

Abstract
To make a safe, long-lasting gene delivery vehicle, we developed a hybrid vector that leverages the relative strengths of adenovirus and Epstein-Barr virus (EBV). A fully gene-deleted helper-dependent adenovirus (HDAd) is used as the delivery vehicle for its scalability and high transduction efficiency. Upon delivery, a portion of the HDAd vector is recombined to form a circular plasmid. This episome includes two elements from EBV: an EBV nuclear antigen 1 (EBNA1) expression cassette and an EBNA1 binding region. Along with a human replication origin, these elements provide considerable genetic stability to the episome in replicating cells while avoiding insertional mutagenesis. Here, we demonstrate that this hybrid approach is highly efficient at delivering EBV episomes to target cells in vivo. We achieved nearly 100% transduction of hepatocytes after a single intravenous injection in mice. This is a substantial improvement over the transduction efficiency of previously available physical and viral methods. Bioluminescent imaging of vector-transduced mice demonstrated that luciferase transgene expression from the hybrid was robust and compared well to a traditional HDAd vector. Quantitative PCR analysis confirmed that the EBV episome was stable at approximately 30 copies per cell for up to 50 weeks and that it remained circular and extrachromosomal. Approaches for adapting the HDAd-EBV hybrid to a variety of disease targets and the potential benefits of this approach are discussed.
One of the foremost challenges of gene replacement therapy is the need for the delivered transgene to be safely and stably maintained in replicating cells, preferably for a lifetime. One approach, used by retrovirus vectors, is to achieve genetic stability via integration into the host cell genome. However, this can give rise to insertional mutagenesis and oncogenesis in both animal models and in human clinical trials (12, 25). Nonintegrating vectors, such as adenovirus-based vectors, are safer in this regard, but they are poorly maintained in replicating cells (10, 13).
As an alternative to integration, gene replacement vectors based on Epstein-Barr virus (EBV) are capable of life-long persistence as a nonintegrating, circular episome (8, 45). Two viral elements mediate this stability: the EBV nuclear antigen 1 (EBNA1) protein and its binding sites in the EBV latent origin (48). This origin contains a series of 21 EBNA1 recognition sites known as the family of repeats (FR). Binding of EBNA1 to the cis-acting FR tethers the episome to host cell chromosomes, thus promoting efficient segregation to both daughter cells during mitosis (41). Plasmid constructs that contain these two EBV elements and an origin of replication are thus maintained in replicating cells at 90 to 95% per cell division without selection (9, 21, 31). EBV episomes have been widely used in animal models to treat diseases as diverse as hemophilia, diabetes, and allergic rhinitis (15, 28, 49). Most of these EBV studies make use of nonviral modalities, such as hydrodynamic injection, to deliver the episome to target tissue, but nonviral approaches are far less efficient in vivo than transduction by viral vectors (3, 24, 37, 40, 42).
Helper-dependent adenovirus (HDAd) vectors, also known as gutless adenoviruses, are vectors in which all viral coding regions have been deleted (18, 34). The absence of viral genes renders these vectors far less immunogenic or cytotoxic than previous generations of adenovirus vectors (10, 29, 30). In addition, HDAd vectors can be manufactured to high titers, have a large cloning capacity, and are efficient at transducing target cells in vivo (24, 33, 37).
Given the relative strengths of HDAd vectors for delivery and EBV episomes for persistence, we developed a hybrid vector that incorporates favorable elements of both. In this system, the therapeutic or reporter transgene is expressed from a circular plasmid that includes the FR and an EBNA1 expression cassette from EBV. This, along with a human replication origin (hORI) (21), confers stability to the episome in replicating mammalian cells. The elements of the EBV episome are delivered to target cells in a linear form in an adenovirus virion we call an “HDAd-EBV hybrid vector.” To facilitate excision and circularization of the EBV episome in target cells, this region is flanked by parallel recognition sites for Cre recombinase (loxP sites). The Cre necessary to excise the EBV episome in target cells is provided by a second vector, HDAd.Cre. Consequently, episome recombination occurs in cells coinfected by both HDAd vectors (9).
Previous studies by our group and others have validated this hybrid vector approach in tissue culture models (9, 19, 20, 22, 43). High rates of episome formation were observed in cotransduced, cultured cells. Once recombined, the hybrid circular episomes persisted significantly better than linear HDAd episomes in continuously dividing cells (9). In the present study, we extend these in vitro results to an animal model. Hepatocytes in mice were transduced by a single intravenous injection of the HDAd-EBV vector. The transduction efficiency was evaluated by confocal microscopy of liver sections and by flow cytometric analysis of single cell suspensions of hepatocytes. Transgene expression was monitored by bioluminescent imaging in living animals. Circularization, maintenance, and integration state of the episome were assayed by quantitative PCR (qPCR). Taken together, these results demonstrate the potential utility of the HDAd-EBV hybrid vector for robust delivery of a nonintegrating, persistent expression vector for in vivo transgene expression.
MATERIALS AND METHODS
Vector propagation.
Construction of the HDAd vectors as plasmids was described in detail by Dorigo et al. (9). HDAd.Cre and HDAd-EBV.RL vectors were propagated by using the AdLC8cluc helper virus and 293Cre4 cells as described by Parks et al. (34). HDAd-EBV.RL vectors were propagated by using FL helper virus and 293FLPe cells as described by Umana et al. (47). In brief, purified vector DNA was transfected into 293Cre4 or 293FLPe cells by means of calcium phosphate (Promega, Madison, WI). After 24 h, the cells were infected with helper virus at a multiplicity of infection of five infectious genome units (IGU) per cell (see below for titration of viruses). Progeny virions were harvested after 48 to 72 h by freeze-thaw lysis of the infected cells. This crude cell lysate was then used to reinfect fresh 293 cells along with additional helper virus. Vectors were amplified in this manner in increasing numbers of cells over the course of seven to 10 passages. The titer of helper virus and HDAd vector were monitored at each passage by qPCR. After the final step, vectors were purified by successive rounds of CsCl step-gradient ultracentrifugation (1.2 over 1.4 g of CsCl/ml, 36 krpm for 1.5 h in an SW-40 rotor). Banded virions were dialyzed into three changes of 20 mM Tris (pH 8) and then formulated by the addition of NaCl to 25 mM and glycerol to 2.5%. Virus stocks were stored in small aliquots at −80°C until use.
Vector titration by qPCR.
All virus stock titers were determined by a novel method in which viral DNA is harvested from the nuclei of infected cells and quantified by qPCR. This method is similar to an approach previously published by Puntel et al. (36). HeLa cells (American Type Culture Collection, Manassas, VA) were grown in high-glucose Dulbecco modified Eagle medium supplemented with 10% bovine calf serum. Cells were seeded onto six-well tissue culture plates 1 day before infection. Once the cells were 80 to 95% confluent, wells were infected, in duplicate, with 5 μl of vector stock diluted in 500 μl of medium. After 30 min of incubation at 37°C, an additional 2 ml of medium was added to each well. Plates were allowed to incubate for 3 to 6 h. The medium was removed, and cells were washed twice with phosphate-buffered saline (PBS) and aspirated. Cell lysis was performed by the addition of 1 ml of freshly made NP-40 lysis buffer containing 0.65% NP-40 substitute (Calbiochem, San Diego, CA), 150 mM NaCl, 10 mM Tris-HCl, and 1.5 mM MgCl2. Cells were incubated for 5 to 10 min at room temperature to allow cell lysis to occur. The lysate was pipetted up and down multiple times to aid in lysis and then transferred from the plate to an Eppendorf tube. Nuclei were pelleted by spinning three minutes at 2,000 × g in a microfuge. The supernatant was decanted, and an additional 1 ml of NP-40 lysis buffer was added. The pellet of nuclei in fresh buffer was vortex mixed briefly before an additional 3-min spin at 2,000 × g. Supernatant was decanted, and nuclei were suspended in 200 μl of PBS.
