

Abstract
β(1,3)-glucans represent 40% of the cell wall of the yeast Candida albicans. The dectin-1 lectin-like receptor has shown to recognize fungal β(1,3)-glucans and induce innate immune responses. The importance of β-glucan-dectin-1 pathways for the recognition of C. albicans by human primary blood cells has not been firmly established. In this study we demonstrate that cytokine production by both human peripheral blood mononuclear cells and murine macrophages is dependent on the recognition of β-glucans by dectin-1. Heat killing of C. albicans resulted in exposure of β-glucans on the surface of the cell wall and subsequent recognition by dectin-1, whereas live yeasts stimulated monocytes mainly via recognition of cell-surface mannans. Dectin-1 induced cytokine production through the following 2 pathways: Syk-dependent production of the T-helper (Th) 2-type anti-inflammatory cytokine interleukin-10 and Toll-like receptor-Myd88-dependent stimulation of monocyte-derived proinflammatory cytokines, such as tumor necrosis factor-α. In contrast, stimulation of Th1-type cytokines, such as interferon-γ, by C. albicans was independent of the recognition of β-glucans by dectin-1. In conclusion, C. albicans induces production of monocyte-derived and T cell-derived cytokines through distinct pathways dependent on or independent of dectin-1.
Understanding the mechanisms through which the host immune system recognizes and eliminates pathogens has become increasingly important because a new resurgence of infectious diseases has occurred, owing to the increased impact of immunocompromising medical treatments (e.g., chemotherapy and transplantation surgery), as well as the global AIDS pandemic. Invasive Candida albicans infections are a serious clinical threat in patients who are immunosuppressed and those who have undergone major surgical procedures, with mortality reaching 30%-40% despite the availability of new classes of antifungal drugs [1, 2]. Thus, new therapies for fungal infection are urgently needed, and adjunctive immunotherapy is one promising yet unfulfilled treatment strategy.
Much has been done to elucidate the host defense mechanisms against systemic candidiasis. The importance of phagocytosis and the elimination of C. albicans by cells of the innate immune system, especially neutrophils, monocytes, and macrophages [3-6], as well as the modulation of the activity of these cells by proinflammatory and anti-inflammatory cytokines, has been firmly established [7-9]. Recently, it has been shown that recognition of microbial structures, referred to as “pathogen-associated molecular patterns,” by pattern-recognition receptors (PRRs) is essential for the effective activation of host defense mechanisms in general and for production of cytokines in particular. Earlier studies by our group and by others described the role of several PRRs in recognizing components of the C. albicans cell wall, including mannans by mannose receptor (MR) and mannoproteins by Toll-lie receptor (TLR) 4 [10-15], phospholipomannan (PLM) by TLR2 [16], and β-mannosides by galectin-3 [17]. β(1,3)-glucans and β(1,6)-glucans are important structural components of the C. albicans cell wall, and recent work has described dectin-1 as the C-type lectin PRR responsible for their recognition [18]. Several studies have established the capacity of dectin-1 to recognize β-glucans if zymosan is present[19, 20]. However, recent studies have generated contrasting evidence for the importance of dectin-1 in anticandidal host defenses in mice [21, 22]. Little is known about the relative importance of β-glucan recognition by dectin-1 for the activation of host defense mechanisms by C. albicans in primary cells of the innate immune system in humans.
In previous studies, we demonstrated that O-linked and N-linked mannans of the cell surface of C. albicans were immunostimulatory molecules [14]. It has also been shown that disruption of the integrity of the native cell wall by heat killing or deletion of glycosylation genes led to enhanced signaling in leukocytes via dectin-1 [14]. We therefore set out to establish the relative contribution of these various immunostimulatory pathways in intact, live cells and in heat-killed cells. In addition, we aimed to establish whether recognition of β-glucan by dectin-1 is a nonredundant pathway for C. albicans-associated stimulation of cytokine production in human peripheral blood mononuclear cells (PBMCs). It was recently demonstrated that dectin-1 engagement induces cell activation through 2 independent pathways. One pathway involves amplification of TLR-MyD88-dependent signals; a second, TLR-independent mechanism relies on signals transmitted through the Syk-adaptor molecule. A further aim of the study was to evaluate the differential involvement of these 2 activation pathways in C. albicans-induced stimulation of proinflammatory and anti-inflammatory cytokine production.
