

Abstract
Poxviruses express proteins that limit host immune responses to infection. For example, the molluscum contagiosum virus MC160 protein inhibits tumor necrosis factor alpha (TNF-α)-induced NF-κB activation. This event correlates with MC160-induced IKK1 protein degradation, suggesting a mechanism for the above-mentioned phenotype. IKK1 is stabilized when it associates with the cellular heat shock protein 90 (Hsp90). Here, Hsp90 overexpression restored IKK1 levels in MC160-expressing cells, suggesting that MC160 competitively interacted with Hsp90. In support of this, further investigation showed that a mutant MC160 protein comprising only the C-terminal region (C protein) immunoprecipitated with Hsp90. In contrast, Hsp90 IP with a mutant MC160 protein consisting of only the N-terminal tandem death effector domains (DEDs) (N protein) was dramatically decreased. Since cells expressing either the N or C mutant MC160 protein remained similarly resistant to TNF-α-induced NF-κB activation, the N mutant protein probably utilized a different mechanism for inhibiting NF-κB. One likely mechanism for the N protein lies in its association with the DED-containing procaspase-8 protein, a cellular apoptosis precursor protein that regulates NF-κB activation. Here, IPs revealed that this association relied on the presence of the DED-containing N terminus of the MC160 protein but not the C-terminal portion. These interactions appear to have relevance with NF-κB activation, since the expression of the viral DEDs strongly inhibited procaspase-8-mediated NF-κB activation, an event not substantially altered by the C protein. Thus, the MC160 protein utilizes at least two distinct mechanisms for impeding NF-κB activation, association with Hsp90 to result in IKK1 protein degradation or interaction with procaspase-8.
Molluscum contagiosum virus (MCV) is a dermatotropic poxvirus that infects only humans (16). It induces the formation of persistent, benign neoplasms in the skin and is a common infection in children and sexually active young adults. Like other poxviruses, the MCV genome encodes multiple immunoevasion products, neutralizing the effects of cytokines, chemokines, and apoptosis (37). These immunoevasion proteins likely contribute to the persistence of an MCV infection. Unlike better-characterized poxviruses, such as vaccinia virus, MCV has yet to be successfully grown in cell culture. Thus, the functions of MCV immunomodulatory proteins have been identified by expressing MCV open reading frames (ORFs) independent of infection or by surrogate poxvirus systems (15, 39, 52, 58).
Tumor necrosis factor alpha (TNF-α) is a central antiviral immune defense. When bound to one of its receptors, TNF receptor 1 (TNF-R1), it can activate the host NF-κB transcription factor to upregulate the expression of immune response genes (31) and can also induce apoptosis to eliminate virus-infected cells (10). MCV expresses two gene products that modulate TNF-α-induced NF-κB activation, MC160 and MC159 (39, 41). While each viral protein possesses two tandem death effector domains (DEDs; DED1 and DED2), the MC160 product possesses a unique C-terminal region (47). These proteins possess different mechanisms for their NF-κB inhibitory functions: MC160 protein production correlates with a reduction of IKK1 protein levels (see below) (41), an event not observed in MC159-expressing cells (41).
The canonical pathway of TNF-α-induced NF-κB activation is initiated by the binding of TNF-α to TNF-R1, resulting in receptor clustering. Subsequently, the TNF-R-associated death domain protein (TRADD), receptor-interacting protein 1 (RIP1), and TNF-R-associated factor 2 (TRAF2) migrate to the TNF-R1 cytoplasmic tail, forming a signalsome complex (31). Afterwards, the I kappa kinase (IKK) complex, consisting of at least two catalytic subunits (IKK1, IKK2) and an essential regulatory subunit (IKKγ), migrates to the signalsome (reviewed in reference 45). The IKK1 and IKK2 subunits convert to their activated kinase forms and, in turn, phosphorylate IκBα (17, 36). The modified IκBα protein is then polyubiquitinated, resulting in its dissociation from NF-κB and its ultimate degradation by the host 26S proteosome (3, 12, 46). Consequently, the now-exposed nuclear localization signal triggers NF-κB translocation to the nucleus. The p65 subunit of the classical NF-κB dimer is acetylated and phosphorylated (phospho-p65), thereby enabling NF-κB to initiate the transcription of target genes (43).
Proteins other than the classical signalsome components, such as heat shock protein 90 (Hsp90), regulate the IKK complex to affect TNF-R1-induced NF-κB activation (45). For example, Hsp90 is recruited preferentially to IKK1 by the Cdc37 cochaperone (9, 22). Hsp90 is necessary for the stabilization of the IKK1 protein, thereby allowing mature IKK complexes to migrate to the TNF-R1 signalsome (9). Treatment of cells with geldanamycin (GA; an agent that inhibits Hsp90 function) abrogates Hsp90-IKK interactions, resulting in IKK1 subunit degradation and the mitigation of TNF-α-induced NF-κB activation (6, 9).
Procaspase-8 (7, 25), the uncleaved form of the proapoptotic caspase-8 enzyme (40), also affects NF-κB. In cells containing procaspase-8 small interfering RNA, TNF-α exposure no longer activates NF-κB, suggesting that this molecule is involved in the TNF-R1 signaling pathway (28). It is still unclear how procaspase-8 may function on a molecular level to exert its effects. In immune cells, procaspase-8 enzymatically cleaves the cellular FLICE-like inhibitory protein (cFLIPL) into a p43 product, which is further processed to p22, with p22 then binding to and activating IKKγ (21). Thus, one possibility is that procaspase-8 may similarly function to activate the IKK complex during TNF-R1-induced NF-κB activation. Alternatively, during lipopolysaccharide-Toll-like receptor 4 interactions, procaspase-8 coprecipitates with the IKK complex (30). Whether these same interactions are necessary for TNF-R1-induced NF-κB activation is unknown.
Previously, it was observed that MC160 protein expression correlated with IKK1 protein degradation, suggesting that the MC160 protein alters the stability of the IKK complex, thereby preventing TNF-α-induced NF-κB activation (41). However, the mechanism responsible for this degradation was unknown. The IKK1 degradation phenotype of MC160-containing cells is similar to that observed in cells treated with GA (9). Thus, the MC160 protein may alter Hsp90-IKK1 interactions, ultimately altering IKK complex stability to inhibit NF-κB activation. An alternative mechanism for the inhibitory effect of the MC160 protein may be based on its observed interaction with procaspase-8 (51), most likely through their corresponding DEDs, to affect IKK activation and subsequent NF-κB activation.
To query these possibilities, several expression vectors were generated, each carrying different mutated MC160 ORFs that express proteins only containing either one or both of its DEDs or its unique C-terminal region. Since MCV cannot be grown in cell culture, these mutant proteins were instead ectopically expressed in human embryonic kidney 293T (293T) cells and then analyzed for their ability to inhibit TNF-α-induced NF-κB activation based on a luciferase reporter assay. The effect of each altered protein on IKK1 protein stability and on its association with either Hsp90 or procaspase-8, as shown by immunoprecipitation (IP) assays, was also examined. Data demonstrated that both the DED2 and the C-terminal regions inhibited TNF-α-induced NF-κB activation as determined by a luciferase reporter assay. Further, these two regions appear to effect their suppression by using different mechanisms as follows: the MC160 C terminus as a result of its interaction with Hsp90 and the MC160 N terminus region through its association with procaspase-8.
MATERIALS AND METHODS
Cells and plasmids.
The human embryonic kidney 293T cell line was obtained from the American Type Culture Collection and was maintained in Eagle's minimal essential medium supplemented with 10% fetal bovine serum (HyClone), 100 U/ml penicillin, and 100 mg/ml streptomycin. This well-characterized cell line has been utilized by multiple laboratories to study the TNF-R1 signal transduction pathway because it expresses low levels of TNF-R2 and is easily transfectable (8, 23, 24).
Derivatives of the pCI eukaryotic expression vector (Promega) contained either an unmodified MC160 ORF (pMC160), an unmodified MC159 ORF (pMC159), an ORF that encodes the MC160 protein with an influenza virus hemagglutinin (HA) epitope at its amino terminus (pHA-MC160), or a similarly tagged MC159 protein (pHA-MC159); each viral ORF was under the control of a cytomegalovirus promoter as described previously (51). In addition, plasmids that encode either an HA-tagged human Hsp90 (pHA-Hsp90) (60) or a FLAG-labeled human IKK1 (pIKK1) (38) were kindly provided by Chen Wang (Chinese Academy of Sciences) and Ulrich Siebenlist (National Institutes of Health), respectively. pHA-TRAF2 contains cDNA for an HA epitope-tagged TRAF2 molecule (33), while pRIP encodes a nonmodified RIP1 protein (8). We also utilized pProcaspase-8 C360S (51), which contains a mutated form of procaspase-8 that does not induce apoptosis but still activates NF-κB (7, 49). pHA-K1L expresses the vaccinia virus K1L ORF under the control of a cytomegalovirus (CMV) promoter and has been described elsewhere (20).
To ascertain the importance of the MC160 DEDs and the C-terminal region for NF-κB inhibitory function (48), five plasmids, each containing a different mutated MC160 ORF lacking coding sequences for one or more of these domains and under CMV promoter regulation, were created by using recombinant PCR (Fig. 1). pN, which encodes MC160 cDNA for amino acid residues 1 to 220 (Fig. 1), was constructed as follows: MC160 nucleotides 1 to 660 and the HA epitope tag at its N terminus were PCR amplified from pHA-MC160 using primers NFOR (5′-CGAGAATTCGCCACCATGTATCCA-3′) and NREV (5′-CCGTCCGCTCTAGATTACCCCGCGGA-3′). The NFOR and NREV oligonucleotides possessed the EcoRI and XbaI restriction enzymes, respectively (underlined DNA sequences). The resultant purified amplicon was restricted with EcoRI and XbaI and subsequently inserted into a similarly restricted pCI.
FIG. 1.