Total DNA was isolated and purified by means of a DNeasy Blood & Tissue kit (Qiagen, Valencia, CA) using the protocol for purification of total DNA from animal blood or cells. Briefly, 4 μl of RNase was added to the pelleted nuclei, and samples were incubated for 5 min at room temperature. Then, 20 μl of proteinase K and 200 μl of Qiagen buffer AL were added, and samples were incubated for 10 min at 70°C. Next, 200 μl of ethanol was added, and DNA was immobilized on a DNeasy column by centrifugation. After two wash steps with the Qiagen buffers AW1 and AW2, the DNA was eluted with two 200-μl volumes of Qiagen buffer AE and pooled.
Quantification of the viral DNA by qPCR occurred as follows. Each vector DNA preparation was assayed in duplicate or triplicate in optical-grade, 96-well plates (Applied Biosystems, Foster City, CA). Each 25-μl reaction contained 12.5 μl of TaqMan Universal Master Mix (Applied Biosystems), 5 μl of purified DNA template, primers (900 nM final concentration), and probe (250 nM final concentration). PCR runs were performed on an ABI 7500 real-time thermocycler using SDS software v.1.3 (Applied Biosystems). Reaction conditions were as follows: 10 min at 94°C, followed by 40 cycles of 10 s at 94°C and 30 s at 60°C. The number of viral genomes in each reaction was determined by comparison to a standard curve. This standard curve was generated by the serial dilution of a plasmid containing the PCR template.
HDAd-EBV vector titers were determined by using the following primers: forward, AAACCAGGGAGGCAAATCTACTC; reverse, GACAAAGCCCGCTCCTACCT; and probe, FAM-ATCGTCAAAGCTGCACACAGTCACCC-BHQ1A. The HDAd.RL titer was determined with the forward primer AATGCGATGCAATTTCCTCAT, the reverse primer TGCCTTCCTTGACCCTGG, and the probe HEX-TTAGGAAAGGACAGTGGGAGTGGCACC-BHQ1A. The HDAd.Cre titer was determined with the forward primer TGGAAAATGCTTCTGTCCGTTT and the reverse primer CGCGCCTGAAGATATAGAAGATAAT. Mouse hepatocytes were quantified by using the forward primer CAAGGGTTGAGTACTTGTTTAGGGTTA, the reverse primer GGTGGGTAGAGAGAAGAAATATCTGACT, and the probe TAMRA-AGGACAATGGCCTTGGCTGGACAA-BHQ2A. Unrecombined HDAd-EBV vector was assayed by using the forward primer CGACAAGCCCAGTTTCTATTGG, the reverse primer CGTAGTAGTGTGGCGGAATCG, and the probe HEX-CCAACTTCTGCAGCGGCCGCTCT-BHQ1A. Recombined HDAd-EBV vector was assayed by using the forward primer CGACAAGCCCAGTTTCTATTGG, the reverse primer TGGTTTAGTGAACCGTCTGATCTC, and the probe HEX-CCAACTTCTGCAGCGGCCGCTCT-BHQ1A.
Animal handling.
All animals were handled according to institutional guidelines and under the approval of the UCLA Office for the Protection of Research Subjects. Experiments were performed in adult, female Nu-Foxn1nu nude mice (Charles River, Wilmington, MA). Mice were maintained in sterile Department of Laboratory Animal Medicine facilities on a 12-h light/dark cycle and given acidified water and irradiated rodent chow ad libitum. Mice that showed signs of illness, mainly low body weight and/or hunched posture, were given amoxicillin at 50 mg/kg/day in the drinking water. Mice that did not show improvement and mice at the endpoint of analysis were sacrificed by CO2 inhalation.
Vector injections.
Injections were performed under a general anesthetic cocktail of ketamine (100 mg/kg) and xylazine (10 mg/kg) injected into the peritoneal cavity. Mice were given ophthalmic ointment to prevent desiccation of the cornea, and a warm pad was used to prevent hypothermia. Six hours prior to vector injections, mice were preinjected with 6 mg of mouse immunoglobulin G to prevent sequestration of vectors in the Kupffer cells of the liver (44). All injections were performed intravenously via the tail vein by using a 0.5-ml tuberculin syringe fitted with a 0.5-in. 29 gauge needle. Vectors were injected at the indicated titers as assayed by the IGU protocol. Dilutions were made in sterile PBS as needed.
Confocal microscopy.
Mice were injected with HDAd-EBV.RL vector at the indicated doses as described above. At 6 days postinjection, mice were sacrificed, and liver tissues were harvested. Pieces of tissue ∼3 mm3 in size were fixed in 4% paraformaldehyde for 30 min at 37°C. The tissue was then embedded in agarose and sectioned into 50-μm-thick slices by using a Vibratome (Vibratome, St. Louis, MO). Slices were stained with TO-PRO-3 nuclear stain (Invitrogen, Carlsbad, CA) and mounted on slides by hardset Vectorshield (Vector Laboratories, Burlingame, CA). Imaging was performed on a Leica TCS SP2 AOBS single photon confocal microscope (Leica Microsystems, Inc., Bannockburn, IL) using a ×63 1.4-numerical aperture oil immersion objective lens.
Fluorescence microscopy.
The same mouse tissue harvested for confocal microscopy was also used for nonconfocal epifluorescence light microscopy. Here, unfixed tissue ∼3 mm3 in size was embedded in optimal cutting temperature medium and flash frozen in liquid nitrogen. Then, 5-μm- thick slices were cryosectioned and mounted on slides. Microscopy was performed on an Olympus CK40 inverted light microscope (Olympus America, Inc., Center Valley, PA) illuminated by a mercury short arc lamp. Images were acquired with an Olympus Camedia C-4000 digital camera mounted to the microscope. All images were taken using identical exposure settings, and all image adjustments were performed in parallel.
Flow cytometry.
The same mouse tissue harvested for confocal microscopy was also analyzed by analytical flow cytometry. Here, pieces of unfixed tissue ∼3 mm3 in size were broken down into single cell suspensions by means of a digestion in 1 mg of collagenase/ml, 100 mM HEPES (pH 7.5), and 300 nM CaCl2 for 1 h at 37°C. The cell suspension was filtered through a 100-μm-pore-size nylon cell strainer (Becton Dickinson Biosciences, San Jose, CA) by gravity feed and then fixed with 4% paraformaldehyde for 30 min at 37°C. Cells were washed and finally resuspended in PBS for cytometry.