METHODS
C. albicans strains and growth conditions
C. albicans strain CAI-4 was shaken at 200 rpm at 30°C in Sabouraud broth (1% mycological peptone and 4% glucose) overnight, transferred to fresh medium, and incubated for 4 h. The cells were harvested by centrifugation, and the pellets were washed twice in 20 -L sterile PBS and resuspended to a density of 1 × 108 cells/mL before heat killing at 56°C for 30 min. In separate experiments, live C. albicans were washed, resuspended in RPMI 1640 at a concentration of 1 × 106 cfu/mL, and used for the stimulation of cytokine production. To create ultraviolet (UV) radiation-killed C. albicans, cells were grown and processed as described above and then exposed in a thin liquid suspension to 4 doses of UV radiation (100 mJ/cm2) in a UV-DNA crosslinker. To create heat-killed C. albicans, cells were grown and processed as described above and then stored at 56°C for 30 min.
Animals
Dectin-1 knock-out (dectin-1-/-) mice were generated as described elsewhere [21]. All mice weighed 20-25 g and were 6-8 weeks old. The mice were fed sterilized laboratory chow and water ad libitum. The experiments were approved by the ethics committee on animal experiments of Cape Town University.
Antibodies
The monoclonal anti-TLR4 HTA125 antibody was kindly provided by Dr. Kensuke Miyake (Saga Medical School). The monoclonal mouse antihuman MR antibody and the isotype-matched IgG antibody used as control in all experiments were purchased from Sigma. Glucan phosphate, a specific inhibitor of dectin-1, was kindly provided by Dr. David Williams (University of Tennessee).
Stimulation of cytokine production in human PBMCs
PBMCs were isolated as described elsewhere [23]. A 100-μL volume of PBMCs (concentration, 5 × 105 PBMCs/mL) was added to round-bottomed 96-well plates (Greiner) and incubated with 100 μL of live, UV radiation-killed, or heat-killed C. albicans (concentration, 1 × 106 yeast cells/mL), unless otherwise indicated. In receptor-blocking studies, PBMCs were preincubated for 1 h at 37°C with 10 μg/mL of either glucan phosphate or 1 of 3 monoclonal antibodies (anti-TLR4, anti-MR, and control IgG) before stimulation with C. albicans. In addition, 50 nmol/L of 3-(1-methyl-1H-indol-3-yl-methylene)-2-oxo-2,3-dihydro-1H-indole-5-sulfonamide (Calbiochem) was used for the pharmacological inhibition of Syk in cytokine-stimulation assays. After incubation at 37°C for 24 h (durations varied between 2 and 48 h in the kinetics experiments), the PBMC-C. albicans suspensions were centrifuged; supernatants were collected and stored at -70°C until assayed. Human tumor necrosis factor (TNF)-α concentrations were determined by specific radioimmunoassays as described previously [24]. Interleukin (IL)-6, IL-10, and interferon (IFN)-γ concentrations were measured by commercial ELISA kits (Pelikine Compact; Sanquin).
Cytokine production by murine peritoneal macrophages
Resident peritoneal macrophages from the dectin-1-/- and control mouse strains were harvested by injecting 4 mL of sterile PBS containing 0.38% sodium citrate [25]. After centrifugation and washing, the cells were resuspended in RPMI 1640. Cells were cultured in 96-well plates and stimulated with 100 μL of the various C. albicans strains (concentration, 1 × 106 yeast cells/mL) for 24 h at 37°C [24]. Murine TNF-α was determined by a specific radioimmunoassay (lower limit of detection, 20 pg/mL) [26].
Statistical analyses
Two experiments, each involving triplicate samples of PBMCs obtained from distinct groups of 8 volunteers, were performed. The differences between groups were analyzed by the Mann-Whitney U test. The level of significance between groups was taken as P < .05. The data are given as mean values ± SD.