Representation of WT and mutant MC160 proteins. Schematic representation of the 371-amino-acid WT MC160 protein, with its two tandem DEDs at residues 5 to 79 (DED1) and 97 to 175 (DED2) and its C-terminal region (residues 175 to 371). Below the WT MC160 protein diagram are illustrations of each of the mutant MC160 proteins, labeled N, C, D1, D2, and D2C, with the numbers above each construct indicating the first and last residues present. An HA epitope tag was introduced into the N terminus of each protein.
pC contained the MC160 C-terminal ORF (amino acids 224 to 371), thereby eliminating the expression of both N-terminal DEDs (Fig. 1). It was created in a manner similar to pN. Specifically, 441 bp of the MC160 ORF was PCR amplified from pHA-MC160 using primers CFOR (5′-CCGTATGACGTGCCGGACTATGCCGCGGACGGGGCT-3′) and CREV (5′-CGTCTAGACGCTCGCTAGTAGG-3′), with the sequence for the part of the HA epitope tag underlined. Next, DNA encoding the remainder of the HA epitope tag was engineered onto the N terminus of the purified MC160 amplicon by a second, sequential PCR amplification that utilized oligonucleotides CREV and CFOR2 (5′-TCGAATTCGCCACCATGGCGTACCCGTATGACGTG-3′); the HA epitope sequence in CFOR2 is underlined. The CFOR2 and CREV primers also possessed EcoRI and XbaI restriction sites, respectively, thereby enabling the digestion of the purified second amplicon with these enzymes and subsequent ligation into the corresponding restriction sites of pCI. The pC2 plasmid contained an MC160 mutant protein consisting of amino acid residues 168 to 371. This construct also produces an HA epitope-tagged protein. Primer sequences used to amplify this C2 ORF from the wild-type (WT) MC160 ORF are 5′-CCGAATTCGCCACCATGTATCCATATCATCTTCCAGACTATGCTTCGGCAGTAGCACATGGGCAC-3′ and 5′-CGTCTAGACGCTCGCTACTAGG-3′. The EcoRI or XbaI restriction enzyme cleavage sites present are underlined, respectively. PCR amplicons were subsequently digested with the restriction enzymes and cloned into the EcoRI and XbaI sites in the pCI vector.
Three other pCI constructs were created that contained, under CMV promoter control, altered MC160 ORF sequences consisting of amino acid residues 1 to 74 (pD1), 83 to 220 (pD2), or 83 to 371 (pD2C) (Fig. 1). For pD1 (encoded only DED1), MC160 nucleotides 1 to 222 were PCR amplified from pHA-MC160, utilizing primers D1FOR (5′-GAGAATTCGCCACCATGTATCCATATGATG-3′), which contains sequences for an EcoRI restriction site (underlined) and HA epitope, and D1REV (5′-CGCGCGTTCTAGACTAGCGGACC-3′), which possesses an XbaI restriction site (underlined). Thus, after the amplicons were digested with EcoRI and XbaI, they were unidirectionally cloned into their respective restriction sites of pCI. pD2C (encoded DED2 and C terminus) was created by PCR amplification of an 864-bp portion of the MC160 ORF from pHA-MC160, using primers D2CFOR (5′-GAATTCGCCACCATGTATCCATATGATGTTCCAGACTATGCTGGGCGCTTGCTGGGCACG-3′) and D2CREV (5′-CGTCTAGACGCTCGCTAGTAGG-3′). EcoRI and XbaI restriction sites were present in the D2CFOR and D2CREV primers, respectively (underlined), as well as an HA epitope sequence in D2CFOR. Next, gel-purified, restriction enzyme-digested amplicons were inserted into the sites of a similarly restricted pCI. To create pD2 (encoded only DED2), pD2C was digested with EcoRI and SfoI, and the resultant gel-purified, 411-bp fragment was then ligated into the similarly restricted pCI. For all plasmids, sequence analysis of representative plasmids demonstrated the presence of the N-terminal HA epitope tag and the absence of the appropriate regions of the WT MC160 ORF.
Luciferase reporter assays.
The luciferase reporter assay quantifying TNF-α-induced NF-κB activation in MC160 protein-expressing cells was described previously (41). Briefly, subconfluent 293T cell monolayers in 6-well plates were cotransfected with pNF-κBluc (450 ng), pRL-null (50 ng), and 1,500 ng of either pCI, pHA-MC160, pN, pC, pD1, pD2, or pD2C using the FuGene 6 transfection reagent (Roche) as per the manufacturer's instructions. At 24 h posttransfection, cellular monolayers were incubated with fresh medium lacking or containing TNF-α (10 ng/ml medium; Roche). After an additional 4 h, medium was removed, and the cellular monolayers were separately harvested and lysed in passive lysis buffer (Promega) according to the manufacturer's conditions. In an additional experiment using IKK1 overexpression to activate NF-κB, 293T cells were cotransfected with 450 ng pNF-κBluc, 50 ng pRL-null, 500 ng pIKK1, and 500 ng of either pCI, pHA-MC160, pN, pC, pD1, pD2C, or pD2. An additional experiment instead uses 1,000 ng pC or pC2. As a control, a separate set of cells was transfected with 1,000 ng pCI, 450 ng pNF-κBluc, and 50 ng pRL-null and incubated in regular medium. For this set of experiments, 293T cell monolayers were harvested at 16 h posttransfection as described above. In addition, in an experiment in which procaspase-8 C360S overexpression induced NF-κB activation, 293T cells were cotransfected with 225 ng pNF-κBluc, 25 ng pRL-null, 250 ng pProcaspase-8 C360S, and 750 ng of either pCI, pHA-MC160, pN, pC, pD1, or pD2. As a control, a separate set of cells was transfected with 1,000 ng pCI, 225 ng pNF-κBluc, and 25 ng pRL-null. For this experiment, medium was removed at 12 h posttransfection, and cellular monolayers were separately processed as described above. For all experiments, lysates were assayed for sea pansy and firefly luciferase activities by using the Dual-Luciferase reporter assay (Promega). Luciferase activity was measured as relative light units by using the Luminoskan luminometer. All assays were performed in triplicate. For each experimental point, firefly luciferase activity was divided by sea pansy luciferase activity to correct for differences in transfection efficiencies. The resultant ratios were normalized to those of the appropriate control cells, consisting of cells cotransfected with pCI, pNF-κBluc, and pRL-null, and incubated in regular medium, whose value was taken as 1. Results were displayed as relative change in luciferase activity compared to that of pCI-transfected cells. Statistical significance was determined by using Student's t test. Lysates from these luciferase assays also were analyzed for MC160 protein levels by immunoblotting (see below).
Gel mobility shift assays.
For gel mobility shift assays to detect NF-κB nuclear translocation, subconfluent 293T cellular monolayers in 6-well plates were transfected with 1,000 ng of the indicated MC160-containing plasmids. At 24 h postinfection, cells were treated with TNF (10 ng/ml) for 120 min. Next, cells were harvested and lysed according to a previous description (41). Nuclei were isolated from cellular lysates and disrupted in nuclear extraction buffer containing Halt protease inhibitors. Two micrograms of nuclear-extracted proteins from each sample were incubated with 0.35 pmol of 32P-labeled double-stranded oligonucleotides containing binding sites for the NF-κB transcription factor (Promega) in gel shift assay system binding buffer (Promega) as per the manufacturer's directions. Some reactions also included excess molar amounts of nonradiolabeled oligonucleotides that either contained or lacked NF-κB binding sites. Alternatively, in some reactions, 1 μg of monoclonal anti-p65 antiserum (Santa Cruz Biotechnology) was present. Following incubation, reactions were resolved electrophoretically in a 6% acrylamide gel (Invitrogen) under nondenaturing conditions. Dried gels were exposed to a PhosphorImager plate (Molecular Devices), and images were developed and analyzed using the Image Gauge and Image Reader programs, respectively (Fuji).
IP assays.
For IP assays to detect MC160 interactions with the various factors, subconfluent 293T cellular monolayers in 6-well plates were transfected with the following plasmid combinations. To test for MC160 interactions with endogenous Hsp90, 293T cells were transfected with 1 μg of either pCI, pHA-MC160, pN, pC, pD1, pD2, or pHA-K1L. For detection of procaspase-8, 293T cells were cotransfected with 1 μg pProcaspase-8 C360S and 1 μg of either pCI, pHA-MC160, pN, pC, pD1, or pD2. For TRAF2, 293T cells were cotransfected with 1 μg pHA-TRAF2 and 1 μg of either pMC160 or pMC159, while for RIP, cells were cotransfected with 1 μg pRIP and 1 μg of either pCI, pMC160, or pHA-TRAF2. For all transfections, FuGene 6 transfection reagent (Roche) was utilized as per the manufacturer's recommendations. For all experiments, at 16 h posttransfection, cells were harvested by scraping and pelleted by centrifugation at 10,000 × g for 10 s. Next, cells were lysed in 500 μl DED IP lysis buffer (51) containing Halt protease inhibitors (Pierce) during a 30-min incubation at 4°C, after which the lysates were centrifuged at 10,000 × g at 4°C for 10 min, and the supernatants were transferred to fresh tubes (51). Approximately 50 μl of each clarified lysate was added to new tubes containing 5× Laemmli buffer (Pierce) and 5% 2-mercaptoethanol. For each sample, the remaining 450 μl of clarified lysate was incubated with Protein G Sepharose beads (Amersham) that had been previously been bound to either anti-HA antibodies (Sigma Aldrich) or anti-RIP antibodies (Santa Cruz Biotechnology) as per the manufacturer's recommendations. For the MC160-Hsp90 IPs, the HA-MC160 protein was immunoprecipitated with an HA affinity matrix (Roche) according to the manufacturer's directions. After a rotating incubation for 1 h at 4°C, the samples were briefly centrifuged (1,000 × g for 30 s) to collect the protein-bound beads. The beads were washed three times in large volumes (500 μl) of DED IP lysis buffer and then suspended in 30 μl 2× Laemmli buffer (Pierce) containing 5% 2-mercaptoethanol. Both the clarified lysates and immunoprecipitated samples were boiled for 5 min, briefly cooled on ice, and then subjected to sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis (SDS-12% PAGE). Next, the separated proteins were electrophoretically transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore), and the blots were incubated with either rabbit anti-MC160 antiserum (51), rabbit anti-MC159 antiserum (51), rabbit anti-TRAF2 antiserum (Santa Cruz Biotechnology), rabbit anti-Hsp90 antiserum (Cell Signaling), rabbit anti-RIP (BD Transduction), mouse anti-HA, or rabbit anti-human caspase-8 antiserum (R & D Systems) in Tris-buffered saline (pH 7.0) containing 0.5% Tween 20 (TBS-T). The anti-caspase-8 antibody was specific for residues 294 to 391 and, therefore, detects both the precursor (55-kDa) and the cleaved (43-kDa) forms of caspase-8. After subsequent incubation with either horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG; Pierce) or HRP-conjugated goat anti-mouse IgG (Pierce), antigen-antibody reactions on each immunoblot (IB) were detected by using chemiluminescence and autoradiography. For all IBs except for the Hsp90 IBs, the Pierce SuperSignal West Pico substrate was utilized according to the manufacturer's protocols; for the Hsp90 IBs, the Pierce West Femto reagent was used according to the manufacturer's protocols.