Cytometry data acquisition was performed on a Becton Dickinson FACSCalibur analytical flow cytometer. Analysis was performed by using Weasel cytometry analysis software v.2.6.1 (Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia). Events were first gated based on forward and side scatter to distinguish cells from debris. The cells were then sorted by intensity of the enhanced yellow fluorescent protein (EYFP) signal and plotted in histograms. The threshold of EYFP signal indicating transduction was determined relative to the mock-infected samples.
Bioluminescent imaging.
In vivo expression of the Renilla luciferase (RL) transgene was by bioluminescent imaging (2). Mice were first anesthetized with ketamine-xylazine intraperitoneally as described above. The RL substrate, coelenterazine (NanoLight Technology, Pinetop, AZ), was dissolved in methanol to 2 mg/ml and diluted 10 times in sterile PBS. Each mouse was injected intravenously with 20 μg of diluted coelenterazine immediately prior to imaging. All imaging was performed by a cryogenically cooled bioluminescent/optical camera (Xenogen, Alameda, CA). Mice were placed in the supine position on a thermally maintained platform in a lightproof chamber. An optical exposure was taken for 0.2 s, followed by a 5-min bioluminescent exposure. Optical and bioluminescent data were superimposed and analyzed by Living Image Software v2.20 (Xenogen). For each image, a region of interest was drawn over the approximate liver region and used to determine the degree of RL activity in terms of total photons per second.
Integration state of vector DNA.
In order to determine the integration state of the vector in transduced hepatocytes, we performed a modification of the technique developed by Hirt (17). Liver tissue was harvested from vector-transduced mice at various time points as indicated. Samples of liver tissue (approximately 20 to 30 μg each) were digested with 1 mg/ml collagenase (Sigma, St. Louis, MO) in 340 μM CaCl2 and 100 mM HEPES (pH 7.5) for 2 h at 37°C. At this point, 106 copies of an unrelated 35-kb plasmid were added as a low-molecular-weight (LMW) control. Next, the cells were digested with 150 μg of proteinase K in 10 mM Tris, 10 mM EDTA, and 0.6% sodium dodecyl sulfate for 30 min at 37°C. High-molecular-weight (HMW) DNA was selectively precipitated by the addition of 1 M NaCl at 4°C overnight. The precipitated DNA was pelleted at maximum speed in a microfuge at 4°C for 2 h. The DNA was carefully separated into a HMW fraction (the pellet) and a LMW fraction (the supernatant). HMW DNA was resuspended in 200 μl of 10 mM Tris-HCl- 1 mM EDTA (TE). The LMW fraction was ethanol precipitated and resuspended in 200 μl of TE. Both fractions were further processed by the DNeasy Blood & Tissue kit (QIAgen, Valencia, CA) using the protocol for purification of total DNA from animal blood or cells. The resulting purified DNA was analyzed by qPCR as described above.
RESULTS
In vivo transduction efficiency.
In the HDAd-EBV hybrid vector presented here, a circular EBV episome is formed in the nuclei of target cells by Cre-mediated, site-specific recombination (Fig. 1). To prevent this recombination from occurring prior to transduction of target cells, the Cre expression cassette and the loxP-flanked episome sequences are delivered on two independently manufactured HDAd vectors. Formation of a circular EBV episome occurs only in cells that are cotransduced by both vectors (9). In an in vivo context, this means that target tissue must be infected at a high enough multiplicity of infection so that a significant proportion of cells are transduced by both HDAd-EBV and HDAd.Cre.
FIG. 1.
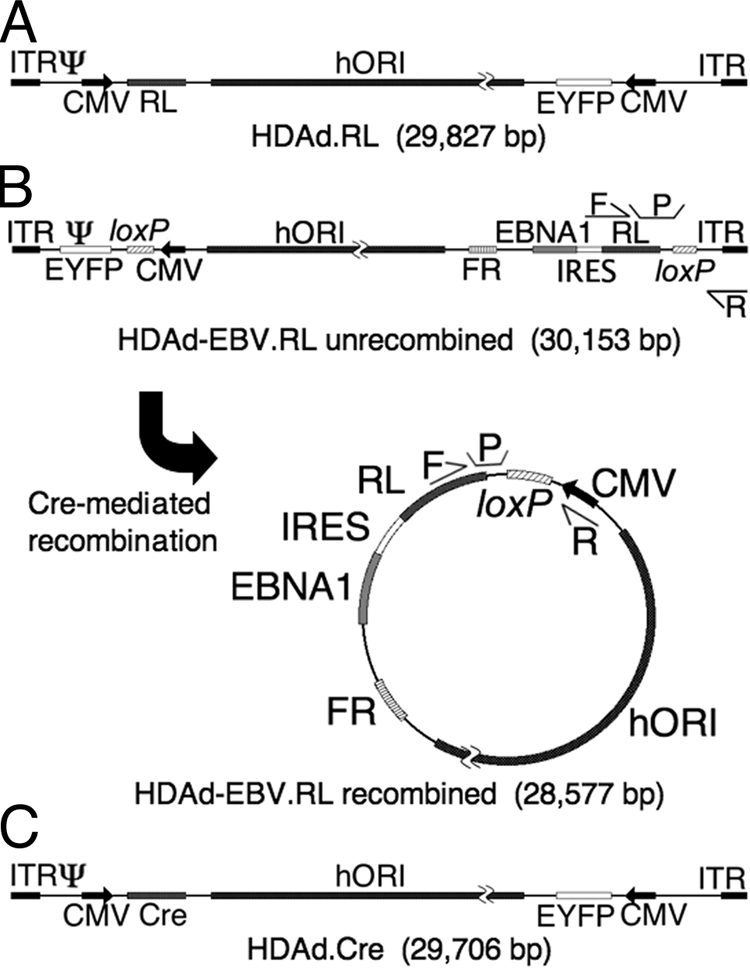
Structure of HDAd vectors. Three vectors were used in the course of these experiments. Each contains an EYFP gene expressed from a constitutive human CMV major immediate-early promoter (CMV); a 19-kb region of human genomic DNA from chromosome 10 for use as a stuffer and as an origin of replication (hORI) (21); and two regions from adenovirus, the inverted terminal repeats (ITR) that function as replication origins and the viral packaging signal (Ψ) that are required in cis for vector packaging and amplification. (A) HDAd.RL is a traditional HDAd vector that was used as a control in these experiments. It expresses RL constitutively from a CMV promoter. (B) HDAd-EBV.RL is a hybrid vector that is designed to deliver a circular EBV episome into target cells by means of a linear HDAd genome. In its linear form, the EBV episome elements are flanked by parallel recognition sites for Cre recombinase (loxP). In the presence of Cre recombinase, the intervening region is excised and circularized as shown. This places a CMV promoter upstream of an expression cassette that contains the RL transgene, an encephalomyocarditis virus internal ribosomal entry site (IRES), and the EBNA1 gene. The episome also caries the FR region that functions with EBNA1 protein to provide a maintenance function to the episome in replicating cells. The relative location of two sets of qPCR primers (F and R) and probes (P) are indicated. One set flanks one of the loxP sites in the linear conformation of the hybrid vector, and a second set flanks the loxP site in the circularized conformation. These were used in qPCR experiments to determine the relative fraction of recombined and unrecombined vector. (C) HDAd.Cre is a traditional HDAd vector that is designed to express Cre recombinase in target cells by means of a CMV promoter. Coinfection of a target cell by both HDAd-EBV.RL and HDAd.Cre is required to form the circular EBV episome.
