mainly as unfolded stretches that are available for interactions with cargoes (11, 27). The central channel harbors a structure, visible by electron microscopy as a dense entity (4), that includes the Nup62 complex composed of Nup62, Nup58, Nup54, and Nup45 (21). This complex plays an important part in the control of selective permeability of the pores (33). The most dramatic, although physiological, alterations of NPC that terminate in their complete dismantling take place during mitosis (43).
Although representatives of enteroviruses, rhinoviruses, and cardioviruses appeared to elicit phenomenologically similar effects on the nucleocytoplasmic transport, they accomplish this by fundamentally different mechanisms. In the case of enteroviruses and rhinoviruses, the key effector protein is viral protease 2Apro (6, 17, 18, 37) which cleaves a number of Nups (Nup62, Nup153, and Nup98). Cardioviruses lack 2Apro-like polypeptide, and alterations of the nucleocytoplasmic transport in cells infected with these viruses is mediated by the viral leader (L) protein (10, 25), which is devoid of any enzymatic activity. The ability of the protein kinase inhibitor staurosporin to prevent cardiovirus-elicited permeabilization of the nuclear envelope (25) suggests that the L protein interacts with some phosphorylation pathway involved in the control of the nucleocytoplasmic exchange. The leader protein was reported to interact with Ran and is supposed to affect the energy-dependent active nuclear transport (41), but it is unclear how, and whether, this interaction could directly explain staurosporin-sensitive permeabilization of the nuclear pores.
It is appropriate to note that phosphorylation was proposed to participate in regulation of the nuclear transport machinery in uninfected cells, although appropriate mechanisms are yet to be elucidated in detail (22, 35). Importantly, some Nups, including Nup62, were shown to be phosphorylated (16, 28, 30, 34). It is well documented that mitotic disassembly of the nuclear envelope and, in particular, of nuclear pores, is associated with changes in the phosphorylation status of certain Nups (16, 30, 34).
In the present study, we investigated the fate (intracellular localization, integrity, and phosphorylation status) of some Nups, in particular Nup62, in cells infected with cardioviruses or their L-deficient mutants. We also compared the behavior of Nup62 in cardiovirus-infected cells, on the one hand, and at early stages of mitosis on the other.
MATERIALS AND METHODS
Cells and viruses.
HeLa-B (a subline of HeLa cells) (51) and HeLa-3E cells (constitutively expressing 3×EGFP-NLS, i.e., three copies of the enhanced green fluorescent protein [EGFP] fused to the simian virus 40 NLS) (6) were grown on petri dishes in Dulbecco modified Eagle medium with 10% fetal bovine serum. The cells were washed with serum-free Eagle medium and infected with appropriate viruses at a multiplicity of infection (MOI) of ∼100 PFU/cell unless indicated otherwise. After 30 min of adsorption with agitation at 18°C, the cells were washed again and incubated with 5% CO2 in serum-free medium at 37°C for various time intervals. Poliovirus type 1 Mahoney strain and mengovirus strain of encephalomyocarditis virus were used. The mengovirus mutants were described previously (19, 56, 57).
Cell synchronization.
To obtain a fraction of mitotic enriched cells, half-confluent monolayers of HeLa-3E cells were incubated with the above medium containing 250 μM thymidine (Sigma) for 16 h and in the absence of thymidine for an additional 11 to 12 h. Cells were detached by gentle shaking and collected by centrifugation at 300 × g for 10 min at 4°C. As judged by DNA staining followed by microscopy, the overwhelming majority of the cells in such preparations were mitotic or early postmitotic. For the immunofluorescence analysis of Nups in a synchronized cell population, the double thymidine block was used. Cells were incubated for 16 h in the presence of thymidine, for 8 h in its absence, for 16 h again with the nucleoside, and finally for 12.5 h in the thymidine-free medium.
Plasmids.
The plasmids encoding mengovirus L protein or its mutants with red fluorescent protein (RFP; dsRed1) fused to their C termini were constructed by insertion of PCR fragments obtained from the full genome DNA copies of appropriate viruses (56, 57) into HindIII-BamHI sites of the pdsRed1-N1 vector (Clontech).
Transient transfection.
For transfection, Lipofectamine 2000 (Invitrogen) was used essentially according to the manufacturer's recommendations. Briefly, the reagent was diluted in Dulbecco modified Eagle medium, mixed with 1 to 2 μg of DNA, incubated for 20 min at room temperature, and added to monolayers of HeLa cells.
Preparation of cell extracts.
HeLa-B cells were grown in 1.5-liter roller bottles until near confluence and then mock infected or infected with mengovirus at an input MOI of ∼30 PFU/cell. After incubation for 4.5 h at 37°C, cells were collected by treatment with EDTA, placed in a hypotonic buffer {50 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], 50 mM KCl, 5 mM EGTA, 2 mM MgCl2, 1 mM dithiothreitol [DTT], 10 μg of cytochalasin B/ml; pH 7), and subjected to three cycles of freezing and thawing, with subsequent centrifugation at 100,000 × g, as previously described (6).
Nuclear envelope permeability assay.
Monolayers of HeLa-3E cells were treated with digitonin (1), as previously described (6). Briefly, the cells were washed consecutively with phosphate-buffered saline (PBS) and permeabilization buffer (50 mM PIPES, 50 mM KCl, 5 mM EGTA, 2 mM MgCl2, 1 mM DTT; pH 7), exposed to the detergent (40 μg/ml for 5 min), and then carefully washed with the digitonin-free permeabilization buffer. The digitonin solution was supplemented with Hoechst 33258 (Sigma) dye, which penetrates through the plasma membrane relatively slowly. Under the conditions used, the permeabilized cells were stained well, whereas the cells with intact plasma membranes were stained poorly, if at all. Permeabilized monolayers were overlaid with appropriate cell extracts, diluted 10-fold in permeabilization buffer. Samples were incubated for 1 h at 37°C and examined under an epifluorescence microscope.
Antibodies.
Monoclonal antibody (MAb) 414 antibodies raised against FG repeats of Nups were obtained from Covance. Anti-Nup62 MAb against the 24- to 178-amino-acid region belonging to the N-terminal FG-containing domain of the protein were from BD Transduction Laboratories. Anti-Nup153 (QE5) were from Abcam, antibodies recognizing phosphorylated serine 10 of H3 histone [p-Histone H3 (Ser10)-R] were from Santa Cruz, and anti-actin antibodies (AC-40) were from Sigma. Horseradish-conjugated anti-mouse and anti-rabbit antibodies were from Promega. Anti-mouse antibodies conjugated with Alexa Fluor 555 were from Invitrogen.
Immunofluorescence.
Subconfluent cell monolayers growing on coverslips were fixed for 20 min with 3.7% paraformaldehyde diluted in PBS, washed, and permeabilized with 0.2% Triton X-100 in PBS for 10 min. Cells were washed three times with PBS for 5 min each time, blocked with 1% bovine serum albumin for 1 h at 37°C, incubated with the primary antibodies in immunofluorescence buffer (0.1% bovine serum albumin and 0.05% Tween 20 in PBS) for 1 h, washed thrice for 5 min each time with this buffer, incubated with the secondary antibodies for 0.5 to 1 h, and washed again three times with the same buffer containing Hoechst 33342 (5 μg/ml). The coverslips were mounted in Mowiol and inspected with Leica DMLS or Zeiss Axiovert 200M microscopes.
32P labeling of WGA-binding proteins.