Immunoblotting.
For experiments involving the detection of IKK1 proteins, subconfluent 293T monolayers in 6-well plates were cotransfected with 500 ng pIKK1 and 1,500 ng of either pCI, pHA-MC160, or pHA-MC159. In an additional experiment, 293T cells were instead transfected with 500 ng pIKK1 and 1,500 ng of either pCI, pHA-MC160, pN, pC, pD1, pD2, or pD2C. For these experiments, transfections were performed using FuGene 6 (Roche). At 16 h posttransfection, cells were incubated in a whole-cell NP-40 lysis buffer (39) containing sodium vanadate (1 mM; Fisher), sodium fluoride (10 mM; Fisher), and dithiothreitol (1 mM; Fisher) for 30 min on ice. Next, the lysates were centrifuged for 10 min at 10,000 × g, and supernatants were removed to new tubes. In experiments in which HA-Hsp90 was expressed ectopically, 293T cells were cotransfected with 500 ng pIKK1, 500 ng pHA-MC160 or pC, and either 100 or 250 ng pHA-Hsp90. Control cells were transfected as described above but with 500 ng pCI substituted for pHA-MC160. For this assay, cellular monolayers were harvested 16 h posttransfection as described above and subsequently lysed in cytoplasmic extraction buffer (42) containing Halt protease inhibitors (Pierce) for 20 min at 4°C. For the additional experiment detecting WT and mutant MC160 proteins, 293T cellular lysates generated from luciferase assays (see above) were utilized.
The protein concentration for each cytoplasmic extract, including those generated for luciferase assays, was determined using a bicinchoninic acid assay (Pierce). Approximately 20 μg of protein from each sample was mixed with 5× Laemmli buffer for a final 1× concentration (Pierce) that contained 5% 2-mercaptoethanol, boiled for 5 min, incubated briefly on ice, and then separated by SDS-12% PAGE. Next, the proteins were electrophoretically transferred to a PVDF membrane (Millipore). Membranes were incubated in TBS-T containing 5% nonfat milk (Fisher Scientific) for at least 1 h at 4°C. Next, blots were incubated with antiserum containing either rabbit anti-IKK1 antibodies (Santa Cruz Biotechnology), rabbit anti-phospho-p65 antibodies recognizing serine residue 536 (Cell Signaling), mouse anti-p65 antibodies (Cell Signaling), mouse anti-HA antibodies (Sigma-Aldrich), rabbit antiactin antibodies (Sigma-Alrich), or rabbit anti-MC160 antibodies (51). After being washed three times with TBS-T to remove unbound antibodies, IBs were incubated with either HRP-conjugated goat anti-rabbit IgG (Fisher Scientific) or HRP-conjugated goat anti-mouse IgG (Fisher Scientific) diluted in TBS-T. Blots were washed as described before to remove unbound antibodies, and antibody-antigen reactions were detected by using the SuperSignal West Pico chemiluminescence reagents (Pierce) as per the manufacturer's recommendations. The IBs that detect IKK1 proteins were routinely reprobed for actin proteins. IBs that detect phospho-p65 were incubated in Restore Western blot stripping buffer (Pierce) to remove bound antibodies as per the manufacturer's directions before being incubated with anti-p65 antibodies.
RESULTS
The C-terminal and DED2 regions of MC160 individually inhibit TNF-α-induced NF-κB activation in 293T cells.
The 371-amino-acid MC160 protein possesses two tandem DEDs in its N-terminal region (residues 5 to 79 comprise DED1 and residues 97 to 175 comprise DED2) and a unique C-terminal region comprised of residues 175 to 371 (Fig. 1) (48). To ascertain the region(s) of the MC160 protein responsible for its ability to alter TNF-α-mediated NF-κB activation (41), mutant MC160 constructs were created (Fig. 1) and inserted into eukaryotic expression vectors. For all constructs, an HA epitope tag was introduced onto the N terminus.
While the predicted molecular mass of an HA epitope-tagged MC160 product is 39 kDa, a 55-kDa product was instead detected from pHA-MC160-expressing cells (Fig. 2). This same observation was reported previously (41, 51). Additional, lower-molecular-weight bands were also detected when analyzing pHA-MC160-transfected lysates. These bands most likely represent as-yet uncharacterized cleaved MC160 products and were observed previously (51). Plasmid pN encoded sequences for both DEDs and a small portion of the C terminus. While the predicted molecular mass for the N protein was 24 kDa, it was instead detected as a 27-kDa protein from pN-transfected cell lysates (Fig. 2B). pC, which contained sequences for amino acids 224 to 371, was created to test the function of the C terminus, a region of the MC160 protein that lacks homology to any known proteins. Its predicted molecular mass was 15 kDa, but its mobility was consistent with a protein of 28 kDa (Fig. 2B).
FIG. 2.

The effect of WT and mutant MC160 proteins on TNF-α-induced NF-κB activation. (A and C) Subconfluent 293T cells were cotransfected with 450 ng pNF-κBluc, 50 ng pRL-null, and either 1,500 ng pCI or 1,500 ng of the following plasmids: pHA-MC160, pN, or pC (A), or pHA-MC160, pD1, pD2, or pD2C (C). At 24 h posttransfection, cellular monolayers were incubated for 4 h in medium lacking [(−)] or containing [(+)] TNF-α (10 ng/ml). After this additional incubation time, the medium was removed, cellular monolayers were lysed, and the lysates were analyzed for firefly and sea pansy luciferase activities. All resultant values were normalized to pCI-transfected cells not treated with TNF-α. An asterisk indicates statistically significant inhibition (P < 0.05). (B and D) 20 μg of each cellular lysate was resolved by SDS-12% PAGE, and the proteins were transferred to a PVDF membrane. The blots were incubated with anti-HA antiserum, and antibody reactions were subsequently detected by using chemiluminescence. Bands indicative of the sizes of WT or mutant (N, C, D1, D2, and D2C) proteins are indicated by corresponding labels on the right side of the panel. The relative positions of the molecular weight markers (MW) are indicated on the left side of the panel. (E) Subconfluent 293T cell monolayers were transfected with 1,000 ng of each of the indicated plasmids. At 24 h posttransfection, cells were incubated in medium either lacking or containing TNF (10 ng/ml). Two hours later, cells were harvested and lysed. Nuclear-extracted proteins were incubated with radiolabeled oligonucleotides containing NF-κB binding sites. For some reactions in which nuclear extracts from TNF-treated, pCI-transfected cells were collected, anti-NF-κB (p65) antiserum or an excess of nonradiolabeled oligonucleotides either containing (cold comp) or lacking (nonsp. comp) NF-κB binding sites was present. Reactions were then analyzed by gel electrophoresis under nondenaturing conditions. Gels were dried, and images were obtained by using phosphorimaging. *, NF-κB-containing band; **, supershifted NF-κB-containing band.
To assess the function of the individual DEDs, plasmid pD1 (DED1, expressing residues 1 to 74) and plasmid pD2 (expressing DED2 and 45 residues of the C terminus; residues 83 to 220) were constructed. The D1 and D2 proteins were predicted to be 8 and 14 kDa, respectively. However, when lysates from either pD1- or pD2-transfected cells were immunoblotted with anti-HA antiserum, 10-kDa and 17-kDa proteins were instead detected (Fig. 2D). Finally, pD2C, which encoded residues 83 to 371, was constructed to assess the function of an MC160 protein deleted from DED1. Its predicted molecular mass was 29 kDa. As seen in Fig. 2D, immunoblotting lysates from cells transfected with pD2C instead yielded a 45-kDa band.
The effect of each mutant MC160 protein compared to that of the WT on TNF-α-mediated induction of an ectopic firefly luciferase gene was assessed. Since this reporter gene is transcriptionally regulated by NF-κB, its expression is reflective of an active form of NF-κB in transfected cells. In control cells (pCI transfected) incubated in medium without TNF-α, the level of luciferase activity was low, indicating that NF-κB was inactive (Fig. 2A). However, luciferase activity levels were significantly increased when control cells were instead treated with TNF-α, presumably due to the activation of NF-κB (Fig. 2A). As in a previous report (41), the MC160 protein hampered this activation since there were significantly lower luciferase activity levels in pHA-MC160-transfected cells (Fig. 2A). It appeared that both MC160 regions could independently inhibit TNF-α-induced NF-κB activation; significant reductions in luciferase activity levels were observed when assaying pN- and pC-transfected cells (Fig. 2A). The observation that the mutant MC160 protein consisting of only the N-terminal tandem DEDs (N protein) and the mutant MC160 protein comprising only the C-terminal region (C protein) were expressed in levels that were similar to that of WT was also notable (Fig. 2B).