To estimate the feasibility of this approach, in vivo transduction experiments were performed in immunocompromised nude mice. Mice were injected intravenously via the tail vein with a range of doses of the HDAd-EBV.RL hybrid vector. We compared threefold dilutions from 5 × 109 IGU (see Materials and Methods) down to 5 × 108 IGU, as well as a mock-infected mouse. The hybrid vector expresses an EYFP reporter gene from a constitutive cytomegalovirus (CMV) promoter (Fig. 1). At 6 days postinjection, the mice were sacrificed, and liver tissue samples were analyzed by a number of techniques to determine the percentage of EYFP-positive hepatocytes (Fig. 2). Confocal microscopy was used to observe the cell-by-cell transduction rate (Fig. 2A). By comparing the number of clearly distinct EYFP-positive cells to the total number of cells indicated by the TO-PRO-3 nuclear stain, it appeared that nearly all visible hepatocytes were transduced at the highest dose. This level of transduction fell significantly at the two lower doses. To evaluate the gross transduction pattern on a larger scale, 5-μm sections taken from the same mice were evaluated by nonconfocal, fluorescence-light microscopy at a lower magnification. These images were consistent with the confocal micrographs in that they show total, albeit nonuniform, transduction at the highest dose. The pattern of expression at the intermediate dose suggested that transduction occurs in patches of hepatocytes. In both sets of images, the transduction efficiency appeared to be low at the lowest dose.
FIG. 2.

Efficiency of hepatocyte transduction in vivo. Adult, female nude mice were injected with HDAd-EBV.RL via the tail vein. Two mice at each of three different vector doses (5.0 × 108, 1.7 × 109, and 5.0 × 109 IGU per mouse as indicated) and a mock-infected mouse were compared. At 6 days postinjection, the mice were sacrificed and liver tissue was harvested for the following analyses. Expression of the EYFP transgene from the vector was used to determine the efficiency of liver transduction. (A) Representative samples of liver tissue from each vector dose are presented. The first and second columns are laser scanning confocal micrographs of 50-μm-thick sections of fixed liver tissue. EYFP indicates vector-transduced cells. TO-PRO-3 was used as a nuclear stain. The third column contains micrographs taken from an epifluorescence- light microscope of 5-μm-thick unfixed sections. Signal intensity was adjusted for image quality in the confocal micrographs but is consistent across all of the fluorescent micrographs. Size bar equals 50 μm in all micrographs. The fourth column displays histograms of EYFP intensity quantified by analytical flow cytometry of single cell suspensions of liver tissue. The vertical line indicates the positive/negative threshold of EYFP as determined by the mock control samples. (B) Aggregate data from the flow cytometry analysis is presented. Each bar represents the mean percentage of EYFP-positive cells at each dose from replicate samplings (n = 3). (C) Additional aggregate cytometry data from the samples described above demonstrate the median signal intensity of EYFP-positive cells as determined from the cytometry histograms. Each bar represents the mean of replicate samplings (n = 3). (D) The number of vector genomes per diploid cell was quantified by qPCR. Each bar represents the mean of replicate samplings (n = 5). Error bars indicate mean ± the SEM.
To quantify the transduction efficiency, single cell suspensions of transduced liver tissue were prepared for analysis by flow cytometry. The intensity of EYFP expression per cell was plotted in histograms for each of the doses (representative samples are presented in Fig. 2A). The results of this analysis performed on triplicate samples of each dose were averaged and presented in Fig. 2B. We calculate that the transduction efficiency at the highest dose to be 96% ± 1% (mean ± the standard error of the mean [SEM]). From the micrographs and from the histograms, it appeared that the transduced cells at the highest dose were brighter than those at the lower doses. To quantify this, we calculated the median intensity of EYFP signal for the positive cells in each histogram and plotted this as an average of triplicate samples for each dose (Fig. 2C). The median brightness at the highest dose was ∼10 times higher than at the lowest dose.
Lastly, we performed qPCR on DNA prepared from liver tissue harvested from the same mice. A primer-probe set specific for HDAd-EBV.RL vector DNA was used to determine the number of vector genomes in a given sample. A second primer-probe set complementary to the mouse Oct4 promoter region was used to determine the number of murine cells in each sample. Assuming a diploid genome, there were 26 ± 7 (mean ± the SEM) copies of the vector per cell at the highest vector dose (Fig. 2D).
In vivo EBV episome delivery.
Since transduction of hepatocytes was found to be highly efficient, we next determined whether Cre-mediated recombination of linear HDAd-EBV vectors into circular EBV episomes occurs in vivo. A total of 16 mice were coinjected with 5 × 109 IGU of HDAd-EBV.RL and 5 × 109 IGU of HDAd.Cre. When these mice were imaged for bioluminescence, expression of RL was observed in the region of the liver (Fig. 3A). As soon as 1 week after injection, the expression of RL was ∼1,000 times higher than the background of the assay as determined with mock-infected mice (Fig. 3B). Over the course of the next several weeks, expression continued to increase to more than 5,000 times higher than that of mock-infected mice. As a control, six mice were injected with 5 × 109 IGU of HDAd.RL. This traditional HDAd vector constitutively expresses RL, so it requires no recombination event. To control for the number of virion particles injected, these mice were injected simultaneously with 5 × 109 IGU of HDAd.Cre, which does not contain a luciferase gene. Expression from the standard HDAd.RL linear vector was comparable to the recombined hybrid vector.
FIG. 3.

EBV episome delivery by HDAd-EBV vectors. Adult, female nude mice were injected intravenously with HDAd vectors and assayed for RL expression by bioluminescent imaging. Four cohorts were compared. Group 1 received 5 × 109 IGU of HDAd-EBV.RL and 5 × 109 IGU of HDAd.Cre (n = 16). Group 2 received 5 × 109 IGU of HDAd.RL and 5 × 109 IGU of HDAd.Cre (n = 6). Group 3 received 1010 IGU of HDAd-EBV.RL (n = 2). To determine the background of the assay, a fourth “mock” group was imaged using the same conditions as the experimental groups but received no vector (n = 8). (A) Representative images from the first three groups at 1 week postinfection. The total number of photons per second, as averaged over a 5-min bioluminescent exposure, was overlaid on an optical image of the mouse. The signal intensity was color coded according to the included scale from 1.5 × 105 to 1.5 × 107 total photons/s. (B) Mice were imaged weekly over a 6-week period after vector injection as in panel A. The number of photons/second detected in the region of the liver was plotted for each cohort over time. The dashed line indicates the background of the assay as determined by the mock cohort. Error bars indicate the mean ± the SEM. (C) Total DNA was prepared from samples of transduced liver tissue for analysis of recombination at the loxP sites by qPCR. Samples ranged from 9 to 30 weeks postinjection. A total of 10 mice injected with HDAd-EBV.RL were analyzed. Eight received HDAd.Cre, and two received no Cre. Each result is presented as a percentage of recombined vector relative to the total. A plasmid with only the unrecombined sequence was used to determine the background of the assay. Error bars indicate the mean ± the SEM.