At the indicated times postinfection (p.i.), mock- and virus-infected cells were washed with phosphate-free medium and incubated in the presence of 300 μCi of [32P]orthophosphate/ml in phosphate-free medium for 2 h at 37°C. The cells were washed, harvested, and pelleted by centrifugation at 300 × g for 5 min at 4°C. The pellets were washed with ice-cold lysis buffer A (250 mM sucrose, 10 mM HEPES [pH 7.4], 50 mM KCl, 2.5 mM MgCl2, 1 mM DTT, 20 μg of cycloheximide/ml, and 5 μg [each] of cytochalasin B, chymostatin, leupeptin, and pepstatin A/ml), resuspended in 20 volumes of lysis buffer B (20 mM HEPES [pH 7.0], 100 mM KCl, 2 mM MgCl2, 1 mM DTT, 5 μg [each] of chymostatin, leupeptin, and pepstatin A/ml), and lysed by three cycles of freezing and thawing with brief vortexing prior to each freezing. Total cell lysates were centrifuged at 300 × g for 15 min at 4°C, and the supernatants represented the cytosolic fraction. The pellets were washed with the buffer B and resuspended in 20 volumes of the same buffer yielding the membrane fraction. The extraction of wheat germ agglutinin (WGA)-binding proteins was performed as previously described (14). Briefly, 1 volume of cytoplasmic and membrane fractions were incubated with 0.5 volume of packed, prewashed WGA-agarose (Sigma) for 1 to 2 h at 4°C with rocking. The WGA-agarose was pelleted (1,000 × g, 30 s), and the supernatant was discarded. The WGA granules were washed extensively with lysis buffer B. WGA-binding proteins were eluted by incubation (30 min, 4°C) with a 0.25 volume of the same buffer containing 0.5 M N-acetylglucosamine. WGA-binding fraction of 32P-labeled proteins, which is known to correspond to Nups (34), was resolved by sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis (SDS-12% PAGE) and analyzed by autoradiography. Prestained protein markers (Fermentas) were used for the molecular mass evaluation.
Immunoprecipitation.
Cells were collected after treatment with versene and pelleted by centrifugation at 300 × g for 10 min at 4°C. All of the following manipulations were performed at 4°C. The pellets were resuspended in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 1 mM EDTA, 10 mM Tris-HCl [pH 7.5]) supplemented with a phosphatase inhibitors cocktail (Sigma) to suppress endogenous phosphatase activity. The lysates were subjected to centrifugation at 12,000 × g for 10 min, and the pellets were discarded. The supernatants were supplemented with 3 to 5 μg of the anti-Nup62 MAb/ml and a RIPA-washed protein A-agarose (Sigma) suspension (20% of the supernatant volume). The control sample received no antibodies. The samples were incubated for 1 to 3 h with rocking, and the agarose-Nup62 complexes were washed twice with RIPA buffer and heated in sample buffer for Western blotting or treated with λ-phosphatase as described below.
Phosphatase treatment.
Two phosphatases, calf intestine alkaline phosphatase (Promega) and λ-protein phosphatase (λ-PPase; New England Biolabs), were used to treat the phosphorylated form of Nup62. In the former case, the mock- and mengovirus-infected cell lysates were dialyzed against alkaline phosphatase buffer (50 mM Tris-HCl [pH 8.9], 5 mM MgCl2, 0.1 mM ZnCl2, 1 mM DTT), and the samples were incubated in the presence of 100 U of the enzyme/ml in the same buffer at 30°C for 30 min. To treat Nup62 with λ-PPase, the immunoprecipitated samples of Nup62 were washed twice with λ-PPase buffer (100 mM NaCl, 2 mM MnCl2, 2 mM dithiothreitol, 0.1 mM EGTA, 0.01% Brij 35, 50 mM Tris-HCl [pH 7.5]; New England Biolabs) and exposed to 1,000 U of the enzyme/ml for 30 min at 30°C. The resulting samples were subjected to SDS-6% PAGE, followed by Western blotting with the use of anti-Nup62 antibodies.
Western blotting.
The cells were lysed by addition of the sample buffer (2% SDS, 50 mM 2-mercaptoethanol, 50 mM Tris-HCl [pH 6.8]), sonicated in ice, and heated at 95°C for 10 min. After separation by electrophoresis in a polyacrylamide gel, the proteins were electrotransferred in the transfer buffer (190 mM glycine, 2.5 mM Tris [pH 8.3]) onto Hybond-P membrane (Amersham) and probed with appropriate antibodies in TBS (114 mM NaCl, 17 mM Tris-HCl [pH 8.0]) containing 5% nonfat milk. The membranes were developed with an ECL+ kit (Amersham) according to the manufacturer's recommendations.
2D electrophoresis.
The cells collected by EDTA treatment or by shaking the culture vessel were pelleted by centrifugation, washed with PBS, resuspended in two-dimensional (2D) electrophoresis sample buffer (9 M urea, 2 mM N-lauroylsarcosine, 5% Bio-Lyte 3/10 [Bio-Rad], 65 mM DTT), lysed by freezing-thawing, and sonicated. In the case of immunoprecipitated Nup62 samples, the preparations were washed twice with 2 mM Tris-HCl (pH 7.5) and then resuspended in another isoelectrofocusing buffer (9 M urea, 5% β-mercaptoethanol, 2% Triton X-100, 2% Bio-Lyte 3/10), releasing the protein from protein A-agarose. The samples were applied to isoelectrofocusing tubes containing the first-dimension gel (9 M urea, 4.5% acrylamide-bisacrylamide [28:2], 4 to 5% Bio-Lyte 3/10 and 0.7 to 1% Bio-Lyte 4/6 [Bio-Rad], 0.6 mM N-lauroylsarcosine, 5% Nonidet P-40, 0.1% N,N,N′,N′-tetramethyl-ethylenediamine, 0.02% ammonium persulfate) and exposed to the electric field (200 V for 2 h, 500 V for 2 h, and finally 800 V for 16 h in 14-cm-long tubes) with 20 mM NaOH as the cathode and 10 mM H3PO4 as the anode running. The gels were carefully pressed out, balanced in transfer buffer (3% SDS, 20 μM bromophenol blue, 70 mM Tris-HCl [pH 8.8]), subjected to electrophoresis on SDS-containing 6% polyacrylamide, and analyzed by Western blotting.
Electron microscopy.
The subconfluent monolayers of HeLa-3E cells growing on coverlsips were fixed with 2.5% glutaraldehyde (Sigma) in 0.1 M phosphate buffer (pH 7.4), postfixed with 1% OsO4 (Sigma), dehydrated in 70% ethanol containing 2% uranyl acetate and acetone to permeabilize cells, and embedded in Epon 812 (Fluka). To obtain preparations of permeabilized cells, the coverslips were treated with 1% Triton X-100 in “condensing” buffer (50 mM triethanolamine, 3 mM MgCl2) for 1 min at 4°C and fixed with 1% glutaraldehyde in the same buffer (23). Ultrathin sections were prepared with an LKB Ultratome-III, stained with lead citrate, and examined with an HU-12 electron microscope (Hitachi).
Image editing.
The color images obtained by microscopy were transformed into grayscale mode by using Adobe Photoshop 7.0. Developed films (for Western and autoradiography assays) were scanned and processed with the same program. When appropriate (e.g., see the upper-row panels of Fig. 5,as well as Fig. 6A and 8B), the brightness and contrast of the grayscale pictures were equally optimized (i.e., to make visible details presented in the color images) by using the “Levels” tool of Adobe Photoshop 7.0.
FIG. 5.

Mengovirus-induced permeabilization of the nuclear envelope is sensitive to protein kinase inhibitors. Permeabilized uninfected HeLa-3E cells were incubated with the lysates from mengovirus-infected (4 h p.i.) and mock-infected HeLa cells for 1 h at 37°C in the absence or presence of the indicated concentrations of a broad-spectrum protein kinase inhibitor staurosporin and two inhibitors of cycle-dependent kinases, roscovitine and olomoucine. An inactive analog of the latter drug, iso-olomoucine, was used as a control.