To identify the DED responsible for the inhibition of NF-κB, TNF-α-mediated luciferase activity was measured in cells transfected with either pD1 (DED1 coding region) or pD2 (DED2 coding region). The DED1 appeared to lack an NF-κB inhibitory function since TNF-α-induced luciferase activity levels in pD1-transfected cells were equal to or greater than those observed in vector-transfected cells (Fig. 2C). In contrast, significant reductions in luciferase activities were observed in pD2-transfected cells receiving TNF-α treatment, inferring that the expression of the DED2 was sufficient to prevent NF-κB activation. The addition of the C-terminal portion of the protein to the DED2, resulting in the 45-kDa D2C protein, maintained the inhibitory effect of the DED2 region, as would be expected (Fig. 2C). D1, D2, and D2C protein expression was detected by immunoblotting, and all three mutant proteins were expressed in amounts lower than that of WT MC160 (Fig. 2D). One argument is that the D1 lack of function could be due to low protein expression. However, the D2 protein, which is expressed at similarly low levels, possessed inhibitory effects, suggesting that low protein expression is not correlative to loss of function. Nevertheless, the D2 and D2C proteins exhibited an inhibitory phenotype despite their lower expression levels. It was also observed that luciferase activity levels differed between the experiments (Fig. 2A and 2C), exemplifying the typical variation seen with this assay. Regardless, the WT protein consistently inhibited TNF-α-induced luciferase activity by approximately 2.5-fold.
A hallmark of NF-κB activation is the translocation of this transcription factor from the cytoplasm to the nucleus. Based on the results from the luciferase assays, we predicted that TNF-induced NF-κB nuclear translocation would be inhibited in cells where MC160 proteins reduced TNF-medicated luciferase activity. To test this hypothesis, nuclear-extracted proteins from TNF-treated, MC160-expressing cells were incubated with radiolabeled oligonucleotides possessing NF-κB binding sites. These reactions were analyzed by gel electrophoresis under nondenaturing conditions to detect protein-oligonucleotide complexes. As shown in Fig. 2E, a unique band was detected in lanes containing extracts from TNF-treated, pCI-transfected cells. This band was confirmed to contain NF-κB, since this band's mobility supershifted if anti-NF-κB antibodies were added to the reaction. Like previous reports (41), MC160 protein expression decreased TNF-induced nuclear translocation, as evidenced by a dramatic decrease in the intensity of the NF-κB-containing band. A similar decrease was also observed in cells expressing the N, C, D2, or D2C proteins but not in cells expressing the D1 proteins. We interpret this result to mean that the reason that the MC160 WT and mutant proteins inhibit TNF-induced luciferase activity is because NF-κB is not active (nuclearly localized) in these cells.
The effect of various mutant MC160 proteins on IKK1 degradation and its function.
Concurring with a previous publication (41), ectopic IKK1 protein levels were no longer detectable in MC160-expressing 293T cells, compared to IKK1 protein levels either in cells expressing the similar MC159 protein or in control cells (Fig. 3A). The IKK1 protein mediates the phosphorylation of the NF-κB p65 subunit at serine residue 536, an event necessary for NF-κB transcriptional activation (44, 53). Thus, it was predicted that MC160-expressing cells would no longer yield phospho-p65 in response to events such as IKK1 overexpression or TNF-α treatment. To test this possibility, p65 phosphorylation was detected by probing blotted cellular lysates with antiserum specific for the phosphorylated serine-536 form of p65. As shown in Fig. 3A, the phospho form of p65 was detected readily in cells overexpressing IKK1, a situation in which NF-κB is active (41). In comparison, in cells coexpressing MC160 and IKK1, the phospho-p65 signal was reduced, concomitant with a decrease in ectopic IKK1 protein levels. This decrease in the phospho-p65 and IKK1 signals was not due to global protein degradation, since similar amounts of unmodified p65 and actin proteins were detected in all samples. The overexpression of the similar MCV MC159 product did not significantly diminish cellular levels of the phospho-65 or IKK1 proteins, as previously published (41), indicating that this phenotype is unique to the MC160 protein. Importantly, similar results were observed if p65 phosphorylation was instead triggered by the addition of TNF-α to transfected cells; phospho-p65 was decreased in pHA-MC160-transfected cells compared to that in pCI-transfected cells (data not shown).
FIG. 3.

The effect of WT and mutant MC160 proteins on IKK1 and phospho-p65 protein levels and on IKK1-induced NF-κB activation. (A) 293T cells were cotransfected with 500 ng pIKK1 and 1,500 ng of either pHA-MC160 or pHA-MC159. At 16 h posttransfection, cells were harvested by scraping, collected by centrifugation, and then lysed in NP-40 lysis buffer. Cytoplasmic extracts (20 μg per lane) were resolved by SDS-12% PAGE, and proteins were electrophoretically transferred to a PVDF membrane. The blots were incubated with the indicated antibodies and developed by using chemiluminescence. A closed arrowhead denotes the position of the HA-MC160 protein, whereas an open arrowhead indicates the position of the HA-MC159 protein. The positions of the molecular weight markers (MW) are indicated on the left side of the panel. −, lacking; +, containing. (B) 293T cells were cotransfected with 500 ng pIKK1 and 1,500 ng of the indicated plasmids. At 16 h posttransfection, cells were harvested and processed as in panel A. Blots were incubated with the indicated antibodies, and the IBs were subsequently developed using chemiluminescence. For the anti-HA IB showing MC160 proteins, bands indicative of the sizes of WT or mutant (N, C, D1, D2, and D2C) proteins are indicated by corresponding labels on the right side of the panel. (C) Subconfluent 293T cells were cotransfected with 450 ng pNF-κBluc, 50 ng pRL-null, 500 ng pIKK1, and 500 ng of either pCI, pHA-MC160, pN, pC, pD1, pD2, or pD2C. Control cells were instead cotransfected with 450 ng pNF-κBluc, 50 ng pRL-null, and 1,000 ng pCI. At 16 h posttransfection, cellular monolayers were lysed, and lysates were analyzed for firefly and sea pansy luciferase activities. All resultant values were normalized to those of cells lacking pIKK1. An asterisk indicates statistically significant inhibition (P < 0.05). Lysates from these assays were subsequently probed for IKK1 protein levels by immunoblotting, with IKK1 proteins detected by chemiluminescence. (D) Subconfluent 293T cells were cotransfected with 450 ng pNF-κBluc, 50 ng pRL-null, 500 ng pIKK1, and 1,000 ng of either pCI, pHA-MC160, pC, or pC2. Control cells were instead cotransfected with 450 ng pNF-κBluc, 50 ng pRL-null, and 2,000 ng pCI. At 16 h posttransfection, cellular monolayers were lysed, and lysates were analyzed for firefly and sea pansy luciferase activities. All resultant values were normalized to those of cells lacking pIKK1.
When examining the effect of the various mutant MC160 proteins on IKK1 protein levels, the DED2 (D2) and the C-terminal regions were sufficient for the degradation phenotype, as IKK1 proteins were detected only in lysates from either pCI- or pD1-transfected cells (Fig. 3B). Not surprisingly, these same D1-containing lysates possessed the highest amount of phospho-p65 protein compared to that of lysates containing the other MC160 protein forms (Fig. 3B). Although phospho-p65 was still detectable in the pC-transfected cell lysates, its levels were diminished compared to those of pCI- or pD1-transfected cells. Since similar amounts of unmodified p65 and actin were detected in each reaction, the differences in the phospho-p65 levels were not due to variations in sample loading. Although the same amount in nanograms of MC160-based plasmids was present in each reaction, the MC160D2 and D1 proteins were expressed at lower amounts than those of other WT and mutant MC160 proteins. Despite this trend, the D2 protein still exerted an effect on IKK1 degradation.
Overexpression of the IKK1 protein activates NF-κB. Thus, a second assay was performed to confirm the modulation of each mutant MC160 protein on NF-κB activation triggered by IKK1 overexpression. Using the luciferase reporter assay, ectopic IKK1 expression was shown to activate NF-κB in the control (pCI) cells, as determined by a 140-fold increase in luciferase activity compared to that in cells devoid of pIKK1 (Fig. 3C). The N terminus of the MC160 protein possessed inhibitory activity, a phenotype ascribed to DED2, as IKK1-induced luciferase activity remained low in the pN-, pD2-, and pD2C-transfected cells but not in the pD1-transfected cells (Fig. 3C). Furthermore, the C terminus of the MC160 protein conferred inhibition that was considered significant, albeit to a lesser degree than that for DED2-containing proteins when cells were transfected with equal amounts of pC and pIKK1 plasmids. Inhibition of IKK1-induced luciferase activity was attributed to lower IKK1 protein levels in the MC160, N, C, D2, and D2C lysates. Additionally, MC160 protein levels in these luciferase assays were comparable to those observed for Fig. 2 and 3B (data not shown).
The inhibitory effect of the pC plasmid was more dramatic when cells were instead transfected with an increased amount of pC (1,000 ng) compared to pIKK1 (500 ng) (Fig. 3D). Additionally, a second plasmid, pC2, expressing the C-terminal residues 168 to 371 of the MC160 protein, also inhibited IKK1-induced NF-κB activation (Fig. 3D). This C2 protein, which is 56 residues longer than the C protein, is expressed at levels similar to those of the C protein and also induces IKK1 degradation comparable to that observed in C-expressing cells (data not shown). Thus, the MC160 C-terminal region has an inhibitory effect independent of the DED N-terminal regions.
MC160-induced IKK1 degradation is reversed by overexpression of Hsp90.