In order to determine whether RL expression in the HDAd-EBV hybrid vector was dependent on Cre-mediated recombination, two mice were injected with 1010 IGU of HDAd-EBV.RL, but no Cre vector. RL activity in these mice was significantly lower than mice coinjected with HDAd.Cre. However, expression of luciferase from this vector in the absence of Cre was significantly higher than the background of the assay.
Cre-mediated recombination of the EBV episome in vivo.
The expression of RL from an HDAd-EBV hybrid vector in the absence of Cre was a surprising one given that there should be no promoter upstream of the RL gene. Our observations suggested two possibilities. The first is that RL expression was driven by the adenoviral inverted terminal repeat at the right end of the vector (14). The second possibility was that a small fraction of the vector recombined in the absence of Cre by an unanticipated mechanism. To distinguish between these models, we determined whether EBV episomes were in fact recombined at the loxP sites in the presence or absence of Cre. Two sets of qPCR primers that anneal at sites immediately flanking loxP were generated to distinguish between recombined and unrecombined forms of the vector (Fig. 1B). These primers were designed so that only recombination at the loxP site would be assayed. The primers were used on purified liver DNA from eight mice that had been coinjected with both HDAd-EBV.RL and HDAd.Cre and from two mice that received only HDAd-EBV.RL. A total of 94% of vector DNA was recombined in the presence of Cre, while 6% was circularized in the absence of Cre (Fig. 3C). A plasmid with only the linear form of the vector DNA was also assayed to determine the background signal for circularized vector in PCRs with only linear DNA. In this analysis, the linear plasmid was assayed as being 2% recombined. Since the value obtained for circularized vector in liver from animals infected with HDAd-EBV.RL without HDAd.Cre (6%) was determined to be above this background value, we conclude that a small percentage of the HDAd-EBV.RL vector sequence was recombined at the loxP sites in the absence of Cre. This was probably due to murine homologous recombination mechanisms acting at the repeated loxP sites in the vector, accounting for the low level of RL expression observed in these animals (Fig. 3B).
In vivo maintenance of vector DNA.
The main purpose of the EBV elements in the HDAd-EBV hybrid vector is to ensure that the vector DNA is well maintained in transduced cells. We next examined the stability of hybrid vector DNA by means of qPCR. Samples of liver tissue were harvested from vector-transduced mice at various times ranging from 12 to 50 weeks postinjection. A total of 13 samples were collected and assayed by qPCR. The quantity of hybrid vector DNA in each sample was normalized to the number of diploid murine cells in the sample determined using primers for a single copy mouse gene as described above. Next, the samples were divided into three categories based on the number of weeks postinjection. Samples in each category were averaged and compared. The qPCR analysis detected an average of 30 ± 3 copies (mean ± the SEM) of the HDAd-EBV hybrid vector per cell (Fig. 4). There was no statistical change in the number of HDAd-EBV vector genomes per cell over the course of the 50-week study.
FIG. 4.

Persistence of viral DNA in vivo. Liver tissue was harvested at various time points from adult, female nude mice cotransduced with 5 × 109 IGU of HDAd-EBV.RL and 5 × 109 IGU of HDAd.Cre. Vector DNA in each tissue sample was quantified by qPCR with a primer-probe set unique to each vector. Values were normalized to the number of murine cells per sample as quantified by a third primer-probe set specific to the mouse genome. To facilitate the evaluation of the data, samples were divided into three groups based on the number of weeks postinjection that the sample was harvested. n, Number of mice in each group. Error bars represent the mean ± the SEM. Variation between the three time frames was determined by one-way Fisher analysis of variance. There was no statistically significant change in the HDAd-EBV hybrid vector (*, P = 0.52), but there was a significant decrease in the linear HDAd vector (**, P = 0.04).
To compare the maintenance of the HDAd-EBV hybrid relative to a traditional linear HDAd vector, we examined the maintenance of HDAd.Cre in the same samples used above. This vector has 85% sequence identity with the HDAd-EBV hybrid vector but lacks the loxP sites needed for circularization or the EBV elements designed to confer stability. Since HDAd.Cre was coinfected with HDAd-EBV.RL at a 1:1 ratio, we were able to perform a direct comparison. Using qPCR primers unique to HDAd.Cre, we observed a modest, but statistically significant decrease in the linear vector from the early to the later time points (Fig. 4).
Episomal versus integrated vector DNA.
A key safety feature of the HDAd-EBV hybrid vector is that it is designed to be maintained without integrating into the genomes of target cells. To determine the integration status of the vector in vivo, DNA was harvested from vector-transduced mouse liver and selectively salt precipitated to distinguish chromosomal DNA from episomal DNA. This protocol, a modification of the technique developed by Hirt (17), separates the DNA into an HMW fraction that includes chromosomal DNA and any integrated vector genomes, and an LMW fraction that includes episomal DNA. Two controls were included in this analysis to demonstrate that chromosomes and episomes could be distinguished by this approach. As a control for the HMW chromosomal DNA, qPCR was performed with a primer set specific for the mouse Oct4 promoter on chromosome 17. As a control for episomal DNA, an unrelated 35-kb plasmid was spiked into the nuclear lysate after harvesting but prior to the salt precipitation. This LMW control was independently assayed by a second set of qPCR primers. Lastly, the vector DNA was quantified by a third qPCR primer set. In each case, the relative number of copies in the HMW fraction was expressed as a percentage of the combined total (Fig. 5).
FIG. 5.

Integration state of vector DNA. A selective salt precipitation was performed to separate the DNA from vector-transduced liver tissue into HMW and LMW fractions. A 35-kb plasmid was added to each sample prior to precipitation as a control for separation of HMW and LMW DNA. The number of copies of chromosomal DNA, plasmid DNA, and HDAd-EBV vector DNA in each fraction were assayed in separate qPCR reactions. Each bar represents the percentage of copies in the high-molecular-weight fraction relative to the total copy number from both fractions. “Week 12” represents nine preparations of DNA from three mice harvested 12 weeks postinjection. “Weeks 30 to 50” represents six preparations from three mice harvested during the later time period. Error bars indicate mean ± the SEM. Variation between the plasmid DNA and HDAd-EBV episome DNA was insignificant as determined by one-way Fisher analysis of variance (*, P = 0.50; **, P = 0.13).
Separation of chromosomal and episomal DNA into separate fractions was highly efficient. Over 95% of the genomic DNA was detected in the HMW fraction, while more than 96% of the plasmid DNA was found in the LMW fraction. Consistent with the premise that the HDAd-EBV episomes had not integrated, the vector DNA fractionated into the LMW fraction precisely with the plasmid control. This assay was performed on liver tissue harvested from mice at an early time point (12 weeks postinjection) and at a late time point (30 to 50 weeks postinjection), but there was no increase of vector in the HMW fraction.