FIG. 6.

Individually expressed leader protein affects nucleocytoplasmic traffic. HeLa-3E cells were transfected with the plasmids encoding leader proteins fused to RFP. (A) Efflux of the nuclear protein marker (3×EGFP-NLS) into the cytoplasm is visible in a proportion of cells transfected with the plasmid encoding wild-type (wt) L protein but not with the one encoding inactive C19A/C22A (Zn) mutant. Cells were stained with Hoechst 33342 DNA stain and captured 24 h posttransfection. (B) Percentage of the cells transfected with wild-type (wt) and mutant L-expressing plasmids, demonstrating 3×EGFP-NLS fluorescence in the cytoplasm. The transfected cells were incubated in the presence of 100 μM zVAD(OMe).fmk to prevent apoptosis, which could complicate interpretation of the results.
FIG. 8.
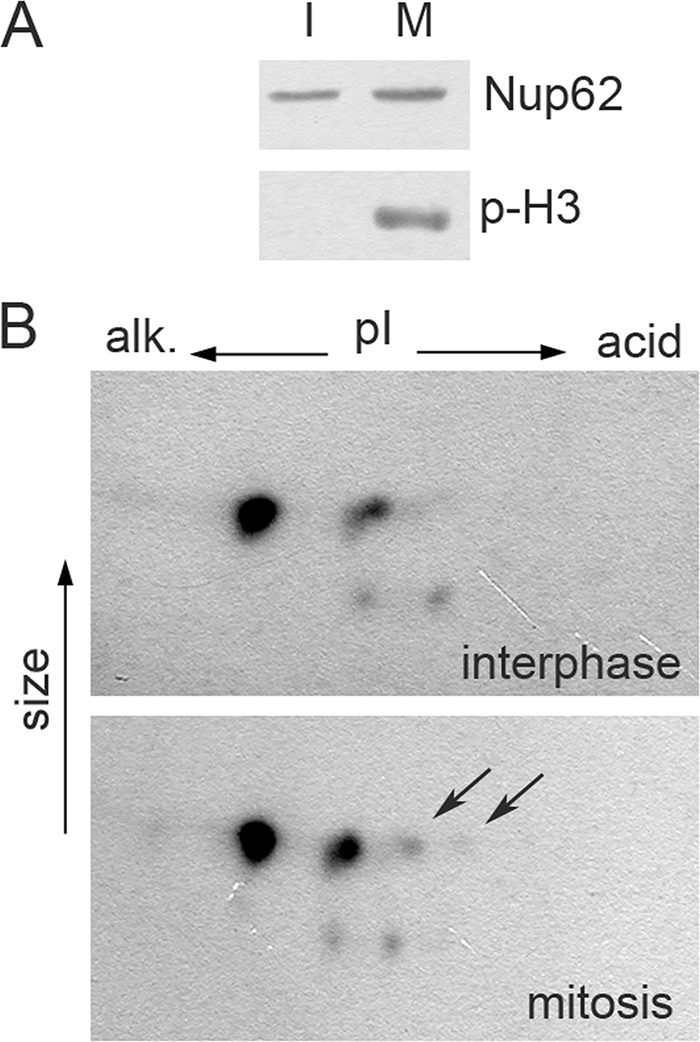
Nup62 is phosphorylated in mitotic HeLa cells. (A) Control for synchronization. Samples of interphase (I) and mitotic (M) cells (see Materials and Methods) were analyzed by Western blotting for Nup62 and Ser-10 phosphorylated H3 histone (p-H3). (B) The same preparations were analyzed by 2D electrophoresis, followed by Western blotting for Nup62. Acidic spots are visible in the preparations of mitotic cells, but those of interphase cells are marked by arrows.
RESULTS
Nup62 and Nup153 remained associated with the NPC in cardiovirus-infected cells.
The increase in nuclear envelope permeability in cardiovirus-infected cells was ascribed to changes in the NPC, the sole gates to and out of the nucleus (25). However, in contrast to infections with poliovirus, coxsackievirus B3, and rhinovirus (17, 18, 25), no degradation of Nup62 and Nup153 could be observed in cardiovirus-infected cells (25).
To gain more detailed information about the fate of these Nups, their localization in mengovirus-infected HeLa-3E cells (i.e., expressing a high-molecular-mass NLS-containing fluorescent marker protein) was investigated by means of immunofluorescence. Staining of these cells with antibodies against Nup62, Nup153, and FG-repeat motifs revealed that they mainly remained associated with the nuclear envelope even at late (6 h) stages of the cardiovirus infection (Fig. 1B), when cytoplasmic exit of the nuclear marker protein did occur in the majority of the cells (Fig. 1A). In the poliovirus-infected cells, in contrast, the antigens recognized by the anti-Nup153 and anti-FG antibodies were to a significant extent detached from the nuclear rim already at 4 h p.i. (Fig. 1B), most probably as a result of their proteolytic cleavage (17). Positive signals (although apparently somewhat weaker) in the nuclear rim region were observed, however, in poliovirus-infected cells stained with anti-Nup62 antibodies, suggesting that an antibody-recognized cleavage product of this Nup remained, at least partly, associated with the nuclear envelope.
FIG. 1.

Localization of Nups in cells infected with mengovirus and poliovirus. HeLa-3E cells were infected with the viruses with an input MOI of ∼100 PFU/cell and fixed at indicated times. (A) Exit of the nuclear marker protein into the cytoplasm. (B) Detection of FG-repeat-containing proteins, Nup153, and Nup62 by immunostaining. Images in a given row were made with equal expositions.
In sum, no significant differences in the distribution of Nup62 and Nup153 could be revealed between uninfected and cardiovirus-infected cells at the time when the permeability of the nuclear envelope had already enhanced, whereas poliovirus infection was accompanied with a significant loss of these Nups from the nuclear envelope.
Electron microscopic comparison of NPC in mengovirus- and poliovirus-infected cells.
Previously, we demonstrated preservation of the integrity of Nup62 and Nup153 upon cardiovirus infections (25). As shown above, these Nups were retained in the nuclear envelope under these conditions. These facts appeared to be in some conflict with our previous electron microscopic observations, demonstrating that, while the pore complexes did not seem to be significantly destroyed, their central channels (presumably containing Nup62) appeared to be more electron transparent in cardiovirus-infected cells than in uninfected ones (25).
To resolve this discrepancy and to characterize further the events occurring in the nuclear envelope of cardiovirus-infected cells, we performed additional electron microscopic studies. A comparison of the NPC in cardiovirus- and poliovirus-infected cells by using direct in situ fixation, i.e., the approach similar to that exploited in previous studies (6, 25), confirmed the already described features of the nuclear envelope in cells infected with both viruses: disruption of the bar-like structures in many central channels seen in cross-sections (Fig. 2A). However, in tangential sections, these channels looked somewhat different in cells infected with mengovirus and poliovirus. While in the former case, the channels looked nearly empty (as reported previously [6]), irregular granules, possibly reflecting products of proteolytic destruction of Nups, could be discerned within some channels in the poliovirus-infected cells (Fig. 2A).
FIG. 2.

Electron microscopy of nuclear pores in infected cells. HeLa-3E cells were infected with mengovirus and poliovirus and fixed at 4 h p.i. The upper rows in panels A and B correspond to cross-sectioned nuclear envelopes, the middle rows are tangentially cut nuclear envelopes in plane, and the lower rows represent the twofold-enlarged examples of NPC from the appropriate middle rows. Scales bars correspond to 200 nm. The nuclear sides in cross-sections are marked as “nuc,” and the cytoplasmic ones are marked as “cyt.” Regions of NPC are marked with asterisks. (A) In situ fixation. The bar-like structures visible in NPC cross-sections of healthy cells are absent from a number of pores after poliovirus and mengovirus infections. In tangential section, control NPC display an electron-dense granule in the central channel, which is totally or partially lost in infected cells. (B) Fixation after dissolution of membranes with Triton X-100. NPC attached to the lamina are visible in cross-sections of the control envelope, and these complexes appeared to be only slightly disorganized in the mengovirus-infected sample. On the other hand, they are significantly destroyed by poliovirus infection. In tangential sections, the pores in control and mengovirus-infected cells looked very similar, whereas the NPC from poliovirus-infected cells displayed a much higher electron transparency and are less structured.