The cellular Hsp90 is part of the IKK complex (6, 9, 22). Treatment of cells with GA, a drug that binds to the ATP-binding pocket of Hsp90 (54, 57), prevents Hsp90-IKK1 interactions, resulting in IKK1 degradation (6). Since ectopic MC160 protein expression rendered a phenotype similar to that of GA-treated cells (41), it was postulated that the MC160 protein altered Hsp90 function. If this assumption were correct, then the overexpression of an epitope-tagged Hsp90 in MC160-expressing cells would reverse IKK1 degradation in a dose-dependent manner, an event measurable by immunoblotting. Indeed, IKK1 protein levels were detected in cells cotransfected with pHA-Hsp90 and pHA-MC160 in a dose-dependent manner but not in cells transfected with pHA-MC160 alone (Fig. 4). As would be expected, overexpression of HA-Hsp90 in the absence of MC160 lacked deleterious effects on IKK1 protein levels. Thus, the decrease in IKK1 protein levels was specific to the presence of the MC160 protein and was not due to global protein degradation, as similar amounts of actin were detected in each sample.
FIG. 4.

IKK1 protein levels in cells coexpressing MC160 and Hsp90. Subconfluent 293T cells were cotransfected with 500 ng pIKK1 and 500 ng of either pCI or pHA-MC160. As indicated, some cellular monolayers were simultaneously cotransfected with either 100 ng or 250 ng pHA-Hsp90. At 16 h posttransfection, the cells were collected, lysed, and processed for IB as described before. Blots were incubated with anti-IKK1 antiserum, and the reactive proteins were detected by using chemiluminescence. After being washed, the blot was reprobed with antiactin antibodies, and the reactive proteins were detected as mentioned above. An identical blot was probed with anti-HA antibodies to detect HA-MC160 (closed arrowhead) and HA-Hsp90 (open arrowhead); NS indicates additional, nonspecific proteins recognized by the anti-HA antiserum. The positions of the molecular weight markers (MW) are indicated on the left side of the panel. −, lacking; +, containing.
The C terminus of the MC160 protein immunoprecipitates with endogenous Hsp90.
We posited that MC160 proteins competitively bind to Hsp90 molecules, an event that would presumably disrupt Hsp90-IKK1 interactions and subsequently render IKK1 proteins susceptible to destabilizing conditions. To test this hypothesis, MC160 and associated proteins were immunoprecipitated from pHA-MC160-transfected cells and prepared for immunoblotting with anti-Hsp90 antiserum. The MC160 protein was found to interact with the cellular Hsp90, as determined by the presence of a 90-kDa Hsp90-containing band (Fig. 5A). A band, albeit fainter, of similar mobility was detected in lanes containing immunoprecipitates from vector-only transfected cells (pCI) or from cells overexpressing the vaccinia virus K1L ORF (pHA-K1L), suggesting that MC160-Hsp90 interactions were specific. Similarly, Hsp90 IP was not observed when antibodies lacking reactivity to MC160 were substituted (data not shown). Since similar quantities of Hsp90 were detected in each clarified lysate (Fig. 5A, lysates), ectopic MC160 expression did not appear to affect the endogenous production of Hsp90.
FIG. 5.

The MC160 protein immunoprecipitates with Hsp90. (A and B) Subconfluent 293T cells were transfected with 1 μg of one of the following plasmids: pCI, pHA-MC160, or pHA-K1L (A), or pCI, pHA-MC160, pN, pC, pD1, or pD2 (B). At 16 h posttransfection, cellular monolayers were harvested by scraping, collected by centrifugation, and lysed in DED buffer. A portion of the clarified lysates was incubated for 1 h at 4°C with Protein G Sepharose beads that were conjugated previously to anti-HA. Next, the immunoprecipitated samples and the remaining clarified lysates were individually subjected to SDS-12% PAGE, and the resolved proteins were transferred to a PVDF membrane. Blots were probed with either anti-Hsp90 or anti-HA antibodies, and reactive proteins were detected on each IB by using chemiluminescence. (B) For the IB showing MC160 proteins, bands indicative of the sizes of WT or mutant proteins are indicated by corresponding labels on the right side of the panel. As shown in panel B, right, a longer exposure of the IB revealed the D2 mutant protein (bottom). IgG HC refers to the 50-kDa heavy chain of IgG, while IgG LC refers to the 25-kDa light chain. Positions of the molecular weight markers (MW) are indicated on the left side of the panel. (C) 293T cells were cotransfected with the indicated plasmids: either 100 or 250 ng pHsp90, 1 μg pC or pCI, and 500 ng pIKK1. At 16 h posttransfection, cells were lysed in cytoplasmic extraction buffer, and 20 μg protein from each sample was separated by gel electrophoresis. IBs were incubated with either anti-IKK1, anti-HA, or antiactin antibodies. A double asterisk indicates the phospho form of IKK1, while a single asterisk indicates the unmodified form of this protein. −, lacking; +, containing.
To determine the MC160 region interacting with Hsp90, IP assays were performed using the lysates from cells expressing mutant MC160 proteins. As mentioned above, immunoprecipitates were assessed for the presence of Hsp90 by immunoblotting. As shown in Fig. 5B, the C terminus of the MC160 protein was responsible for interaction with Hsp90; Hsp90 was immunoprecipitated from cells expressing either WT or C viral proteins but not from cells expressing either the D1 or D2 proteins. Although Hsp90 was detected in IP assays with pN-transfected cells, the amount was dramatically lower than those observed for WT- or C-expressing cells. As would be expected, Hsp90 also immunoprecipitated with D2C, a mutant possessing the C terminus of the MC160 protein (data not shown). Hsp90 was equivalently detected in the lysates from all samples, indicating that a lack of Hsp90 in immunoprecipitated reactions was not due to decreased Hsp90 synthesis (Fig. 5B, lysates).
All MC160 variants were detected in immunoprecipitated samples and in lysates when probing IBs with anti-HA antiserum (Fig. 5B). Since the anti-HA antibody used for this IB was also used for IP assays, bands representative of the 50-kDa IgG heavy chain and 25-kDa IgG light chain were present in each lane. While all mutant proteins were detected in immunoprecipitated samples, a longer exposure of the immunoblotted lysates was required for adequate detection of the D2 protein (Fig. 5B, bottom).
As further proof that the C protein-Hsp90 interactions were biologically relevant, we repeated the IKK1 rescue experiment from Fig. 4, substituting pC in place of pMC160. As shown in Fig. 5C, IKK1 protein levels are decreased while the C protein is overexpressed. Cotransfection of cells with pC and pHA-Hsp90 results in a slight decrease in IKK1 protein levels. These data imply that the C terminus of the MC160 protein indeed binds to Hsp90, an event that induces IKK1 degradation. Further, this IKK1 degradation phenotype of the C protein can be reversed if an excess of Hsp90 is present.
The DED-containing N-terminal portion of MC160 interacts with procaspase-8 and is sufficient to protect against procaspase-8-induced NF-κB activation.
The DED-containing portion of the MC160 protein, while able to inhibit TNF-α-induced NF-κB activation (Fig. 2A), did not require Hsp90 binding for its function (Fig. 5B, IP), suggesting a unique mechanism for its inhibitory function. An alternative inhibitory mechanism for this region was posited to be through its interaction with other known members of the TNF-R1-based NF-κB signalsome (namely, RIP and TRAF2). Accordingly, the association of the MC160 protein with either protein was examined by using IP assays. It appeared that the MC160 protein did not directly interact with the HA-tagged TRAF2 protein, since the WT MC160 protein (lacking an HA epitope tag) was not detected in cotransfected samples in which HA-TRAF2 molecules were immunoprecipitated using anti-HA antibodies (Fig. 6A, IP). Conditions were conducive for detecting TRAF2 binding partners since the MC159 protein, previously reported to associate with TRAF2 (39), was detected using this method. In cells cotransfected with pRIP and pMC160, MC160-RIP interactions also were undetectable by IP assay (Fig. 6B, IP), although similar conditions enabled the detection of known RIP-TRAF2 interactions.
FIG. 6.

Association of WT or mutant MC160 products with either TRAF2, RIP, or procapase-8 proteins. (A to D) 293T cells were transfected with 1 μg of each of the indicated plasmids. At 16 h posttransfection, cellular monolayers were harvested, collected by centrifugation, and lysed in DED buffer. A portion of clarified lysates was incubated with Protein G Sepharose beads that had been conjugated to anti-HA (A, C, and D) or anti-RIP (B). Immunoprecipitated samples and the remaining clarified lysates were individually subjected to SDS-12% PAGE, and the resolved proteins were transferred to a PVDF membrane. Blots were probed with the indicated antibodies, and reactive proteins on each IB were detected by using chemiluminescence. −, lacking; +, containing. (C and D) WT and mutant MC160 proteins are indicated on the right side of the IB. (D) A longer exposure of the anti-HA IB to film was required for visualization of the D1 and D2 proteins (bottom). IgG HC refers to the 50-kDa heavy chain. Positions of the molecular weight markers (MW) are indicated on the left side of the panel.
While procaspase-8 overexpression mediates NF-κB activation (7, 28), the molecular role it utilizes for TNF-R1-based NF-κB activation is still unclear. Interestingly, the MC160 protein was shown to immunoprecipitate with the 65-kDa, unprocessed procaspase-8 (51), and verified in this study (Fig. 6C, IP), suggesting that these interactions may be important for the MC160 protein's regulation of NF-κB activation. Given that the MC160 N terminus possesses homologous DEDs, it was not surprising to observe that the mutant N protein interacted with procaspase-8, as evidenced by the 65-kDa procaspase-8 protein immunoprecipitating with the HA-tagged N protein (Fig. 6C, IP). This associative property appeared to reside only in the N terminus: procapase-8 was no longer detected in immunoprecipitates from cells expressing the MC160 C protein, ruling out the possibility that procaspase-8 was binding nonspecifically to the MC160 protein or the agarose beads. Since the HA-tagged WT and mutant MC160 proteins were present with comparable levels in both the cellular lysates (Fig. 6C, lysates) and the immunoprecipitated samples (data not shown), this lack of a detectable association between procaspase-8 and the MC160 C terminus was not due to poor protein expression. Furthermore, either DED1 or DED2 was sufficient for this interaction (Fig. 6D, IP). This effect was presumably not due to inequivalent amounts of MC160 protein, since the D1 and D2 proteins were detected at similar levels in their respective cytoplasmic lysates (Fig. 6D). A 42-kDa cleavage product of procaspase-8 that is reported to be detected by this antibody in normal cells was also observed in immunoprecipitates and lysates.