Although we did not detect integrated vector above the background of the assay, this approach is limited by the efficiency of the fractionation of the DNA into the HMW and LMW fractions. We conservatively estimate that the sensitivity of the assay, as based on the results from the plasmid DNA (mean + one standard deviation), is such that we would have detected integrated vector DNA if 4% or more of the vector DNA had integrated into cellular chromosomal DNA.
DISCUSSION
Effectiveness of the HDAd-EBV hybrid approach.
Previously, we reported that a hybrid HDAd-EBV vector system could be used to efficiently deliver circular EBV episomes into the nuclei of transduced cells in culture (9, 43). These circular episomes, once formed, were genetically stable. Here, we evaluate these same vectors in an in vivo system. Our goal was to demonstrate that EBV episomes, which have been shown to be highly stable in both tissue culture and in vivo experiments, could be delivered to target tissue with high efficiency by an HDAd hybrid vector.
Our results clearly demonstrate that episome delivery and recombination by our vector are highly efficient in vivo. Following the systemic intravenous injection of reasonable titers of HDAd-EBV hybrid vectors into mice, we observed approximately 30 copies of the vector DNA per hepatocyte. This is consistent with the 96% transduction efficiency that we observed by flow cytometry. Circularization of the EBV episome was also found to be rapid and robust. Within a matter of weeks, 94% of the vector DNA in transduced tissue was recombined, as shown by qPCR.
These vectors were designed so that the transgene has no promoter until after Cre-mediated circularization of the EBV episome. The high level of RL transgene expression as early as 1-week postinjection corroborates the qPCR result that the vector DNA underwent site-specific Cre-mediated recombination rapidly and efficiently. Further, expression from the recombined, circularized HDAd-EBV hybrid vector DNA was comparable to a traditional HDAd vector, which required no coinfection or recombination. EBV episomes delivered by the HDAd-EBV hybrid were stable and remained episomal. Although we observed a diminution in RL expression over the course of the experiment (data not shown), we attribute this to properties of the CMV promoter used for transcription of the transgene. This promoter frequently undergoes epigenetic silencing in vivo (5, 6, 27). In contrast to reporter gene expression, the vector DNA was in fact stable over the course of 50 weeks. Future experiments will address the silencing issue by replacing the CMV promoter with alternate promoters that are less susceptible to epigenetic silencing (32).
Stability.
Since hepatocytes in healthy, adult mice replicate infrequently (1), we expect that the model system chosen here was less than ideal for addressing the issue of DNA stability in our hybrid vector system. We did, however, observe a modest benefit in stability with the HDAd-EBV vector relative to a traditional HDAd vector over the course of the year-long experiment presented in Fig. 4. We expect that this slight difference would be come increasingly significant over the course of many years in a longer-lived organism. Furthermore, there is ample evidence that EBV episomes similar to those presented here are highly stable in proliferating cells both in vivo and in tissue culture (9, 21, 42). Future work will address this issue directly by retargeting the vector to replicating cell populations such as hematopoietic stem cells.
Safety.
A primary goal of this project was to create a vector that would be genetically stable without genomic integration and the attendant risk of insertional mutagenesis. From our comparison of HMW and LMW DNA, we found that the vector DNA sequences segregated in parallel with a plasmid added to the cell lysate. This was true at both an intermediate and a late time point. This indicates that the at least 96% of the vector DNA remained episomal.
As for the risks posed by the vector itself, we anticipate that the HDAd-EBV hybrid vector would have a safety profile similar to other HDAd vectors. In other words, it would be safe for clinical trials below a certain threshold. HDAd vectors have been shown to be significantly safer than earlier generations of adenovirus vectors due to the lack of viral open reading frames (10, 29, 30). In fact, the only virally derived gene remaining in the HDAd-EBV hybrid is EBNA1. This gene is unlikely to pose a significant safety issue since it is constitutively expressed in EBV-transduced B cells in ca. 90% of human adults with little consequence (35, 46). Further, EBNA1 protein has evolved to be resistant to major histocompatibility complex I presentation, so it is unlikely to cause cytotoxic-T-lymphocyte-mediated clearance of transduced cells (23).
Capacity and potential disease models.
The HDAd-EBV vector presented here includes a 20-kb region from human chromosome 10 (21). This region functions as an origin of replication to ensure that the episome will replicate during S phase along with cellular chromosomes. Krysan et al. determined that most large human genomic fragments (i.e., >16 kb) will mediate this replication function (21). We propose that the hybrid vector presented here could be adapted to a number of experimental models by replacing this region from chromosome 10 with any number of other human genomic regions that express a therapeutic gene. Given the packaging limitations of HDAd vectors, genomic regions of up to 29 kb could be incorporated into the hybrid design. The potential advantage of this cloning capacity is that therapeutic transgenes could be delivered to target tissue with their endogenous regulatory elements intact. Studies done in the context of both adenovirus vectors (39) and EBV episomes (42) have shown that transgene expression often is both higher and more stable when a gene is expressed from genomic DNA compared to cDNA.
Comparison to other EBV episome approaches.
Most studies of EBV episome-mediated gene therapy utilize nonviral approaches for delivery of the plasmid to target cells in vivo. These methods include cationic lipid formulations, electrotransfer, and hydrodynamic delivery (28, 40, 49). A recurring problem in these studies has been the low rate of transduction. Hydrodynamic tail vein injection (HTVI) is arguably the most effective nonviral technique for the transduction of hepatocytes in mouse. Transduction efficiencies in liver can be as high as 40% (26). However, this is relatively inefficient compared to the nearly 100% transduction we observed with our adenovirus-based approach. Furthermore, the level of transduction by HTVI decreases as the plasmid size increases, so the benefits of using a genomic transgene are difficult to realize. For example, Stoll et al. (42) observed almost 40-fold fewer copies of a 28-kb plasmid relative to a 5-kb plasmid in the hepatocytes of mice after HTVI. Further, HTVI is mostly limited to the transduction of hepatocytes (16). In contrast, adenovirus has been adapted and retargeted to be capable of robust in vivo transduction of liver (100%) (24), skeletal muscle (99%) (37), myocardiocytes (78%) (38), cardiac endothelium (81%) (4), and a wide variety of cancers (11).
Lastly, most EBV plasmids are made by traditional cloning techniques in prokaryotes and contain bacterially derived DNA. It has been shown that these sequences can dramatically restrict transgene expression. For example, expression levels of human alpha-1 antitrypsin were found to be 560 times higher from a plasmid devoid of bacterial sequences versus one containing them (7). The EBV episome delivered by our hybrid vector lacks bacterial sequences and should benefit from this relative to EBV plasmids manufactured by traditional methods.
In conclusion, while EBV episomes seem promising for safe, long-term expression of therapeutic transgenes in vivo, their application has been constrained by inefficient delivery modalities. Our HDAd-EBV hybrid vector approach leverages the high transduction efficiency of adenovirus-based vectors to mitigate this limitation. We have shown that this method is highly robust in a mouse model for transduction of hepatocytes. Taken as a proof of principle, these studies demonstrate the potential of the HDAd-EBV vector approach.
Acknowledgments
This study was supported by grants R37-CA025235 and T32-GM07104 from the National Cancer Institute, National Institutes of Health. Additional funding was provided by the Warsaw Family Fellowship. Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by NIH awards CA-16042 and AI-28697 and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA. Cryosectioning was performed by the UCLA JCCC Translational Pathology Core Laboratory.