Another approach consisted of electron microscopic investigation of virus-infected cells after destruction of their membranes with Triton X-100. In this case, the integrity of the NPC should not be stabilized by their anchoring to the nuclear membrane and was expected to depend only on internucleoporin interactions, as well as on the binding of Nups to nuclear lamina and chromatin (54). It should be noted that a relatively high concentration of Mg2+ (3 mM) in the permeabilization buffer was used in these experiments to preserve the nuclei (47). The results of such an approach shown in Fig. 2B were strikingly different from those depicted in Fig. 2A. In poliovirus-infected cells, the NPC appeared to be significantly destroyed in cross-sections and were highly translucent in tangential sections, pointing out to the weakening of inter-NPC and/or NPC-lamina interactions. On the other hand, the pores in cardiovirus-infected cells looked very similar to those in control preparations, displaying some disorganization (although much less prominent than in poliovirus-infected cells) visible only in cross-sections.
Thus, the results of electron microscopy strongly suggested that alterations of the NPC structure caused by cardiovirus and poliovirus infections were clearly different. Although interpretation of this difference was not straightforward, the observations were consistent with the assumption that structural changes in the nuclear pores in cardiovirus-infected cells were not accompanied by a significant impairment of inter-NPC and NPC-lamina bonds.
Nup62 is phosphorylated in mengovirus infection.
Taking into account that alterations of the nucleocytoplasmic traffic upon cardiovirus infection could be prevented by treatment with a broad-spectrum protein kinase inhibitor staurosporin (25), we decided to test the hypothesis that these alterations were caused by changes in the phosphorylation status of Nups. As a first step, we made use of WGA-tagged beads known to adsorb several Nups, in particular Nup62, as the only major class of phosphoproteins that possesses WGA-binding glycosyl residues (9, 29, 30, 34). Cardiovirus-infected cells were labeled with inorganic [32P]phosphate, and electrophoretic analysis of WGA-binding membrane fraction from these revealed a glycoprotein of ∼70 kDa, presumably Nup62, with a significantly increased level of phosphorylation compared to control cells (Fig. 3A, arrow). Enhanced phosphorylation of Nup62 upon cardiovirus infection was confirmed by immunoprecipitation (Fig. 3B). A Western blot assay revealed that the Nup62 band from unlabeled virus-infected cells was significantly more heterogeneous compared to that from uninfected cells and contained some material with a lower electrophoretic mobility (Fig. 3C, arrows). This material was clearly detectable by 4 h p.i., and its amount increased during the next 2 h of infection. Such a material was absent from the sample from poliovirus-infected cells, which instead contained, in line with the published data (17, 37), a low-molecular-mass component, most likely a product of proteolytic degradation of Nup62 (arrowheads). The low-mobility form of Nup62 disappeared nearly completely from the cardiovirus-infected sample after the treatments with alkaline phosphatase (Fig. 3D, upper panel) or λ-PPase (Fig. 3D, lower panel), suggesting that the electrophoretic mobility-changing modification could be phosphorylation. Finally, the existence of differently phosphorylated forms of Nup62 in cardiovirus-infected cells was demonstrated by 2D electrophoresis, followed by Western blot analysis. As shown in Fig. 3E and F, this Nup existed in infected cells in multiple forms (varying in different experiments from four to eight forms), whereas only two such forms, a major and a minor form, could usually be discerned in uninfected samples. The additional spots disappeared after treatment with λ-PPase (Fig. 3F). It may be noted that a more acidic minor species of Nup62 in the mock-infected samples (and a similar form in the virus-infected samples) exhibited partial sensitivity to the λ-PPase treatment (Fig. 3F), suggesting that it might also correspond to a phosphorylated form of Nup62, but its actual nature was not further investigated here. We conclude that cardiovirus infection was accompanied by hyperphosphorylation of Nup62.
FIG. 3.

Nup62 is hyperphosphorylated in mengovirus-infected cells. (A) [32P]phosphate incorporation in WGA-binding proteins upon cardiovirus infection. HeLa-3E cells were labeled with [32P]orthophosphate during the first 2 h of infection, lysed, and subfractionated into cytosolic (c) and membrane (m) fractions as described in Materials and Methods. Nups were precipitated with WGA-beads and analyzed by PAGE and autoradiography. The first lane represents the control probe incubated for 2 h in ice. The arrow points out to a ∼70-kDa band appearing in mengovirus-infected cells. (B) Immunoprecipitation of 32P-labeled Nup62 from mengovirus-infected cells. Infected and mock-infected HeLa-3E cells were labeled with inorganic [32P]phosphate during the time periods indicated. The cold probe was exposed to the label for 6 h in ice. The cells were lysed and Nup62 was immunoprecipitated and subjected to electrophoresis as described in Materials and Methods (upper row). The amounts of Nup62 in the samples were controlled by Western blotting (lower row). (C) Modifications of Nup62 by different picornaviruses. HeLa-3E cells were infected with mengovirus or poliovirus for indicated times, and their extracts were investigated by PAGE, followed by Western blotting with anti-Nup62 antibodies. The lower mobility component of Nup62 from mengovirus-infected cells are marked with arrows; higher-mobility bands from poliovirus-infected cells corresponding to degradation products are marked with triangles. (D) The low-mobility forms of Nup62 disappeared from mengovirus-infected cell after alkaline phosphatase (upper row) and λ-PPase (lower row) treatments. Lysates from mock- and mengovirus-infected cells made at 4 h p.i. in the case of the former enzyme and immunoprecipitated preparations of Nup62 from such lysates in the case of the latter were treated with the phosphatases for 30 min at 30°C and analyzed by Western blotting. (E) Detection of hyperphosphorylated forms of Nup62 in mengovirus-infected cells. Cell lysates were prepared as in panel C and were subjected to 2D electrophoresis followed by Western blotting. (F) The additional acidic spots of Nup62 disappeared after treatment with λ-PPase. Samples 1 to 4 from the lower row of panel D were subjected to 2D electrophoresis, followed by Western blotting.
Mutations of mengovirus L protein similarly affect permeabilization of the nuclear envelope and hyperphosphorylation of Nup62.
Mengovirus L mutants with either a disrupted Zn-finger motif (C19A/C22A) or a disrupted phosphorylation site (T47A) were previously shown to be unable to trigger nuclear efflux of an NLS containing marker protein or did so with a considerable delay, respectively (25). The capacity of these mutants to elicit hyperphosphorylation of Nup62, as judged by the appearance of material with a lower electrophoretic mobility, was changed very similarly: the former mutant did not demonstrate this capacity, whereas the latter enhanced Nup62 phosphorylation to a significantly lesser extent and only at a late step of infection (i.e., 6 h) (Fig. 4A). On the other hand, infection with the L mutant harboring a substitution mimicking L phosphorylation (T47E) and known to retain the full capacity to permeabilize the nuclear envelope (25), resulted in efficient and fast hyperphosphorylation of Nup62. It could be argued that the deficiency of the Zn finger and T47A mutants to induce Nup62 phosphorylation could be explained just by their less efficient multiplication. To eliminate this possibility, an input MOI of ∼100 PFU/cell was used for these mutants compared to ∼20 PFU/cell for the wild-type and T47E viruses. Under such conditions, the yield of the former mutants at 6 h p.i. was significantly higher than that of the latter viruses at 4 h p.i (Fig. 4B), a time when the enhanced Nup62 phosphorylation was already clearly visible in the cells infected with wild-type and T47E viruses. These data suggested that a less efficient growth of the two mutants was hardly directly responsible for their failure to trigger hyperphosphorylation of Nup62. Thus, mengovirus mutants impaired in their ability to disrupt the nucleocytoplasmic barrier were also inefficient in enhancing phosphorylation of Nup62.