RESULTS
Differential stimulation of cytokine production by heat-killed, UV radiation-killed, and live C. albicans
It has been suggested that heat killing of C. albicans results in exposure of β-glucans on the surface of the cell wall [27]. We investigated the differential induction of cytokines by live and heat-killed C. albicans and the role played by β-glucans in these processes. Heat-killed C. albicans induced significantly greater levels of TNF-α (figure 1A), IL-6 (figure 1B), IL-10 (figure 1C), and IFN-γ (figure 1D) than live C. albicans did, as shown by the dose-dependent production of cytokines by freshly recovered human PBMCs. The kinetics of cytokine production did not differ between live and heat-killed C. albicans, and at all time points, the absolute levels of TNF-α and IL-6 released by PBMCs after stimulation with live C. albicans were 5%-20% of the levels induced by heat-killed yeasts (P < .01). For both live and heat-killed cells, the TNF-α concentration peaked after stimulation for 8-24 h, and then levels decreased sharply, whereas the IL-6 level increased during the same period of stimulation, plateauing after 24-48 h. UV radiation-killed yeasts induced less TNF-α production than did heat-killed C. albicans after 24 h of stimulation (2.4 ± 0.3 vs. 8.2 ± 1.2 ng/mL), but these levels were significantly higher than that in live C. albicans (0.9 ± 0.2 ng/mL; P < .05).
Figure 1.

Mean levels of tumor necrosis factor (TNF)-α (A), interleukin (IL)-6 (B), IL-10 (C), and interferon (IFN)-γ (D) produced in peripheral blood mononuclear cells after incubation with heat-killed (HK) or live Candida albicans. All results are pooled triplicate data from 2 separate experiments, with a total of 8 volunteers per group. Whisker bars, SDs. *P < .05; **P < .01.
Role of dectin-1 in the recognition of C. albicans by human PBMCs
To investigate whether the differences in cytokine stimulation between live and heat-killed C. albicans were due to differential recognition of β-glucan, we blocked dectin-1 recognition by preincubating PBMCs with excess glucan phosphate before exposure to C. albicans. Blocking of dectin-1 recognition strongly suppressed production of TNF-α, IL-6 (figure 2A), and IL-10 (115 ± 34 pg/mL in control cells vs. 42 ± 23 pg/mL in cells preincubated with glucan phosphate; P < .05) in PBMCs exposed to heat-killed C. albicans; no such suppression was observed in PBMCs exposed to live C. albicans. In contrast, blocking of dectin-1 recognition did not significantly influence the production of IFN-γ in PBMCs exposed to heat-killed C. albicans (366 ± 121 in control cells vs. 330 ± 98 pg/mL in cells preincubated with glucan phosphate; P > .05), suggesting that dectin-1 plays little role in the stimulation of this Th1 cytokine. Similar to the stimulation observed in human cells, a greater level of TNF-α was produced by mouse peritoneal macrophages exposed to heat-killed C. albicans, compared with those exposed to live yeast cells. In addition, dectin-1-/- macrophages produced fewer cytokines than did dectin-1+/+ cells when stimulated with heat-killed C. albicans; no such difference was observed after stimulation with live C. albicans (figure 2B).
Figure 2.