To examine if NF-κB inhibition by the MC160 protein was related to its association with procaspase-8, the effect of mutant or WT MC160 proteins on procaspase-8-induced NF-κB activation was studied. To this end, a mutant procaspase-8 protein was ectopically overexpressed in cells, and luciferase activity was used as an indicator of NF-κB activation. The mutant procaspase-8 protein (procaspase-8 C360S) possesses a serine residue at position 360 in place of cysteine. We utilized this construct to avoid procaspase-8-induced apoptosis, thereby minimizing potentially confounding effects of cell death in these assays. As shown in Fig. 7, transfection of pProcaspase-8 C360S alone induced the luciferase activity level to 85-fold above that of vector-only (pCI) transfected cells, implying that NF-κB was activated similarly to other reports (7, 28). In contrast, the luciferase activity level was 2.8-fold lower in lysates from cells coexpressing MC160 and procaspase-8, indicating that the WT MC160 protein blocked this means of NF-κB activation. Luciferase activity levels were decreased in lysates from either pN-, pD1-, or pD2-transfected cells (2.3-, 2.7-, and 4.1-fold decreases, respectively). Thus, either the MC160 DED1 or DED2 regions could mediate this inhibitory effect and do not appear to act in a synergistic manner. Furthermore, both DED regions inhibited NF-κB activation to a greater extent than the MC160 C protein, which inhibited luciferase activity by only 1.3-fold, inferring that MC160 C terminus interaction with procaspase-8 is not central to the role of this inhibitory phenotype (Fig. 7).
FIG. 7.

The effect of WT or mutant MC160 proteins on procaspase-8-induced NF-κB activation. Subconfluent 293T cells were cotransfected with 225 ng pNF-κBluc, 25 ng pRL-null, 250 ng pProcaspase-8 C360S, and 750 ng of either pCI, pHA-MC160, pN, pC, pD1, or pD2. Control cells were instead cotransfected with 250 ng pNF-κBluc, 25 ng pRL-null, and 1,000 ng pCI. At 12 h posttransfection, cellular monolayers were lysed. Cellular lysates were assayed for firefly and sea pansy luciferase activities. All values were normalized to those of pCI-transfected cells devoid of MC160 and pProcaspase-8 C360S proteins. An asterisk indicates statistically significant inhibition (P < 0.05). Lysates were also probed for procaspase-8 and MC160 proteins by immunoblotting. Blots were incubated with the indicated antisera and visualized by chemiluminescence. Bands corresponding to the indicated MC160 mutant proteins are indicated to the right side of the panel. Relative positions of the molecular weight markers (MW) are labeled on the left side of the panel.
DISCUSSION
Previously, it was identified that the MCV MC160 protein impedes TNF-α-induced NF-κB activation, an event that coincides with IKK1 protein degradation (41). Since IKK1 was considered dispensable for this cytokine-mediated event (fibroblasts from IKK1-deficient mice remain responsive to TNF-α [26, 32]), it was initially perplexing as to how the MC160 protein's effect on IKK1 altered TNF-R1 signaling. More recent publications show other IKK1 functions during TNF-R1-induced NF-κB activation: TNF-α treatment directs IKK1 to the nucleus, where IKK1 is proposed to function as an activator of NF-κB-directed genes (1, 59). Additionally, IKK1 mediates NF-κB p65 subunit phosphorylation, an event necessary for TNF-α-induced NF-κB activation (44, 53). As observed here, MC160 expression in cells prevented this posttranslational modification. Thus, it is now conceivable how the MC160 protein's effect on IKK1 protein levels could affect TNF-α-mediated signaling events. Other studies have identified IKK1 as important for processing the NF-κB p100 subunit to activate an alternate NF-κB pathway (4). Although not experimentally tested, it is predicted that expression of the MC160 protein would also inhibit this nonclassical NF-κB activation pathway. It should also be mentioned that similar effects of IKK2 degradation are observed in MC160-expressing cells (data not shown). Therefore, the MC160 effect on inhibiting TNF-mediated nuclear translocation, as detected by electrophoretic mobility shift assays, was expected.
Hsp90 stabilization of IKK1 is important for the maintenance of TNF-R1-mediated NF-κB activation; GA-induced IKK1 degradation inhibits TNF-α-mediated NF-κB activation (6, 9). Since MC160-expressing cells exhibited a similar phenotype (41), we posited that MC160 affected Hsp90-IKK1 interactions. In support of this hypothesis, we detected MC160-Hsp90 associations by IP assays. Furthermore, this interaction was mainly reliant upon the presence of the C-terminal region of the MC160 protein. Thus, our current model is that the MC160 C protein, in binding to Hsp90, induces IKK1 degradation to inhibit TNF-α-induced NF-κB activation. Whether the MC160-Hsp90 interactions are direct or mediated via other proteins, such as the Hsp90-binding CDC37 protein, is the focus of future studies. The C-terminal portion of the MC160 protein has no obvious motifs or similarities to other known Hsp90 binding partners, making it difficult to identify potential MC160 protein binding partners. Interestingly, known Hsp90-RIP interactions, which stabilize the RIP protein, are unaffected in MC160-expressing cells, indicating that the MC160 effect is specific for IKK1 (data not shown).
A second MC160 region, DED2, also induced IKK1 degradation but did not associate with Hsp90, as detected by IP assays. It is unknown how the D2 protein would induce IKK1 degradation when D2-Hsp90 interactions were not detected. We noticed that D2 protein expression is lower in D2-expressing cells than in WT-expressing cells. Thus, we cannot rule out that a lack of D2-Hsp90 interactions is due to low protein levels. In an effort to solve this problem, we constructed a chimeric MC160 protein expressing two tandem DED2 regions. Unfortunately, this DED2-DED2 protein was expressed at levels lower than that of the D2 protein. Future experiments will assess D2 Hsp90 interactions via yeast two-hybrid analysis to verify a lack of interactions. Another possibility is that only minimal binding of D2 to Hsp90, which would be undetectable by IP assays, may be sufficient to mediate IKK1 degradation. It is unlikely that D2 directly interacts with members of the IKK complex, since MC160-IKK interactions were not detected by IP assays (41). Instead, the MC160 DED2 region may affect the presence of other, as yet uncharacterized proteins necessary for stabilizing the IKK complex. Alternatively, since a recent report identifies procaspase-8 interaction with the IKK complex, D2-procaspase-8 interactions may destabilize the IKK complex (30). Additional studies with the D2 protein will include a point mutational analysis to determine if the IKK1 degradation phenotype and the procaspase-8 binding phenotype are related or separate.
One could argue that D1's lack of effect on TNF-induced luciferase activity is also a result of lower levels of protein expression. To address this, we created a chimeric protein expressing two tandem D1 domains. Protein expression levels are higher than those observed for D1 proteins. It also provided no protection against TNF-induced NF-κB activation, as measured by luciferase reporter assays (data not shown).
Previously, the MC160 protein was observed to immunoprecipitate with procaspase-8 (51), an enzyme whose cleaved form was identified originally as a mediator of apoptosis (40) and whose uncleaved form is an activator of NF-κB (7). Thus, an attractive hypothesis was that the DED-containing portion of the MC160 protein interacted with procaspase-8 to inhibit NF-κB activation. This hypothesis was bolstered by the following observations: (i) the WT MC160 protein inhibited procaspase-8-induced NF-κB activation, (ii) only the MC160 DED regions immunoprecipitated with procaspase-8, and (iii) DED-containing MC160 proteins inhibited procaspase-8-induced NF-κB activation to a greater extent than the C protein. Why the MC160 DED1 inhibited procaspase-8-induced, but not TNF-α-induced, NF-κB activation remains unclear. One explanation is that NF-κB activation mediated by an external stimulus is an overlapping, but not identical, pathway as that triggered intracellularly during procaspase-8 overexpression. These two mutant proteins, D1 and D2, also bind to procaspase-8 to the same extent as the N protein (Fig. 6D). Thus, it is possible that the D1 and D2 proteins function by each binding to procapase-8. In this manner, one would expect then that the D1 and D2 proteins would inhibit procaspase-8-induced NF-κB activation no better than the N protein.
Recent publications indicate mechanisms for procaspase-8 activation of NF-κB. One report observes procaspase-8-TRAF2 interactions, via the FLASH protein, with this complex then triggering IKK activation (13, 28). Thus, one prediction is that MC160-procaspase-8 interactions would affect signalsome formation. A second report demonstrates procaspase-8-induced cleavage of cFLIPL to a p22 form, with p22 then associating with IKKγ to render an active IKK complex (21). In this case, the MC160 protein would be expected to prevent cFLIPL cleavage and subsequent IKK activation. Interestingly, we observed that MC160 prevented cFLIPL-mediated NF-κB activation, as triggered by ectopic overexpression of cFLIPL, providing indirect evidence in support of this second mechanism (data not shown). A third report identifies procaspase-8 IP with the IKK complex when B cells are stimulated with lipopolysaccharides (30). It must be reiterated that it is unknown if procaspase-8 utilizes these, or other novel, mechanisms when aiding in TNF-R1-induced NF-κB activation. Thus, the model proposed in Fig. 8 is speculative and relies upon the delineation of the inhibitory mechanism used by this cellular zymogen. As an aside, given the role of procaspase-8 in T-cell receptor-induced NF-κB activation, we would predict that expression of the MC160 protein would prevent this event in immune cells.
FIG. 8.