FIG. 4.

Effect of L protein mutations on Nup62 phosphorylation. (A) HeLa-3E cells were infected with wild-type (wt) and T47E mengoviruses at an MOI of ∼20 PFU/cell and with the C19A/C22A (Zn) and T47A mutants at an MOI of ∼100 PFU/cell to compensate for their relative replicative deficiencies. After incubation, the cells were observed under epifluorescence microscope (results not shown) and harvested for Western blotting and titration. (B) Growth curves of mengovirus mutants at the MOIs indicated above.
Protein kinase inhibitors suppress mengovirus-induced leakiness of the nuclear envelope.
Previously, we have reported that protein kinase inhibitor staurosporin prevented leakiness of the nuclear envelope of digitonin-permeabilized uninfected HeLa-3E cells caused by extracts from mengovirus-infected cells, while a tyrosine kinase inhibitor genistein and casein kinase inhibitor 5,6-dichlorobenzimidazole riboside failed to do so (25). By using the same assay, we now showed that exit from the nucleus of an NLS-containing protein marker was also prevented by two drugs known to inhibit cyclin-dependent protein kinases: roscovitine (100 μM) and olomoucine (1 mM) (Fig. 5). A derivative of the latter, iso-olomoucine, which does not exhibit protein kinase inhibitor activity (52), was also inactive in preventing permeabilization of the nuclear envelope.
When tested in vivo in the cardiovirus-infected cells, roscovitine and olomoucine at the same concentrations failed to prevent the efflux of nuclear marker (not shown), apparently because of the inability of the drugs to accumulate within the cells at sufficiently high concentrations.
Individually expressed mengovirus leader protein induces leakiness of the nuclear envelope.
Although the results reported here and previously (25) strongly suggested that increased permeability of NPC in cardiovirus-infected cells was related to activity of the viral L protein, we decided to test this notion directly. The wild-type and C19A/C22A leader proteins were subcloned into an expressing vector with RFP fused to their C termini. In the mutant-transfected HeLa-3E cells that displayed some red fluorescence, no redistribution of the nuclear protein marker into the cytoplasm could be observed (Fig. 6). On the other hand, a significant proportion of the wild-type L-transfected cells exhibited enhanced permeability of the nuclear envelope but demonstrated only a very faint, if any, signal corresponding to RFP, possibly due to the instability and/or toxicity of the peptide. The difference in the ability of wild-type and mutant L to enhance permeability of the nuclear envelope is fully retained in the presence of a broad-spectrum caspase inhibitor zVAD(OMe).fmk (100 μM) (Fig. 6B), suggesting that this phenomenon is not coupled to apoptosis. These results demonstrated that the cardiovirus L protein alone was sufficient to trigger the cascade of host reactions, leading to the increased permeability of the nuclear envelope.
Comparison of nuclear envelope leakiness elicited by cardiovirus infection and occurring at early prophase.
Since cardioviruses do not encode any protein kinases, phosphorylation of Nups observed during the viral infection should be accomplished by some host enzymatic system. It is known that disassembly of the nuclear pores and nuclear membrane takes place during mitosis. Phosphorylation of Nups is known to be involved in these alterations (13, 30, 34), allowing us to hypothesize that cardioviruses may hijack certain host mechanisms to enhance the leakiness of NPC in the infected cells. However, as shown above, mengovirus infection was not accompanied by a complete disassembly of the NPC. Therefore, we decided to test whether there was a step during early prophase of uninfected HeLa cells (i.e., before the complete dissolution of the nuclear envelope), mimicking the pattern observed during mengovirus infection, that is, Nup phosphorylation and retention in the nuclear envelope, accompanied by enhanced permeabilization of NPC.
To this end, HeLa-3E cells were stained for Nups, and the mitotic cycle stage of individual cells was determined by the pattern of chromatin condensation with the use of Hoechst 33342 stain as described previously (23). As shown in Fig. 7, FG-motif-containing Nups, i.e., Nup62 and Nup153, were largely retained in the nuclear rim in a proportion of the prophase HeLa-3E cells exhibiting exit of the nuclear marker into the cytoplasm. On the other hand, cells entering the subsequent steps of mitosis, prometaphase and metaphase, demonstrated partial and complete redistribution of the Nups throughout the whole cytoplasm.
FIG. 7.

Localization of Nups and efflux of the nuclear protein marker into cytoplasm in prophase and metaphase cells. Uninfected HeLa-3E cells at different stages of mitosis, as judged by DNA staining, were analyzed with respect to localization of Nups and distribution of the fluorescent marker. Prophase cells displaying 3×EGFP-NLS in cytoplasm possessed Nups mainly associated with the nuclear envelope.
Since the proportion of prophase cells in a nonsynchronized population was relatively small, synchronization by means of double thymidine block was undertaken. At 12.5 h after removal of the drug, when the preparations were markedly enriched with mitotic cells, the proportion of cells retaining Nups in the nuclear envelope and displaying nuclear marker in the cytoplasm was markedly increased and corresponded to 34% of all prophase cells (data not shown). Thus, an increase in the permeability of the nuclear envelope might occur in prophase HeLa cells without a significant structural rearrangement of NPC, just as appeared to be the case in mengovirus-infected cells.
The status of phosphorylation of Nup62 in mitotic HeLa-E3 cells was also investigated. Monolayers of HeLa-3E cells were synchronized with a 16-h thymidine block, and mitotic enriched fractions were harvested 11 to 12 h thereafter by gentle shaking and subsequent centrifugation. The collected pools consisted predominantly of mitotic and early postmitotic cells, as judged by DNA staining followed by fluorescence and phase-contrast microscopy. Western blot assay with an antibody recognizing the phosphorylated Ser10 of histone H3, a specific mitotic marker (42), confirmed preponderance of mitotic cells in this fraction (Fig. 8A). The overwhelming majority of these cells demonstrated efflux of 3×EGFP-NLS into the cytoplasm (data not shown). When the cells of this fraction were subjected to 2D electrophoresis followed by Western blotting, additional spots of phosphorylated Nup62 could reproducibly be revealed compared to the interphase cells (Fig. 8B), although the level of phosphorylation of this protein (the number of distinct spots) was not as strong as in the mengovirus-infected cells (cf. Fig. 8B and 3D).
Thus, permeabilization of the nuclear envelope in prophase HeLa cells appeared to be accompanied with a certain level of phosphorylation of Nup62 prior to complete dissolution of the NPC. This pattern resembled that observed upon cardiovirus infection, although it remained an open question as to whether both conditions had the same, or similar, underlying mechanisms. This point will be discussed below in greater detail.
DISCUSSION
Integrity and retention of Nup62 and Nup153 in the nuclear pores.
It has been already demonstrated that cardiovirus-elicited permeabilization of the nuclear envelope (10, 25) differed from a similar phenomenon observed in cells infected with enteroviruses and rhinoviruses with respect to the key viral proteins involved: 2A protease in the latter case and nonenzymatic leader (L) protein in the former (5, 6, 17, 18, 37). In the present study, we attempted to identify L-triggered cellular changes accompanying the nucleocytoplasmic transport disorder. We focused on the fate and status of Nups, in particular Nup62 and Nup153, because, on the one hand, the former is known to be a component of the complex intimately involved in the control of selective permeability of the nuclear pore (33, 45) and, on the other, both of these Nups are targets of 2Apro during enterovirus and rhinovirus infections (17, 18).