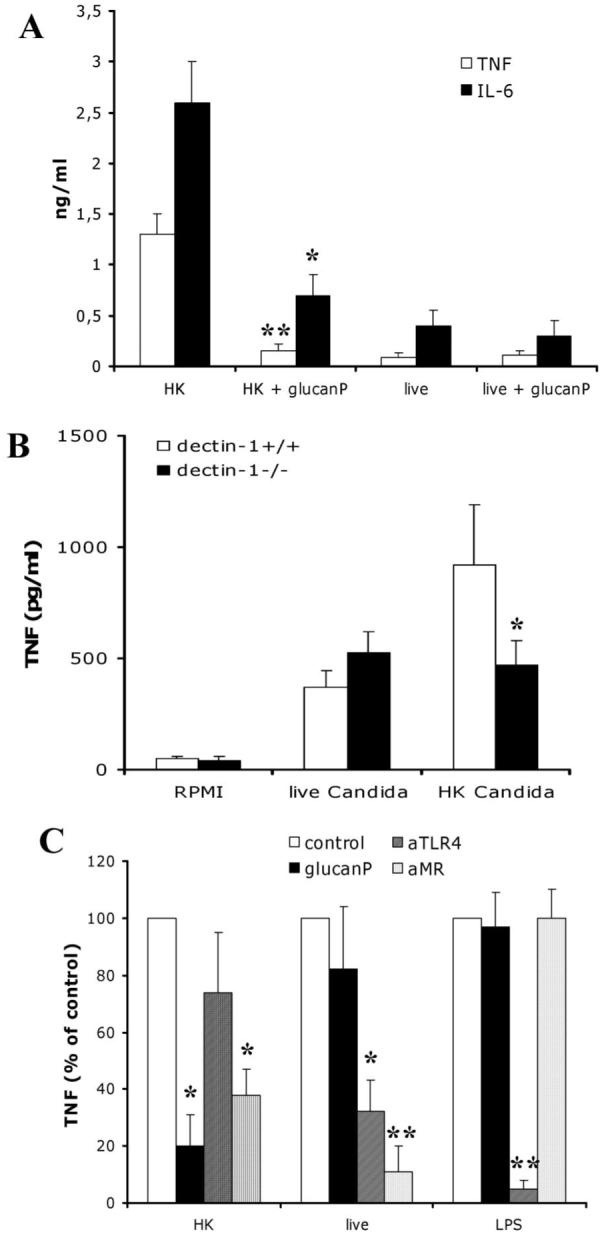
A, Mean levels of tumor necrosis factor (TNF)-α and interleukin (IL)-6 in peripheral blood mononuclear cells (PBMCs), according to the presence or absence of dectin-1 blocking by glucan phosphate (glucanP), after incubation with live or heat-killed (HK) Candida albicans. B, Mean levels of TNF-α in murine peritoneal macrophages from dectin-1+/+ mice and dectin-1-/- mice after incubation with RPMI 1640 (control), live C. albicans, or HK C. albicans. C, Mean levels of TNF-α in PBMCs in which dectin-1 receptor, Toll-like receptor (TLR) 4, and mannose receptor (MR) were blocked by glucanP, anti-TLR4 antibody (aTLR4), and anti-MR antibody (aMR), respectively. TNF-α levels in PBMCs stimulated with lipopolysaccharide (LPS) in growth medium were included as a control. All results are pooled triplicate data from 2 separate experiments, with a total of 6 volunteers per group. Whisker bars, SDs. *P < .05; **P < .01.
In the presence of the dectin-1 blockade, levels of TNF-α and IL-6 induced by heat-killed C. albicans were the same as those induced by live yeasts, demonstrating that the increased induction of TNF-α and IL-6 by heat-killed C. albicans was entirely due to the interaction between β-glucan and dectin-1. Consequently, the importance of TLR4 and MR in the recognition of live C. albicans was greater than their importance in the recognition of heat-killed yeast cells and LPS-stimulated cells (positive control) (figure 2C). These data demonstrate that recognition of live C. albicans is based mainly on TLR4 and MR, whereas heat-killed C. albicans is also recognized by these 2 receptors but with an increased role for dectin-1-mediated recognition.
Differential stimulation of IL-6, IL-10, and TNF-α by dectin-1 through Syk-dependent and Syk-independent mechanisms
The effects of dectin-1 are induced by the adaptor molecule Syk [28] or mediated by the interaction between dectin-1 and the TLR2-MyD88 pathway [19, 20]. Although dectin-1 inhibition by glucan phosphate significantly inhibited production of all analyzed cytokines except, notably, IFN-γ, pharmacological inhibition of Syk only modulated IL-6 and IL-10 production and did not inhibit synthesis of TNF-α induced by heat-killed C. albicans (figure 3). Syk inhibition had no significant effect on TNF-α production in cells stimulated with live C. albicans (TNF-α level, 345 ± 88 pg/mL in Syk-inhibited cells vs. 411 ± 104 pg/mL in control cells; P > .05). Similarly, no effect of Syk inhibition on C. albicans-induced IFN-γ production was observed, consistent with the absence of the effect of dectin-1 blocking on IFN-γ synthesis. These data imply that, although the interaction between dectin-1 and C. albicans β-glucans stimulates production of both proinflammatory and anti-inflammatory cytokines, the cellular pathway involved in induction of TNF-α (a Syk-independent pathway) differs from that for induction of IL-6 and IL-10 (a Sky-dependent pathway).