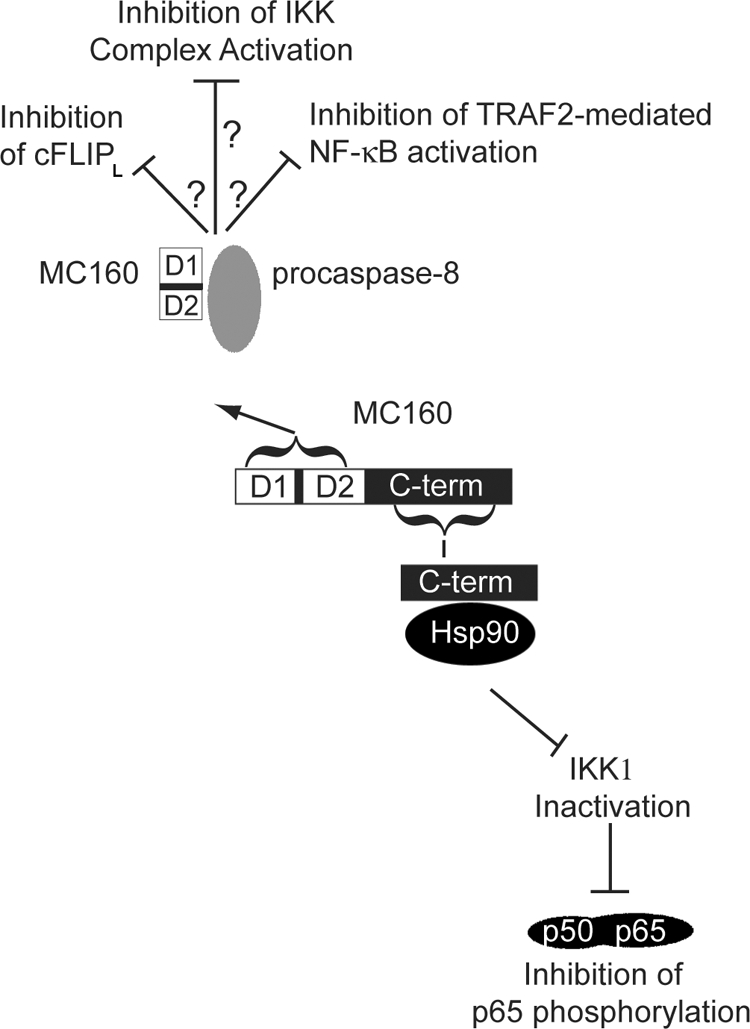
An illustration of the proposed molecular mechanism for MC160 protein interaction with Hsp90 and procaspase-8. The MC160 protein consists of DED1 (D1), DED2 (D2), and a unique C-terminal region (C-term). The proposed partite mechanism for NF-κB inhibition by the MC160 protein is as follows. The MC160 protein, by virtue of its DED-containing N terminus, binds to procapase-8 to inhibit NF-κB activation. Whether this interaction prevents cFLIPL cleavage or FLASH-TRAF2 interactions is unknown. In conjunction with this first means, or alternatively, the MC160 C-terminal region, through its interaction with Hsp90, leads to a degradation of IKK1, an event that inhibits phosphorylation of the p65 NF-κB subunit and subsequent NF-κB transcriptional activation.
How might this dual-functioning MC160 protein operate during an MCV infection? We observed previously that the MC160 protein inhibits NF-κB nuclear translocation soon after TNF-α treatment (41), while MC160 prevents IKK1 degradation at a later time point of about 8 h posttransfection (data not shown). Thus, one model is that the MC160-procaspase-8 binding (ascribed to the MC160 N-terminal region) is important for the initial inhibition of TNF-α-mediated NF-κB nuclear translocation and activation (Fig. 8), perhaps via the inhibition of cFLIPL cleavage or of TRAF2-FLASH-procaspase-8 complex formation, as described above. (It should be noted that we have not determined the region of the procaspase-8 molecule that binds to either D1 or D2. However, it is assumed that these viral DEDs would likely bind to one of the procaspase-8 DEDs.) Upon prolonged MC160 protein expression, as may occur during a persistent MCV infection, MC160-Hsp90 interactions (ascribed to the MC160 C-terminal region) would prevent Hsp90-IKK1 interactions (Fig. 8). Interestingly, Hsp90 is required not only for the IKK complex's activation but also for the stabilization of nascent IKK subunits (6, 9). Therefore, by binding Hsp90, MC160 may both inhibit the activation of preexisting IKK complexes and prevent the formation of mature IKK complexes.
MCV is a dermatotropic poxvirus, inducing the formation of persistent, benign neoplasms in the skin (16). How this virus alters the host cell environment to cause these long-lasting lesions is largely uncharacterized. There is a correlation between a reduction of IKK1 proteins and the formation of skin papillomas and carcinomas (34). Additionally, epidermal growth is dysregulated in mice lacking the IKK1 gene (26, 27, 32, 56). One speculation is that MC160-IKK1 protein interactions, in addition to protecting against the proinflammatory effects of cytokines, may also induce a host cell environment conducive for neoplastic growth. At least one other viral homolog of MC160, the Kaposi's sarcoma virus K13 product, possesses transformative properties when expressed in lymphocytes, albeit via constitutively activating the IKK complex (19, 35, 55).
During an MCV infection in humans, TNF-α is highly expressed in molluscum lesions and in epidermal cells adjacent to the site of infection (29). The importance of TNF-α's presence in controlling an MCV infection is further illustrated by the observation that the depletion of TNF-α in human patients coincides with MCV lesion development (2, 14). Unfortunately, MCV has yet to be successfully propagated in cell culture or animal models, making it difficult to assess the effect of MC160 protein expression during an infection of keratinocytes in either a cell culture or an animal model system. Because poxviruses express multiple NF-κB inhibitory proteins (5, 11, 18, 20, 50), it is expected that the effect of MC160 protein expression would be masked if it were expressed in a surrogate virus, such as vaccinia virus. Regardless, if an animal model system for MCV were available, an expectation is that viruses lacking the MC160 ORF would no longer protect their host cell against the proinflammatory effects of TNF-α. In this scenario, the host antiviral immune response would be effective at eliminating virus-infected cells, resulting in an attenuated infection.
Acknowledgments
We thank Amy MacNeill, Gail Scherba, and Lin-Feng Chen for helpful suggestions.
This work was supported by a grant to J.L.S. (grant AI055530) from the National Institutes of Health.
Footnotes
Published ahead of print on 21 January 2009.
REFERENCES
- 1.Anest, V., J. L. Hanson, P. C. Cogswell, K. A. Steinbrecher, B. D. Strahl, and A. S. Baldwin. 2003. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature 423659-663. [DOI] [PubMed] [Google Scholar]
- 2.Antoniou, C., M. G. Kosmadaki, A. J. Stratigos, and A. D. Katsambas. 2008. Genital HPV lesions and molluscum contagiosum occurring in patients receiving anti-TNF-alpha therapy. Dermatology 216364-365. [DOI] [PubMed] [Google Scholar]
- 3.Baldi, L., K. Brown, G. Franzoso, and U. Siebenlist. 1996. Critical role for lysines 21 and 22 in signal-induced, ubiquitin-mediated proteolysis of I kappa B-alpha. J. Biol. Chem. 271376-379. [DOI] [PubMed] [Google Scholar]
- 4.Bonizzi, G., and M. Karin. 2004. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 25280-288. [DOI] [PubMed] [Google Scholar]
- 5.Bowie, A., E. Kiss-Toth, J. A. Symons, G. L. Smith, S. K. Dower, and L. A. O'Neill. 2000. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 9710162-10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broemer, M., D. Krappmann, and C. Scheidereit. 2004. Requirement of Hsp90 activity for IkappaB kinase (IKK) biosynthesis and for constitutive and inducible IKK and NF-kappaB activation. Oncogene 235378-5386. [DOI] [PubMed] [Google Scholar]
- 7.Chaudhary, P. M., M. T. Eby, A. Jasmin, A. Kumar, L. Liu, and L. Hood. 2000. Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene 194451-4460. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhary, P. M., A. Jasmin, M. T. Eby, and L. Hood. 1999. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 185738-5746. [DOI] [PubMed] [Google Scholar]
- 9.Chen, G., P. Cao, and D. V. Goeddel. 2002. TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol. Cell 9401-410. [DOI] [PubMed] [Google Scholar]
- 10.Chen, G., and D. V. Goeddel. 2002. TNF-R1 signaling: a beautiful pathway. Science 2961634-1635. [DOI] [PubMed] [Google Scholar]
- 11.Chen, R. A., G. Ryzhakov, S. Cooray, F. Randow, and G. L. Smith. 2008. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 4e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, Z., J. Hagler, V. J. Palombella, F. Melandri, D. Scherer, D. Ballard, and T. Maniatis. 1995. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 91586-1597. [DOI] [PubMed] [Google Scholar]
- 13.Choi, Y. H., K. B. Kim, H. H. Kim, G. S. Hong, Y. K. Kwon, C. W. Chung, Y. M. Park, Z. J. Shen, B. J. Kim, S. Y. Lee, and Y. K. Jung. 2001. FLASH coordinates NF-kappa B activity via TRAF2. J. Biol. Chem. 27625073-25077. [DOI] [PubMed] [Google Scholar]
- 14.Cursiefen, C., M. Grunke, C. Dechant, C. Antoni, A. Junemann, and L. M. Holbach. 2002. Multiple bilateral eyelid molluscum contagiosum lesions associated with TNFalpha-antibody and methotrexate therapy. Am. J. Ophthalmol. 134270-271. [DOI] [PubMed] [Google Scholar]
- 15.Damon, I., P. M. Murphy, and B. Moss. 1998. Broad spectrum chemokine antagonistic activity of a human poxvirus chemokine homolog. Proc. Natl. Acad. Sci. USA 956403-6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Damon, I. K. 2007. Poxviruses, p. 2947-2976. In D. Knipe and P. Howley (ed.), Fields virology, 5th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 17.DiDonato, J. A., M. Hayakawa, D. M. Rothwarf, E. Zandi, and M. Karin. 1997. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388548-554. [DOI] [PubMed] [Google Scholar]
- 18.DiPerna, G., J. Stack, A. G. Bowie, A. Boyd, G. Kotwal, Z. Zhang, S. Arvikar, E. Latz, K. A. Fitzgerald, and W. L. Marshall. 2004. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by Toll-like receptors. J. Biol. Chem. 27936570-36578. [DOI] [PubMed] [Google Scholar]
- 19.Field, N., W. Low, M. Daniels, S. Howell, L. Daviet, C. Boshoff, and M. Collins. 2003. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 1163721-3728. [DOI] [PubMed] [Google Scholar]