An important finding was the apparent retention of Nup62 and Nup153 in the nuclear envelope, as revealed by immunofluorescence staining of the mengovirus-infected cells that exhibited relocation of a nuclear marker protein into the cytoplasm (Fig. 1). Because this observation appeared to somewhat contradict the results of our previous electron microscopy of the virus-infected cells showing relative translucence of the central channel in tangential sections (25), additional electron microscopic studies were performed, which appeared to resolve the contradiction, at least partially. When a different methodological approach was used, the central channel of mengovirus-infected cells looked quite similar to that of uninfected cells. Although the exact reason for the discordant results obtained by the two methods is yet to be determined, we believe that the lack of gross alterations in the central channel revealed by one of them may be interpreted as evidence for the preservation of the general architecture of the nuclear pore in mengovirus-infected cells. Taking into account the results of detection of Nup by immunofluorescence, we suggest that changes in the electron transparency of the central channel in mengovirus-infected cells observed upon in situ fixation were due largely to alterations in the conformation of the Nup complexes or individual Nups rather than to their loss. As far as the irregular granules visible in the central channels in poliovirus-infected cells are concerned, we hypothesize that they were caused, at least in part, by the retention, in nuclear pores, of products of proteolytic degradation of some Nups. The absence of such granules in the Triton X-100-treated sample may be explained by the loss of protein-lipid interactions possibly involved in the retention of these degradation products.
Hyperphosphorylation of Nup62 in mengovirus-infected cells as a possible mechanism of cardiovirus-induced permeabilization of the nuclear pores.
The results discussed in the preceding section suggested that the Nup62 complex, or at least Nup62 itself, were retained in the NPC but underwent some modification. This modification most likely involved hyperphosphorylation of Nup62 and possibly of other Nups. The evidence for Nup62 hyperphosphorylation was obtained by detection of Nup62 forms possessing different negative charges and by direct demonstration of enhanced incorporation of phosphate. We propose that this modification might be a key factor in the permeabilization of the nuclear pores. This proposal is supported by the capacity of protein kinase inhibitors to suppress permeabilization of the nuclear envelope caused by the addition of extracts from cardiovirus-infected cells to digitonin-treated uninfected cells (25; the present study). A correlation between the impaired capacities of L-deficient mutants of mengovirus to enhance permeabilization of the nuclear pores, on the one hand, and to elicit hyperphosphorylation of Nup62, on the other, could also support this notion. Admittedly, the above mutants exhibited a somewhat decreased level of reproduction in HeLa cells compared to their wild-type parent, but this defect was hardly directly responsible for their failure to trigger Nup62 phosphorylation, as suggested by the data presented in Fig. 4B. Furthermore, we made an attempt to investigate whether a correlation between the efflux of nuclear proteins into cytoplasm existed also in BHK-21 cells, where reproduction of mengovirus was shown to be less dependent on the functional integrity of L protein (25, 56). This attempt met only with partial success. Although wild-type mengovirus did induce permeabilization of the nuclear envelope in these cells, it did so markedly less efficiently than in HeLa cells. Accordingly, the phosphorylation of Nup62 in the mengovirus-infected BHK-21 cells was barely, if at all, detectable (data not shown). The very low level of Nup62 phosphorylation elicited by wild-type virus did not allow us to reliably ascertain the effect of mutations of leader protein on this process.
An important question concerns the nature of protein kinase(s) executing Nup phosphorylation during cardiovirus infection. The information on this point is thus far inadequate to make any definite conclusions. We know only that three protein kinase inhibitors could suppress the virus-elicited permeabilization of the nuclear envelope. We cannot say, however, that these inhibitors, or even at least one of them, suppressed Nup62 phosphorylation directly. The signal transduction pathway may well involve more than one phosphorylation events. Also, it should be admitted that the concentrations at which roscovitine and olomoucine did suppress efflux of the nuclear marker protein caused by the virus-infected cell extract were rather high.
It has been reported that Nup62 phosphorylation took place in neurons stimulated by angiotensin II, that this reaction is accomplished by MAP kinase, and that it resulted in enhanced protein transport through nuclear pores (28). On the other hand, Nup62 was demonstrated to be phosphorylated in vitro by protein kinase A and glycogen synthase kinase 3a (34). It is unclear, however, how these effects are related to the observations reported here.
Furthermore, the possibility that changes in the phosphorylation status of Nup62 depends on inactivation of a protein phosphatase(s) rather than on activation of a protein kinase(s) should not be neglected. Such enzymes are known to be important participants of diverse physiological pathways, including those involved in disassembly and assembly of the nuclear envelope (8, 36).
Variety of effects of picornaviruses on nucleocytoplasmic traffic.
Thus, representatives of several picornavirus genera induce dramatic alterations in nucleocytoplasmic traffic, although fundamentally different mechanisms may be responsible for these alterations. A relatively widespread occurrence of this phenomenon suggests that it may confer some selective advantage to the viruses. One group of likely favorable (for the virus) consequences of enhanced permeability of the nuclear envelope may consist in facilitating utilization of nuclear proteins for translation and replication of cytoplasmic viruses. Numerous examples of such host nuclear “factors” involved in viral reproduction are known (7, 20, 31, 32, 39, 49, 53). Another group of advantages viruses may get from the entry into the nucleus of viral toxic products (e.g., proteases) able to interfere at transcriptional and posttranscriptional levels with the host innate immunity (3, 55). The exact contribution of the permeabilization of the nuclear pores to efficient viral reproduction is yet to be elucidated. It would not be surprising, if this contribution varies depending on the peculiarities of virus-cell systems, as well as on physiological and environmental factors.
In this regard it should be noted that not all of picornaviruses appeared to be endowed with this specific anti-host armament. For example, cells infected with the Zakarpatsky strain of porcine teschovirus (kindly donated by V. V. Kurinnov, Russian National Research Institute for Veterinary Virology and Microbiology, Pokrov) or parechovirus Ljungan virus (derived from an infectious cDNA clone kindly donated by M. Lindberg, University of Kalmar, Kalmar, Sweden) did not exhibit any marked alterations in the nucleocytoplasmic transport, at least as judged by the efflux of a nuclear marker protein (unpublished results). It is yet an open question whether inactivity of teschoviruses in this respect is causally related to the fact that IRES of these viruses was shown to require a minimal number of protein cofactors for its function (40).
The facts that the picornaviruses that are able to permeabilize the nuclear envelope are achieving this “goal” by exploiting different molecular mechanisms and that there exist picornaviruses devoid of this capacity deserves a special comment. In our view, it strongly suggests that the relevant functions (and consequently cardiovirus L and entero/rhinovirus 2Apro) were acquired independently and perhaps relatively recently in the course of picornaviral evolution, providing an illuminating example of functional but not structural convergence.
Apparent similarity of permeabilization of nuclear envelope during cardiovirus infection and early prophase.
A novel pattern of NPC rearrangement has been discovered in the present study: while retaining Nups, the nuclear envelope becomes permeable and may exist in this state for several hours. The key trigger of this rearrangement appears to be the cardiovirus leader protein. Since this protein is devoid of any known enzymatic activity, it is expected to work by exploiting (“hijacking”) some cellular pathway. A likely candidate for such a pathway is the one that leads to the NPC permeabilization prior to its disassembly during mitosis (24). Taking into account this fact, we compared events occurring in mengovirus-infected and mitotic cells. It turned out that under both of these conditions, efflux of the nuclear marker protein into the cytoplasm occurred prior to massive loss of Nups from the NPC. Also, we could detect enhanced phosphorylation of Nup62 during the prophase, although its level was not as high as in the case of cardiovirus infection. This lower level could either reflect real differences in the efficiency of the Nup phosphorylation under these two conditions or merely indicate that mitotic phosphorylation of Nup62 is confined to a specific relatively short-duration step of prophase and that cells at this step are underrepresented in our sample of mitotic cells.