We thank Carol Eng, Jennifer Woo, Michael Balamotis, and RoseMarie Tsoa for excellent technical assistance.
Footnotes
Published ahead of print on 21 January 2009.
REFERENCES
- 1.Berry, M. N., and A. M. Edwards (ed.). 2000. The hepatocyte review. Kluwer Academic Publishers, Boston, MA.
- 2.Bhaumik, S., and S. S. Gambhir. 2002. Optical imaging of Renilla luciferase reporter gene expression in living mice. Proc. Natl. Acad. Sci. USA 99377-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bloquel, C., E. Fabre, M. F. Bureau, and D. Scherman. 2004. Plasmid DNA electrotransfer for intracellular and secreted proteins expression: new methodological developments and applications. J. Gene Med. 6(Suppl. 1)S11-S23. [DOI] [PubMed] [Google Scholar]
- 4.Brauner, R., M. Nonoyama, H. Laks, D. C. Drinkwater, Jr., S. McCaffery, T. Drake, A. J. Berk, L. Sen, and L. Wu. 1997. Intracoronary adenovirus-mediated transfer of immunosuppressive cytokine genes prolongs allograft survival. J. Thorac. Cardiovasc. Surg. 114923-933. [DOI] [PubMed] [Google Scholar]
- 5.Brooks, A. R., R. N. Harkins, P. Wang, H. S. Qian, P. Liu, and G. M. Rubanyi. 2004. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J. Gene Med. 6395-404. [DOI] [PubMed] [Google Scholar]
- 6.Chen, P., J. Tian, I. Kovesdi, and J. T. Bruder. 2008. Promoters influence the kinetics of transgene expression following adenovector gene delivery. J. Gene Med. 10123-131. [DOI] [PubMed] [Google Scholar]
- 7.Chen, Z. Y., C. Y. He, A. Ehrhardt, and M. A. Kay. 2003. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol. Ther. 8495-500. [DOI] [PubMed] [Google Scholar]
- 8.Conese, M., C. Auriche, and F. Ascenzioni. 2004. Gene therapy progress and prospects: episomally maintained self-replicating systems. Gene Ther. 111735-1741. [DOI] [PubMed] [Google Scholar]
- 9.Dorigo, O., J. S. Gil, S. D. Gallaher, B. T. Tan, M. G. Castro, P. R. Lowenstein, M. P. Calos, and A. J. Berk. 2004. Development of a novel helper-dependent adenovirus-Epstein-Barr virus hybrid system for the stable transformation of mammalian cells. J. Virol. 786556-6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehrhardt, A., and M. A. Kay. 2002. A new adenoviral helper-dependent vector results in long-term therapeutic levels of human coagulation factor IX at low doses in vivo. Blood 993923-3930. [DOI] [PubMed] [Google Scholar]
- 11.Glasgow, J. N., M. Everts, and D. T. Curiel. 2006. Transductional targeting of adenovirus vectors for gene therapy. Cancer Gene Ther. 13830-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hacein-Bey-Abina, S., C. Von Kalle, M. Schmidt, M. P. McCormack, N. Wulffraat, P. Leboulch, A. Lim, C. S. Osborne, R. Pawliuk, E. Morillon, R. Sorensen, A. Forster, P. Fraser, J. I. Cohen, G. de Saint Basile, I. Alexander, U. Wintergerst, T. Frebourg, A. Aurias, D. Stoppa-Lyonnet, S. Romana, I. Radford-Weiss, F. Gross, F. Valensi, E. Delabesse, E. Macintyre, F. Sigaux, J. Soulier, L. E. Leiva, M. Wissler, C. Prinz, T. H. Rabbitts, F. Le Deist, A. Fischer, and M. Cavazzana-Calvo. 2003. LMO2-associated clonal T-cell proliferation in two patients after gene therapy for SCID-X1. Science 302415-419. [DOI] [PubMed] [Google Scholar]
- 13.Harui, A., S. Suzuki, S. Kochanek, and K. Mitani. 1999. Frequency and stability of chromosomal integration of adenovirus vectors. J. Virol. 736141-6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatfield, L., and P. Hearing. 1991. Redundant elements in the adenovirus type 5 inverted terminal repeat promote bidirectional transcription in vitro and are important for virus growth in vivo. Virology 184265-276. [DOI] [PubMed] [Google Scholar]
- 15.He, J., T. Wang, L. Yao, A. Chen, B. Zhou, H. Yu, R. Jia, C. Cheng, L. Huan, and Z. Zhang. 2006. Construction and delivery of gene therapy vector containing soluble TNFα receptor-IgGFc fusion gene for the treatment of allergic rhinitis. Cytokine 36296-304. [DOI] [PubMed] [Google Scholar]
- 16.Herweijer, H., and J. A. Wolff. 2007. Gene therapy progress and prospects: hydrodynamic gene delivery. Gene Ther. 1499-107. [DOI] [PubMed] [Google Scholar]
- 17.Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26365-369. [DOI] [PubMed] [Google Scholar]
- 18.Kochanek, S., P. R. Clemens, K. Mitani, H. H. Chen, S. Chan, and C. T. Caskey. 1996. A new adenoviral vector: replacement of all viral coding sequences with 28 kb of DNA independently expressing both full-length dystrophin and beta-galactosidase. Proc. Natl. Acad. Sci. USA 935731-5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreppel, F., and S. Kochanek. 2004. Long-term transgene expression in proliferating cells mediated by episomally maintained high-capacity adenovirus vectors. J. Virol. 789-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krougliak, V. A., N. Krougliak, and R. C. Eisensmith. 2001. Stabilization of transgenes delivered by recombinant adenovirus vectors through extrachromosomal replication. J. Gene. Med. 351-58. [DOI] [PubMed] [Google Scholar]
- 21.Krysan, P. J., S. B. Haase, and M. P. Calos. 1989. Isolation of human sequences that replicate autonomously in human cells. Mol. Cell. Biol. 91026-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leblois, H., C. Roche, N. Di Falco, C. Orsini, P. Yeh, and M. Perricaudet. 2000. Stable transduction of actively dividing cells via a novel adenoviral/episomal vector. Mol. Ther. 1314-322. [DOI] [PubMed] [Google Scholar]
- 23.Levitskaya, J., M. Coram, V. Levitsky, S. Imreh, P. M. Steigerwald-Mullen, G. Klein, M. G. Kurilla, and M. G. Masucci. 1995. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 375685-688. [DOI] [PubMed] [Google Scholar]
- 24.Li, Q., M. A. Kay, M. Finegold, L. D. Stratford-Perricaudet, and S. L. Woo. 1993. Assessment of recombinant adenoviral vectors for hepatic gene therapy. Hum. Gene Ther. 4403-409. [DOI] [PubMed] [Google Scholar]
- 25.Li, Z., J. Dullmann, B. Schiedlmeier, M. Schmidt, C. von Kalle, J. Meyer, M. Forster, C. Stocking, A. Wahlers, O. Frank, W. Ostertag, K. Kuhlcke, H. G. Eckert, B. Fehse, and C. Baum. 2002. Murine leukemia induced by retroviral gene marking. Science 296497. [DOI] [PubMed] [Google Scholar]