Figure 3.

Mean levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6, interferon (IFN)-γ, and IL-10 produced in peripheral blood mononuclear cells after incubation with Candida albicans in the absence of inhibitors (control; open bars), in the presence of glucan phosphate (glucanP; solid bars), or in the presence of Syk inhibitor (Syk-inh; hatched bars). Results are pooled triplicate data from 2 separate experiments, with a total of 8 volunteers per group. Whisker bars, SDs. aLevel of IL-10 is ×10 ng/mL.
DISCUSSION
In this study, we demonstrated that recognition of C. albicans β-glucans by dectin-1 is important for efficient recognition of C. albicans and subsequent activation of the cytokine response. Although earlier studies demonstrated that dectin-1 is the main β-glucan receptor, we are the first to show that the β-glucan-dectin-1 pathway is a nonredundant recognition mechanism for mediating cytokine stimulation in human primary PBMCs after stimulation by C. albicans and that this pathway is not compensated by alternative recognition mechanisms (e.g., by TLRs or other lectin-like receptors, such as MR or DC-SIGN). In addition, we demonstrated that cytokine production induced by dectin-1 is mediated through both Syk-dependent and Syk-independent pathways.
The studies to date on the recognition of β-glucans by dectin-1 have used zymosan as a stimulus of cytokine production. Zymosan is a product of the cell wall of Saccharomyces cerevisiae, which is composed of β(1,3)-glucans and β(1,6)-glucans but also contains α-mannans and mannoproteins. Cytokine stimulation by zymosan relies on β(1,3)-glucan recognition by dectin-1, which in turn amplifies TLR2 signals induced by an as yet unidentified ligand [19, 20]. Pathways other than those mediated by TLR2 were suggested in 2 recent studies, which showed that dectin-1 can also independently induce IL-2 and IL-10 synthesis through a Syk-CARD9-dependent mechanism [28, 29]. Although the importance of β-glucan recognition by dectin-1 for zymosan-induced cytokines has been established in overexpression experiments involving cell lines and murine cells from genetically modified mice, the present study extends this work by demonstrating that β-glucan-dectin-1 interaction is important for the recognition of C. albicans by human primary PB-MCs.
The cell wall of C. albicans is a complex and dynamic structure consisting of a skeletal layer of chitin and β-glucans embedded in a matrix of mannoproteins and PLM [30]. It is conceivable that if one component of the cell wall is not recognized, alternative recognition mechanisms based on other pathogen-associated molecular patterns might compensate, especially in the case of primary cells that express multiple receptor complexes. In the present study, we demonstrated that this is not the case for dectin-1 recognition of β-glucans. When dectin-1 recognition was blocked by a specific antagonist (i.e., glucan phosphate) in human PBMCs or because of genetic modification in mice, stimulation of the proinflammatory cytokines TNF-α and IL-6 and the anti-inflammatory cytokine IL-10 by heat-killed C. albicans was strongly suppressed. This demonstrates that dectin-1 is a nonredundant pathway for C. albicans recognition in humans. Although this was true for heat-killed or UV radiation-killed C. albicans, live yeast cells stimulated cytokine production through a dectin-1-independent pathway via TLR4 and MR [14]. These findings further support a recent report showing that β-glucans of live yeasts are normally masked by a mannan-mannoprotein layer, precluding recognition by dectin-1 [27, 31].