Importantly, our observations were in line with the published data on Nup62 phosphorylation by mitotic oocytes of Xenopus laevis (34). Moreover, the protein kinase inhibitor roscovitine not only prevented permeabilization of the nuclear envelope by extracts of virus-infected cells (see above) but also suppressed mitotic disassembly of the NPC in Drosophila embryos (36). However, as already mentioned, relatively high concentrations of the inhibitors were required to exert their effects in our system.
Although we focused on the phosphorylation of Nup62 in the present study, other Nups may also undergo a similar modification. It should be kept in mind that a growing list of Nups harbor sites for mitotic kinases, including Nup96, Nup107, Nup116, Nup133, Nup160, and Nup214 (16, 30).
A distinctive feature of permeabilization of the nuclear envelope without significant loss of Nups in mengovirus-infected cells was its duration: the relevant situation might be maintained for several hours. In contrast, dissociation of Nups from the NPC during mitosis is a very rapid process taking 8 to 10 min (12). It may be speculated that, if mengovirus infection switches on the physiological program similar to that operating during the mitotic disassembly on nuclear membranes, implementation of this program is somehow interrupted or suppressed at an early step.
We believe that our data are consistent with the hypothesis assuming involvement of mitotic machinery in the virus-induced NPC rearrangements, but this hypothesis is awaiting more direct confirmation. One could also not exclude that Nup phosphorylation in cardiovirus-infected cells might represent another, still unknown, process of nucleocytoplasmic transport regulation in uninfected cells.
Acknowledgments
We thank A. P. Gmyl, E. V. Khitrina, L. I. Kovalev, G. G. Karganova, and T. M. Dmitrieva for their expert assistance.
This study was supported by grants from the NWO-RFBR (to V.I.A.), the Russian Foundation for Basic Research (to P.V.L. and V.I.A.), the Scientific School Support Program (to V. I. A.), and the Russian President's Grant for Young Scientists (to P.V.L.).
Footnotes
Published ahead of print on 14 January 2009.
REFERENCES
- 1.Adam, S. A., S. R. Marr, and L. Gerace. 1990. Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. J. Cell Biol. 111807-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agol, V. I. 2002. Picornavirus genome: an overview, p. 127-148. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 3.Almstead, L. L., and P. Sarnow. 2007. Inhibition of U snRNP assembly by a virus-encoded proteinase. Genes Dev. 211086-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck, M., F. Förster, M. Ecke, J. M. Plitzko, F. Melchior, G. Gerisch, W. Baumeister, and O. Medalia. 2004. Nuclear pore complex structure and dynamics revealed by cryoelectron tomography. Science 3061387-1390. [DOI] [PubMed] [Google Scholar]
- 5.Belov, G. A., A. G. Evstafieva, O. V. Mikitas, A. B. Vartapetian, and V. I. Agol. 2000. Early alteration of nucleocytoplasmic traffic induced by some RNA viruses. Virology 275244-248. [DOI] [PubMed] [Google Scholar]
- 6.Belov, G. A., P. V. Lidsky, O. V. Mikitas, D. Egger, K. A. Lukyanov, K. Bienz, and V. I. Agol. 2004. Bidirectional increase in permeability of nuclear envelope upon poliovirus infection and accompanying alterations of nuclear pores. J. Virol. 7810166-10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunner, J. E., J. H. Nguyen, H. H. Roehl, T. V. Ho, K. M. Swiderek, and B. L. Semler. 2005. Functional interaction of heterogeneous nuclear ribonucleoprotein C with poliovirus RNA synthesis initiation complexes. J. Virol. 793254-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceulemans, H., and M. Bollen. 2004. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol. Rev. 841-39. [DOI] [PubMed] [Google Scholar]
- 9.Davis, L. I., and G. Blobel. 1987. Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc. Natl. Acad. Sci. USA 847552-7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delhaye, S., V. van Pesch, and T. Michiels. 2004. The leader protein of Theiler's virus interferes with nucleocytoplasmic trafficking of cellular proteins. J. Virol. 784357-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denning, D. P., S. S. Patel, V. Uversky, A. L. Fink, and M. Rexach. 2003. Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proc. Natl. Acad. Sci. USA 1002450-2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dultz, E., E. Zanin, C. Wurzenberger, M. Braun, G. Rabut, L. Sironi, and J. Ellenberg. 2008. Systematic kinetic analysis of mitotic dis- and reassembly of the nuclear pore in living cells J. Cell Biol. 180857-865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Favreau, C., H. J. Worman, R. W. Wozniak, T. Frappier, and J. C. Courvalin. 1996. Cell cycle-dependent phosphorylation of nucleoporins and nuclear pore membrane protein Gp210. Biochemistry 358035-8044. [DOI] [PubMed] [Google Scholar]
- 14.Finlay, D. R., and D. J. Forbes. 1990. Reconstitution of biochemically altered nuclear pores: transport can be eliminated and restored. Cell 6017-29. [DOI] [PubMed] [Google Scholar]
- 15.Fried, H., and U. Kutay. 2003. Nucleocytoplasmic transport: taking an inventory. Cell. Mol. Life Sci. 601659-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glavy, J. S., A. N. Krutchinsky, I. M. Cristea, I. C. Berke, T. Boehmer, G. Blobel, and B. T. Chait. 2007. Cell-cycle-dependent phosphorylation of the nuclear pore Nup107-160 subcomplex. Proc. Natl. Acad. Sci. USA 1043811-3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gustin, K. E., and P. Sarnow. 2001. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 20240-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gustin, K. E., and P. Sarnow. 2002. Inhibition of nuclear import and alteration of nuclear pore complex composition by rhinovirus. J. Virol. 768787-8796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hato, S. V., C. Ricour, B. M. Schulte, K. H. W. Lanke, M. de Bruijni, J. Zoll, W. J. G. Melchers, T. Michiels, and F. J. M. van Kuppeveld. 2007. The mengovirus leader protein blocks interferon-α/β gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 92921-2930. [DOI] [PubMed] [Google Scholar]
- 20.Hellen, C. U., G. W. Witherell, M. Schmid, S. H. Shin, T. V. Pestova, A. Gil, and E. Wimmer. 1993. A cytoplasmic 57-kDa protein that is required for translation of picornavirus RNA by internal ribosomal entry is identical to the nuclear pyrimidine tract-binding protein. Proc. Natl. Acad. Sci. USA 907642-7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu, T., T. Guan, and L. Gerace. 1996. Molecular and functional characterization of the p62 complex, an assembly of nuclear pore complex glycoproteins. J. Cell Biol. 134589-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kehlenbach, R. H., and L. Gerace. 2000. Phosphorylation of the nuclear transport machinery downregulates nuclear protein Import in vitro. J. Biol. Chem. 27517848-17856. [DOI] [PubMed] [Google Scholar]