- 20.Gedey, R., X. L. Jin, O. Hinthong, and J. L. Shisler. 2006. Poxviral regulation of the host NF-kappaB response: the vaccinia virus M2L protein inhibits induction of NF-kappaB activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J. Virol. 808676-8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golks, A., D. Brenner, P. H. Krammer, and I. N. Lavrik. 2006. The c-FLIP-NH2 terminus (p22-FLIP) induces NF-kappaB activation. J. Exp. Med. 2031295-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinz, M., M. Broemer, S. C. Arslan, A. Otto, E. C. Mueller, R. Dettmer, and C. Scheidereit. 2007. Signal responsiveness of IkappaB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J. Biol. Chem. 28232311-32319. [DOI] [PubMed] [Google Scholar]
- 23.Hsu, H., J. Huang, H. B. Shu, V. Baichwal, and D. V. Goeddel. 1996. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4387-396. [DOI] [PubMed] [Google Scholar]
- 24.Hsu, H., J. Xiong, and D. V. Goeddel. 1995. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 81495-504. [DOI] [PubMed] [Google Scholar]
- 25.Hu, W. H., H. Johnson, and H. B. Shu. 2000. Activation of NF-kappaB by FADD, Casper, and caspase-8. J. Biol. Chem. 27510838-10844. [DOI] [PubMed] [Google Scholar]
- 26.Hu, Y., V. Baud, M. Delhase, P. Zhang, T. Deerinck, M. Ellisman, R. Johnson, and M. Karin. 1999. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science 284316-320. [DOI] [PubMed] [Google Scholar]
- 27.Hu, Y., V. Baud, T. Oga, K. I. Kim, K. Yoshida, and M. Karin. 2001. IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature 410710-714. [DOI] [PubMed] [Google Scholar]
- 28.Jun, J. I., C. W. Chung, H. J. Lee, J. O. Pyo, K. N. Lee, N. S. Kim, Y. S. Kim, H. S. Yoo, T. H. Lee, E. Kim, and Y. K. Jung. 2005. Role of FLASH in caspase-8-mediated activation of NF-kappaB: dominant-negative function of FLASH mutant in NF-kappaB signaling pathway. Oncogene 24688-696. [DOI] [PubMed] [Google Scholar]
- 29.Ku, J. K., H. J. Kwon, M. Y. Kim, H. Kang, P. I. Song, C. A. Armstrong, J. C. Ansel, H. O. Kim, and Y. M. Park. 2008. Expression of Toll-like receptors in verruca and molluscum contagiosum. J. Korean Med. Sci. 23307-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemmers, B., L. Salmena, N. Bidere, H. Su, E. Matysiak-Zablocki, K. Murakami, P. S. Ohashi, A. Jurisicova, M. Lenardo, R. Hakem, and A. Hakem. 2007. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J. Biol. Chem. 2827416-7423. [DOI] [PubMed] [Google Scholar]
- 31.Li, H., and X. Lin. 2008. Positive and negative signaling components involved in TNFalpha-induced NF-kappaB activation. Cytokine 411-8. [DOI] [PubMed] [Google Scholar]
- 32.Li, Q., Q. Lu, J. Y. Hwang, D. Buscher, K. F. Lee, J. C. Izpisua-Belmonte, and I. M. Verma. 1999. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 131322-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, X., Y. Yang, and J. D. Ashwell. 2002. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 416345-347. [DOI] [PubMed] [Google Scholar]
- 34.Maeda, G., T. Chiba, S. Kawashiri, T. Satoh, and K. Imai. 2007. Epigenetic inactivation of IkappaB kinase-alpha in oral carcinomas and tumor progression. Clin. Cancer Res. 135041-5047. [DOI] [PubMed] [Google Scholar]
- 35.Matta, H., Q. Sun, G. Moses, and P. M. Chaudhary. 2003. Molecular genetic analysis of human herpes virus 8-encoded viral FLICE inhibitory protein-induced NF-kappaB activation. J. Biol. Chem. 27852406-52411. [DOI] [PubMed] [Google Scholar]
- 36.Mercurio, F., H. Zhu, B. W. Murray, A. Shevchenko, B. L. Bennett, J. Li, D. B. Young, M. Barbosa, M. Mann, A. Manning, and A. Rao. 1997. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278860-866. [DOI] [PubMed] [Google Scholar]
- 37.Moss, B., J. L. Shisler, Y. Xiang, and T. G. Senkevich. 2000. Immune-defense molecules of molluscum contagiosum virus, a human poxvirus. Trends Microbiol. 8473-477. [DOI] [PubMed] [Google Scholar]
- 38.Muller, J. R., and U. Siebenlist. 2003. Lymphotoxin beta receptor induces sequential activation of distinct NF-kappa B factors via separate signaling pathways. J. Biol. Chem. 27812006-12012. [DOI] [PubMed] [Google Scholar]
- 39.Murao, L. E., and J. L. Shisler. 2005. The MCV MC159 protein inhibits late, but not early, events of TNF-alpha-induced NF-kappaB activation. Virology 340255-264. [DOI] [PubMed] [Google Scholar]
- 40.Muzio, M., A. M. Chinnaiyan, K. C. Kischkel, K. O'Rourke, A. Shevchenko, J. Ni, C. Scaffidi, J. D. Bretz, M. Zhang, R. Gentz, M. Mann, P. H. Krammer, M. E. Peter, and V. M. Dixit. 1996. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85817-827. [DOI] [PubMed] [Google Scholar]
- 41.Nichols, D. B., and J. L. Shisler. 2006. The MC160 protein expressed by the dermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosis factor alpha-induced NF-κB activation via inhibition of I kappa kinase complex formation. J. Virol. 80578-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oie, K. L., and D. J. Pickup. 2001. Cowpox virus and other members of the orthopoxvirus genus interfere with the regulation of NF-kappaB activation. Virology 288175-187. [DOI] [PubMed] [Google Scholar]
- 43.Perkins, N. D. 2006. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 256717-6730. [DOI] [PubMed] [Google Scholar]
- 44.Sakurai, H., H. Chiba, H. Miyoshi, T. Sugita, and W. Toriumi. 1999. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 27430353-30356. [DOI] [PubMed] [Google Scholar]
- 45.Scheidereit, C. 2006. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene 256685-6705. [DOI] [PubMed] [Google Scholar]
- 46.Scherer, D. C., J. A. Brockman, Z. Chen, T. Maniatis, and D. W. Ballard. 1995. Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc. Natl. Acad. Sci. USA 9211259-11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Senkevich, T. G., J. J. Bugert, J. R. Sisler, E. V. Koonin, G. Darai, and B. Moss. 1996. Genome sequence of a human tumorigenic poxvirus: prediction of specific host response-evasion genes. Science 273813-816. [DOI] [PubMed] [Google Scholar]
- 48.Senkevich, T. G., E. V. Koonin, J. J. Bugert, G. Darai, and B. Moss. 1997. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology 23319-42. [DOI] [PubMed] [Google Scholar]
- 49.Shikama, Y., M. Yamada, and T. Miyashita. 2003. Caspase-8 and caspase-10 activate NF-kappaB through RIP, NIK and IKKalpha kinases. Eur. J. Immunol. 331998-2006. [DOI] [PubMed] [Google Scholar]
- 50.Shisler, J. L., and X.-L. Jin. 2004. The vaccinia virus K1L gene product inhibits host NF-κB activation by preventing IκBα degradation. J. Virol. 783553-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shisler, J. L., and B. Moss. 2001. Molluscum contagiosum virus inhibitors of apoptosis: the MC159 v-FLIP protein blocks Fas-induced activation of procaspases and degradation of the related MC160 protein. Virology 28214-25. [DOI] [PubMed] [Google Scholar]
- 52.Shisler, J. L., T. G. Senkevich, M. J. Berry, and B. Moss. 1998. Ultraviolet-induced cell death blocked by a selenoprotein from a human dermatotropic poxvirus. Science 279102-105. [DOI] [PubMed] [Google Scholar]
- 53.Sizemore, N., N. Lerner, N. Dombrowski, H. Sakurai, and G. R. Stark. 2002. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J. Biol. Chem. 2773863-3869. [DOI] [PubMed] [Google Scholar]
- 54.Stebbins, C. E., A. A. Russo, C. Schneider, N. Rosen, F. U. Hartl, and N. P. Pavletich. 1997. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 89239-250. [DOI] [PubMed] [Google Scholar]
- 55.Sun, Q., S. Zachariah, and P. M. Chaudhary. 2003. The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-kappaB activation. J. Biol. Chem. 27852437-52445. [DOI] [PubMed] [Google Scholar]
- 56.Takeda, K., O. Takeuchi, T. Tsujimura, S. Itami, O. Adachi, T. Kawai, H. Sanjo, K. Yoshikawa, N. Terada, and S. Akira. 1999. Limb and skin abnormalities in mice lacking IKKalpha. Science 284313-316. [DOI] [PubMed] [Google Scholar]
- 57.Whitesell, L., E. G. Mimnaugh, B. De Costa, C. E. Myers, and L. M. Neckers. 1994. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 918324-8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang, Y., and B. Moss. 1999. IL-18 binding and inhibition of interferon gamma induction by human poxvirus-encoded proteins. Proc. Natl. Acad. Sci. USA 9611537-11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamamoto, Y., U. N. Verma, S. Prajapati, Y. T. Kwak, and R. B. Gaynor. 2003. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature 423655-659. [DOI] [PubMed] [Google Scholar]
- 60.Yang, K., H. Shi, R. Qi, S. Sun, Y. Tang, B. Zhang, and C. Wang. 2006. Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol. Biol. Cell 171461-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]