These data have important consequences for understanding the way in which C. albicans is recognized by and stimulates cells of the innate immune system and for understanding the pathophysiology of fungal infections. On one hand, we demonstrated that exposure of β-glucans and their recognition by dectin-1 amplifies the cytokine production induced by C. albicans. We also confirmed that mannans have a dominant immunostimulatory role for live C. albicans [14]. It is conceivable that a similar phenomenon takes place in infected organs, where yeast cells elicit and progressively enhance the proinflammatory response because of increased exposure of β-glucans during the progressive digestion of the cell wall by phagocytes. It is known that β-glucans are released during invasive candidiasis, and detection of β-glucans released into the circulation is used as a diagnostic marker [32, 33]. Recent in vivo studies involving dectin-1-/- mice have suggested an increased susceptibility to disseminated candidiasis [21], supporting the hypothesis that recognition of β-glucan fragments released during infection contributes to the activation of host defense. In addition, the important β-glucan-induced stimulatory effect on cytokine production may also suggest the potential of β-glucans as adjuvants for fungal vaccines, a therapeutic approach that is currently being studied.
Initial studies have proposed that dectin-1 stimulates cytokine production through amplification of the TLR2 signaling pathway [19, 20]. However, the presence of an immunoreceptor tyrosine-based activation motif in the intracellular domain of dectin-1 suggests that dectin-1 may also induce TLR-independent signals. Indeed, recent studies have demonstrated TLR-independent stimulation of IL-2 and IL-10 production by β-glucan via a pathway involving Syk and CARD9 [28]. When we stimulated the cells with either live or heat-killed C. albicans, we observed that Syk inhibition had a differential effect on cytokine production. Although Syk inhibition did not influence cytokine production induced by live C. albicans, which is consistent with the lack of β-glucan recognition, it did significantly influence cytokine release induced by heat-killed cells. However, although inhibition of the Syk pathway only decreased the production of IL-6 and IL-10, the presence of Syk inhibitor did not influence stimulation of TNF-α by C. albicans. However, blockade of dectin-1 with glucan phosphate reduced TNF-α production. These data suggest that, although all of the cytokines we analyzed were induced by dectin-1 recognition of β-glucan, different pathways lead to stimulation of different cytokines. Release of IL-6 and IL-10 is mediated by dectin-1-Syk signals, whereas the induction of TNF-α by dectin-1 is Syk independent and may be mediated through amplification of TLR pathways. These findings open up new therapeutic possibilities involving the inhibition of cytokines that may have deleterious effects in certain stages of disease (e.g., specific Syk-dependent blockade of IL-10 production in patients with immunoparalysis due to prolonged systemic candidiasis).
Another interesting finding related to the role of β-glucan recognition by dectin-1 involves the induction of the Th1 cytokine IFN-γ. Although heat-killed C. albicans induced greater IFN-γ production than did live yeasts, this production was independent of dectin-1 blockade and Syk inhibition, demonstrating that dectin-1 has no apparent role in the induction of this Th1-type cytokine by C. albicans. Other structures of the C. albicans cell wall are therefore likely responsible for IFN-γ induction, and our earlier experiments suggested an important role of mannans and mannoproteins for this effect [14].
In conclusion, the present study demonstrates that recognition of C. albicans β-glucan by dectin-1 activates a nonredundant recognition pathway that has an important role in cytokine-mediated stimulation and activation of host defenses. Recent studies involving dectin-1-deficient mice also showed the crucial role played by dectin-1 recognition of β-glucans [21, 22], although they differed in their conclusions regarding the in vivo role of dectin-1 during infections with fungal pathogens, such as C. albicans and Pneumocystis carinii [21, 22]. In contrast, in live cells, mannans assume an important role in immune recognition by PBMCs. However, killing of C. albicans with subsequent exposure of β-glucans is necessary for strong dectin-1-mediated stimulation of cytokine production, and differential dectin-1-dependent pathways are involved in stimulating the release of proinflammatory and anti-inflammatory cytokines. Although amplification of TLR2 signals by dectin-1 has been described earlier, further studies are needed to evaluate whether interactions occur between dectin-1 and other PRRs (e.g., TLR4 and MR), especially in the context of complex recognition of the intact fungal cells by primary immune cells.
Acknowledgments
Financial support: The Netherlands Organization for Scientific Research to (Vidi grant to M.G.N.); Wellcome Trust (06324, 072263, and 080088 to N.A.R.G., A.J.P.B., and F.C.O.); Biotechnology and Biological Sciences Research Council.