- 23.Kireeva, N., M. Lakonishok, I. Kireev, T. Hirano, and A. S. Belmont. 2004. Visualization of early chromosome condensation: a hierarchical folding, axial glue model of chromosome structure. J. Cell Biol. 166775-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lénárt, P., G. Rabut, N. Daigle, A. R. Hand, M. Terasaki, and J. Ellenberg. 2003. Nuclear envelope breakdown in starfish oocytes proceeds by partial NPC disassembly followed by a rapidly spreading fenestration of nuclear membranes. J. Cell Biol. 1601055-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lidsky, P. V., S. Hato, M. V. Bardina, A. G. Aminev, A. C. Palmenberg, E. V. Sheval, V. Y. Polyakov, F. J. M. van Kuppeveld, and V. I. Agol. 2006. Nucleocytoplasmic traffic disorder induced by cardioviruses. J. Virol. 802705-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim, R. Y., U. Aebi, and B. Fahrenkrog. 2008. Towards reconciling structure and function in the nuclear pore complex. Histochem. Cell Biol. 129105-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim, R. Y. H., N.-P. Huang, J. Köser, J. Deng, K. H. A. Lau, K. Schwarz-Herion, B. Fahrenkrog, and U. Aebi. 2006. Flexible phenylalanine-glycine nucleoporins as entropic barriers to nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 1039512-9517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu, D., H. Yang, and M. K. Raizada. 1998. Involvement of p62 nucleoporin in angiotensin II-induced nuclear translocation of STAT3 in brain neurons. J. Neurosci. 181329-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lubas, W. A., M. Smith, C. M. Starr, and J. A. Hanover. 1995. Analysis of nuclear pore protein p62 glycosylation. Biochemistry 341686-1694. [DOI] [PubMed] [Google Scholar]
- 30.Macaulay, C., E. Meier, and D. J. Forbes. 1995. Differential mitotic phosphorylation of proteins of the nuclear pore complex. J. Biol. Chem. 270254-262. [DOI] [PubMed] [Google Scholar]
- 31.McBride, A. E., A. Schlegel, and K. Kirkegaard. 1996. Human protein Sam68 relocalization and interaction with poliovirus RNA polymerase in infected cells. Proc. Natl. Acad. Sci. USA 932296-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meerovitch, K., Y. V. Svitkin, H. S. Lee, F. Lejbkowicz, D. J. Kenan, E. K. Chan, V. I. Agol, J. D. Keene, and N. Sonenberg. 1993. La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J. Virol. 673798-3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melcák, I., A. Hoelz, and G. Blobel. 2007. Structure of Nup58/45 suggests flexible nuclear pore diameter by intermolecular sliding. Science 3151729-1732. [DOI] [PubMed] [Google Scholar]
- 34.Miller, M. W., M. R. Caracciolo, W. K. Berlin, and J. A. Hanover. 1999. Phosphorylation and glycosylation of nucleoporins. Arch. Biochem. Biophys. 36751-60. [DOI] [PubMed] [Google Scholar]
- 35.O'Brien, E. M., D. A. Gomes, S. Sehgal, and M. H. Nathanson. 2007. Hormonal regulation of nuclear permeability. J. Biol. Chem. 2824210-4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onischenko, E. A., N. V. Gubanova, E. V. Kiseleva., and E. Hallberg. 2005. Cdk1 and okadaic acid-sensitive phosphatases control assembly of nuclear pore complexes in Drosophila embryos. Mol. Biol. Cell 165152-5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park, N., P. Katikaneni, T. Skern, and K. E. Gustin. 2008. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J. Virol. 821647-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pemberton, L. F., and B. M. Paschal. 2005. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic 6187-198. [DOI] [PubMed] [Google Scholar]
- 39.Pilipenko, E. V., E. G. Viktorova, S. T. Guest, V. I. Agol, and R. P. Roos. 2001. Cell-specific proteins regulate viral RNA translation and virus-induced disease. EMBO J. 206899-6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pisarev, A. V., L. S. Chard, Y. Kaku, H. L. Johns, I. N. Shatsky, and G. J. Belsham. 2004. Functional and structural similarities between the internal ribosome entry sites of hepatitis C virus and porcine teschovirus, a picornavirus. J. Virol. 784487-4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porter, F. W., Y. A. Bochkov, A. J. Albee, C. Wiese, and A. C. Palmenberg. 2006. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 10312417-12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prigent, C., and S. Dimitrov. 2003. Phosphorylation of serine 10 in histone H3, what for? J. Cell Sci. 1163677-3685. [DOI] [PubMed] [Google Scholar]
- 43.Rabut, G., P. Lénárt, and J. Ellenberg. 2004. Dynamics of nuclear pore complex organization through the cell cycle. Curr. Opin. Cell Biol. 16314-321. [DOI] [PubMed] [Google Scholar]
- 44.Radu, A., M. S. Moore, and G. Blobel. 1995. The peptide repeat domain of nucleoporin Nup98 functions as a docking site in transport across the nuclear pore complex. Cell 81215-222. [DOI] [PubMed] [Google Scholar]
- 45.Schwarz-Herion, K., B. Maco, U. Sauder, and B. Fahrenkrog. 2007. Domain topology of the p62 complex within the 3-D architecture of the nuclear pore complex. J. Mol. Biol. 370796-806. [DOI] [PubMed] [Google Scholar]
- 46.Semler, B. L., and E. Wimmer (ed.). 2002. Molecular biology of picornaviruses. ASM Press Washington, DC.
- 47.Sheval, E. V., and V. Y. Polyakov. 2006. Visualization of the chromosome scaffold and intermediates of loop domain compaction in extracted mitotic cells. Cell Biol. Int. 301028-1040. [DOI] [PubMed] [Google Scholar]
- 48.Stanway, G., T. Hovi, N. J. Knowles, and T. Hyypiä. 2002. Molecular and biological basis of picornavirus taxonomy, p. 17-24. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 49.Svitkin, Y. V., H. Imataka, K. Khaleghpour, A. Kahvejian, H. D. Liebig, and N. Sonenberg. 2001. Poly(A)-binding protein interaction with elF4G stimulates picornavirus IRES-dependent translation. RNA 71743-1752. [PMC free article] [PubMed] [Google Scholar]
- 50.Terry, L. J., E. B. Shows, and S. R. Wente. 2007. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science 3181412-1416. [DOI] [PubMed] [Google Scholar]
- 51.Tolskaya, E. A., L. I. Romanova, M. S. Kolesnikova, T. A. Ivannikova, E. A. Smirnova, N. T. Raikhlin, and V. I. Agol. 1995. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J. Virol. 691181-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Veselý J., L. Havlicek, M. Strnad, J. J. Blow, A. Donella-Deana, L. Pinna, D. S. Letham, J. Kato, L. Detivaud, S. Leclerc, and L. Meijer. 1994. Inhibition of cyclin-dependent kinases by purine analogues. Eur. J. Biochem. 224771-786. [DOI] [PubMed] [Google Scholar]
- 53.Waggoner, S., and P. Sarnow. 1998. Viral ribonucleoprotein complex formation and nucleolar-cytoplasmic relocalization of nucleolin in poliovirus-infected cells. J. Virol. 726699-6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walther, T. C., M. Fornerod, H. Pickersgill, M. Goldberg, T. D. Allen, and I. W. Mattaj. 2001. The nucleoporin Nup153 is required for nuclear pore basket formation, nuclear pore complex anchoring and import of a subset of nuclear proteins. EMBO J. 205703-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weidman, M. K., R. Sharma, S. Raychaudhuri, P. Kundu, W. Tsai, and A. Dasgupta. 2003. The interaction of cytoplasmic RNA viruses with the nucleus. Virus Res. 9575-85. [DOI] [PubMed] [Google Scholar]
- 56.Zoll, J., J. M. Galama, F. J. van Kuppeveld, and W. J. Melchers. 1996. Mengovirus leader is involved in the inhibition of host cell protein synthesis. J. Virol. 704948-4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zoll, J., W. J. Melchers, J. M. Galama, and F. J. M. van Kuppeveld. 2002. The mengovirus leader protein suppresses alpha/beta interferon production by inhibition of the iron/ferritin-mediated activation of NF-κB. J. Virol. 769664-9672. [DOI] [PMC free article] [PubMed] [Google Scholar]