- 26.Liu, F., Y. Song, and D. Liu. 1999. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 61258-1266. [DOI] [PubMed] [Google Scholar]
- 27.Loser, P., G. S. Jennings, M. Strauss, and V. Sandig. 1998. Reactivation of the previously silenced cytomegalovirus major immediate-early promoter in the mouse liver: involvement of NF-κB. J. Virol. 72180-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mei, W. H., G. X. Qian, X. Q. Zhang, P. Zhang, and J. Lu. 2006. Sustained expression of Epstein-Barr virus episomal vector mediated factor VIII in vivo following muscle electroporation. Haemophilia 12271-279. [DOI] [PubMed] [Google Scholar]
- 29.Morral, N., W. O'Neal, K. Rice, M. Leland, J. Kaplan, P. A. Piedra, H. Zhou, R. J. Parks, R. Velji, E. Aguilar-Cordova, S. Wadsworth, F. L. Graham, S. Kochanek, K. D. Carey, and A. L. Beaudet. 1999. Administration of helper-dependent adenoviral vectors and sequential delivery of different vector serotype for long-term liver-directed gene transfer in baboons. Proc. Natl. Acad. Sci. USA 9612816-12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morsy, M. A., M. Gu, S. Motzel, J. Zhao, J. Lin, Q. Su, H. Allen, L. Franlin, R. J. Parks, F. L. Graham, S. Kochanek, A. J. Bett, and C. T. Caskey. 1998. An adenoviral vector deleted for all viral coding sequences results in enhanced safety and extended expression of a leptin transgene. Proc. Natl. Acad. Sci. USA 957866-7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nanbo, A., A. Sugden, and B. Sugden. 2007. The coupling of synthesis and partitioning of EBV's plasmid replicon is revealed in live cells. EMBO J. 264252-4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okuyama, T., R. M. Huber, W. Bowling, R. Pearline, S. C. Kennedy, M. W. Flye, and K. P. Ponder. 1996. Liver-directed gene therapy: a retroviral vector with a complete LTR and the ApoE enhancer-α1-antitrypsin promoter dramatically increases expression of human α1-antitrypsin in vivo. Hum. Gene Ther. 7637-645. [DOI] [PubMed] [Google Scholar]
- 33.Palmer, D., and P. Ng. 2003. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 8846-852. [DOI] [PubMed] [Google Scholar]
- 34.Parks, R. J., L. Chen, M. Anton, U. Sankar, M. A. Rudnicki, and F. L. Graham. 1996. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. USA 9313565-13570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porter, D. D., I. Wimberly, and M. Benyesh-Melnick. 1969. Prevalence of antibodies to EB virus and other herpesviruses. JAMA 2081675-1679. [PubMed] [Google Scholar]
- 36.Puntel, M., J. F. Curtin, J. M. Zirger, A. K. Muhammad, W. Xiong, C. Liu, J. Hu, K. M. Kroeger, P. Czer, S. Sciascia, S. Mondkar, P. R. Lowenstein, and M. G. Castro. 2006. Quantification of high-capacity helper-dependent adenoviral vector genomes in vitro and in vivo, using quantitative TaqMan real-time polymerase chain reaction. Hum. Gene Ther. 17531-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sapru, M. K., K. M. McCormick, and B. Thimmapaya. 2002. High-efficiency adenovirus-mediated in vivo gene transfer into neonatal and adult rodent skeletal muscle. J. Neurosci. Methods 11499-106. [DOI] [PubMed] [Google Scholar]
- 38.Sasano, T., K. Kikuchi, A. D. McDonald, S. Lai, and J. K. Donahue. 2007. Targeted high-efficiency, homogeneous myocardial gene transfer. J. Mol. Cell Cardiol. 42954-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schiedner, G., N. Morral, R. J. Parks, Y. Wu, S. C. Koopmans, C. Langston, F. L. Graham, A. L. Beaudet, and S. Kochanek. 1998. Genomic DNA transfer with a high-capacity adenovirus vector results in improved in vivo gene expression and decreased toxicity. Nat. Genet. 18180-183. [DOI] [PubMed] [Google Scholar]
- 40.Sclimenti, C. R., A. S. Neviaser, E. J. Baba, L. Meuse, M. A. Kay, and M. P. Calos. 2003. Epstein-Barr virus vectors provide prolonged robust factor IX expression in mice. Biotechnol. Prog. 19144-151. [DOI] [PubMed] [Google Scholar]
- 41.Sears, J., M. Ujihara, S. Wong, C. Ott, J. Middeldorp, and A. Aiyar. 2004. The amino terminus of Epstein-Barr virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J. Virol. 7811487-11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stoll, S. M., C. R. Sclimenti, E. J. Baba, L. Meuse, M. A. Kay, and M. P. Calos. 2001. Epstein-Barr virus/human vector provides high-level, long-term expression of α1-antitrypsin in mice. Mol. Ther. 4122-129. [DOI] [PubMed] [Google Scholar]
- 43.Tan, B. T., L. Wu, and A. J. Berk. 1999. An adenovirus-Epstein-Barr virus hybrid vector that stably transforms cultured cells with high efficiency. J. Virol. 737582-7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao, N., G. P. Gao, M. Parr, J. Johnston, T. Baradet, J. M. Wilson, J. Barsoum, and S. E. Fawell. 2001. Sequestration of adenoviral vector by Kupffer cells leads to a nonlinear dose response of transduction in liver. Mol. Ther. 328-35. [DOI] [PubMed] [Google Scholar]
- 45.Thorley-Lawson, D. A., and G. J. Babcock. 1999. A model for persistent infection with Epstein-Barr virus: the stealth virus of human B cells. Life Sci. 651433-1453. [DOI] [PubMed] [Google Scholar]
- 46.Tischendorf, P., G. J. Shramek, R. C. Balagtas, F. Deinhardt, W. H. Knospe, G. R. Noble, and J. E. Maynard. 1970. Development and persistence of immunity to Epstein-Barr virus in man. J. Infect. Dis. 122401-409. [DOI] [PubMed] [Google Scholar]
- 47.Umana, P., C. A. Gerdes, D. Stone, J. R. Davis, D. Ward, M. G. Castro, and P. R. Lowenstein. 2001. Efficient FLPe recombinase enables scalable production of helper-dependent adenoviral vectors with negligible helper-virus contamination. Nat. Biotechnol. 19582-585. [DOI] [PubMed] [Google Scholar]
- 48.Yates, J., N. Warren, D. Reisman, and B. Sugden. 1984. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc. Natl. Acad. Sci. USA 813806-3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoo, H. S., O. Mazda, H. Y. Lee, J. C. Kim, S. M. Kwon, J. E. Lee, I. C. Kwon, H. Jeong, Y. S. Jeong, and S. Y. Jeong. 2006. In vivo gene therapy of type I diabetic mellitus using a cationic emulsion containing an Epstein-Barr virus-based plasmid vector. J. Control Release 112139-144. [DOI] [PubMed] [Google Scholar]