Footnotes
Potential conflicts of interest: none reported.
References
- 1.Gudlaugsson O, Gillespie S, Lee K, et al. Attributable mortality of nosocomial candidemia, revisited. Clin Infect Dis. 2003;37:1172–7. doi: 10.1086/378745. [DOI] [PubMed] [Google Scholar]
- 2.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–17. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 3.van ’t Wout JW, Linde I, Leijh PCJ, van Furth R. Contribution of granulocytes and monocytes to resistance against experimental disseminated Candida albicans infections. Eur J Clin Microbiol Infect Dis. 1988;7:736–41. doi: 10.1007/BF01975039. [DOI] [PubMed] [Google Scholar]
- 4.Kullberg BJ, van ’t Wout JW, van Furth R. Role of granulocytes in enhanced host resistance to Candida albicans induced by recombinant interleukin-1. Infect Immun. 1990;58:3319–24. doi: 10.1128/iai.58.10.3319-3324.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marodi L, Korchak HM, Johnston RB., Jr. Mechanisms of host defense against Candida species. 1. Phagocytosis by monocytes and monocyte-derived macrophages. J Immunol. 1991;146:2783–9. [PubMed] [Google Scholar]
- 6.Qian Q, Jutila MA, Van Rooijen N, Cutler JE. Elimination of mouse splenic macrophages correlates with increased susceptibility to experimental disseminated candidiasis. J Immunol. 1994;152:5000–8. [PubMed] [Google Scholar]
- 7.Netea MG, van Tits LJH, Curfs JH, et al. Increased susceptibility of TNF-α lymphotoxin-α double knockout mice to systemic candidiasis through impaired recruitment of neutrophils and phagocytosis of Candida albicans. J Immunol. 1999;163:1498–505. [PubMed] [Google Scholar]
- 8.Kaposzta R, Tree P, Marodi L, Gordon S. Characteristics of invasive candidiasis in gamma interferon- and interleukin-4-deficient mice: role of macrophages in host defense against Candida albicans. Infect Immun. 1998;66:1708–17. doi: 10.1128/iai.66.4.1708-1717.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tonnetti L, Spaccapelo R, Cenci E, et al. Interleukin-4 and -10 exacerbate candidiasis in mice. Eur J Immunol. 1995;25:1559–65. doi: 10.1002/eji.1830250614. [DOI] [PubMed] [Google Scholar]
- 10.Tada H, Nemoto E, Shimauki H, et al. Saccharomyces cerevisiae- and Candida albicans-derived mannan induced production of tumor necrosis factor alpha by human monocytes in a CD14- and Toll-like receptor 4-dependent manner. Microbiol Immunol. 2002;46:503–12. doi: 10.1111/j.1348-0421.2002.tb02727.x. [DOI] [PubMed] [Google Scholar]
- 11.Netea MG, de Graaf C, Vonk A, Verschueren I, Van der Meer JW, Kullberg BJ. The role of Toll-like receptors in the defense against disseminated candidiasis. J Infect Dis. 2002;185:1483–9. doi: 10.1086/340511. [DOI] [PubMed] [Google Scholar]
- 12.Netea MG, Sutmuller R, Hermann C, et al. Toll-like receptor 2 inhibits cellular responses against Candida albicans through pathways mediated by IL-10 and CD4+CD25+ regulatory T cells. J Immunol. 2004;172:3712–8. doi: 10.4049/jimmunol.172.6.3712. [DOI] [PubMed] [Google Scholar]
- 13.Bellocchio S, Montagnoli C, Bozza S, et al. The contribution of Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol. 2004;172:3059–69. doi: 10.4049/jimmunol.172.5.3059. [DOI] [PubMed] [Google Scholar]
- 14.Netea MG, Gow NA, Munro CA, et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest. 2006;116:1642–50. doi: 10.1172/JCI27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto Y, Klein TW, Friedman H. Involvement of mannose receptor in cytokine interleukin-1β (IL-1β), IL-6, and granulocyte-macrophage colony-stimulating factor responses, but not in chemokine macrophage inflammatory protein 1β (MIP-1β), MIP-2, and KC responses, caused by attachment of Candida albicans to macrophages. Infect Immun. 1997;65:1077–82. doi: 10.1128/iai.65.3.1077-1082.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jouault T, Ibata-Ombetta S, Takeuchi O, et al. Candida albicans phospholipomannan is sensed through Toll-like receptors. J Infect Dis. 2003;188:165–72. doi: 10.1086/375784. [DOI] [PubMed] [Google Scholar]
- 17.Jouault T, El Abed-El Behi M, Martinez-Esparza M, et al. Specific recognition of Candida albicans by macrophages requires galectin-3 to discriminate Saccharomyces cerevisiae and needs association with TLR2 for signaling. J Immunol. 2006;177:4679–87. doi: 10.4049/jimmunol.177.7.4679. [DOI] [PubMed] [Google Scholar]
- 18.Brown GD, Gordon S. A new receptor for β-glucans. Nature. 2001;413:36–7. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 19.Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of β-glucans. J Exp Med. 2003;197:1119–24. doi: 10.1084/jem.20021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197:1107–17. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor PR, Tsoni SV, Willment JA, et al. Dectin-1 is required for β-glucan recognition and control of fungal infection. Nat Immunol. 2007;8:31–8. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saijo S, Fujikado N, Furuta T, et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol. 2007;8:39–46. doi: 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- 23.Endres S, Ghorbani R, Lonnemann G, Van der Meer JWM, Dinarello CA. Measurement of immunoreactive interleukin-1β from human mononuclear cells: optimization of recovery, intrasubject consistency, and comparison with interleukin-1α and tumor necrosis factor. Clin Immunol Immunopathol. 1988;49:424–38. doi: 10.1016/0090-1229(88)90130-4. [DOI] [PubMed] [Google Scholar]
- 24.Drenth JPH, Van Uum SHM, Van Deuren M, Pesman GJ, Van der Ven-Jongekrijg J, Van der Meer JWM. Endurance run increases circulating IL-6 and IL-1ra but downregulates ex vivo TNF-α and IL-1β production. J Appl Physiol. 1995;79:1497–1503. doi: 10.1152/jappl.1995.79.5.1497. [DOI] [PubMed] [Google Scholar]
- 25.Kullberg BJ, van ’t Wout JW, Hoogstraten C, van Furth R. Recombinant interferon-γ enhances resistance to acute disseminated Candida albicans infection in mice. J Infect Dis. 1993;168:436–43. doi: 10.1093/infdis/168.2.436. [DOI] [PubMed] [Google Scholar]
- 26.Netea MG, Demacker PNM, Kullberg BJ, et al. Low-density lipoprotein receptor-deficient mice are protected against lethal endotoxinemia and severe gram-negative infections. J Clin Invest. 1996;97:1366–72. doi: 10.1172/JCI118556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeasts but not filaments. EMBO J. 2005;24:1277–86. doi: 10.1038/sj.emboj.7600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rogers NC, Slack EC, Edwards AD, et al. Syk-dependent cytokine induction by dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 2005;22:507–17. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 29.Gross O, Gewies A, Finger K, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006;442:651–6. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- 30.Klis FM, de Groot P, Hellingwerf K. Molecular organization of the cell wall of Candida albicans. Med Mycol. 2001;39(Suppl 1):1–8. [PubMed] [Google Scholar]
- 31.Wheeler RT, Fink GR. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog. 2006;2:328–39. doi: 10.1371/journal.ppat.0020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obayashi T, Yoshida M, Mori T, et al. Plasma (1-->3)-β-D-glucan measurement in diagnosis of invasive deep mycosis and fungal febrile episodes. Lancet. 1995;345:17–20. doi: 10.1016/s0140-6736(95)91152-9. [DOI] [PubMed] [Google Scholar]
- 33.Odabasi Z, Paetznick VL, Rodriguez JR, Chen E, McGinnis MR, Ostrosky-Zeichner L. Differences in β-glucan levels in culture supernatants of a variety of fungi. Med Mycol. 2006;44:267–72. doi: 10.1080/13693780500474327. [DOI] [PubMed] [Google Scholar]