


Abstract
The selection of modified DNAzymes represents an important endeavor in expanding the chemical and catalytic properties of catalytic nucleic acids. Few examples of such exist and to date, there is no example where three different modified bases have been simultaneously incorporated for catalytic activity. Herein, dCTP, dATP and dUTP bearing, respectively, a cationic amine, an imidazole and a cationic guanidine, were enzymatically polymerized on a DNA template for the selection of a highly functionalized DNAzyme, called DNAzyme 9-86, that catalyzed (M2+)-independent self-cleavage under physiological conditions at a single ribo(cytosine)phosphodiester linkage with a rate constant of (0.134 ± 0.026) min−1. A pH rate profile analysis revealed pKa's of 7.4 and 8.1, consistent with both general acid and base catalysis. The presence of guanidinium cations permits cleavage at significantly higher temperatures than previously observed for DNAzymes with only amines and imidazoles. Qualitatively, DNAzyme 9-86 presents an unprecedented ensemble of synthetic functionalities while quantitatively it expresses one of the highest reported values for any self-cleaving nucleic acid when investigated under M2+-free conditions at 37°C.
INTRODUCTION
SELEX and related combinatorial methods of in vitro selection (1–3), have resulted in the identification of an increasing number of catalytic nucleic acids that catalyze a variety of reactions including ligation (4–6), DNA phosphorylation (7), RNA cleavage (8,9), thymine dimer photoreversion (10), formation of nucleopeptide linkages (11) and carbon–carbon bonds (Diels-Alder and aldol reactions) to name just a few (12,13). Unlike catalytic RNA, catalytic DNA has no precedent in nature (14–17). In the past few years, DNAzymes have emerged as prime candidates for applications ranging from biosensors of metal cations (4,18–23) to therapeutic agents for the sequence-specific destruction of mRNA for gene deactivation (8,24,25).
Generally, the selection for self-cleavage at a ribophosphodiester (RNA) linkage continues to predominate for several reasons including: (i) ease of selection (3,14,26), (ii) the precedence of naturally occurring catalytic RNA (27), (iii) the possibility of allostery for use in sensing (28) and (iv) the anticipated pharmaceutical and biotechnological applications of sequence-specific and catalytic mRNA destruction (25). In regards to this last application, the in vivo utility of ribozymes has been undermined by their relative chemical instability and susceptibility to ribonuclease-mediated degradation (29,30). DNA, being resistant to ribonucleases, represents an increasingly attractive platform for developing anti-mRNA catalysts.
To that end, highly efficient RNA cleaving DNAzymes (e.g. 10–23) have been identified. These generally require at least one divalent metal cation (M2+) for optimal activity and, at 10–25 mM Mg2+ display impressive kcat (∼4 min−1) and kcat/Km (109 M−1 min−1) values. Nevertheless, when the same DNAzymes are investigated under physiological conditions, kcat/Km values fall in the range of 102–104 M−1 min−1 (31). Since the intracellular Mg2+ concentration lies in the range of 0.1–0.2 mM (32–35), the paucity of Mg2+ may seriously undermine the efficacy of DNAzymes as well as ribozymes for catalytic destruction of intracellular mRNAs. We (36,37) and others (38–40) have recognized the value of catalytic nucleic acids that could operate efficiently at low or no Mg2+ so as to overcome this unfortunate limitation inherent to the intracellular milieu.
Examples of M2+-independent RNA-cleaving DNAzymes are rare; reasonably efficient cleavage was observed but twice: (i) kobs ∼ 0.05 min−1 at 5 mM L-histidine in 1 M M+Cl− (41) and (ii) kobs ∼ 1 min−1 at pH 3–5 (42) where adenine and cytosine competently afford general acid/base catalysis as is seen in naturally occurring ribozymes (43) and for a self-cleaving ribozyme selected under the same conditions (44). Three other reports characterized M2+-independent cleavage by DNAzymes at pH ∼7 in 0.25–1 M monovalent cations (Na+, K+). In two cases, 40–50 nt M2+-independent self-cleaving DNA sequences were found to be incapable of turnover (kself-cleav. 10−3–10−4 min−1) (45,46). A more recent report describes a M2+-independent DNAzyme with a similar kobs. that turns over twice in ∼100 h (47). The significance of M2+-independent RNases is further underscored in numerous reports on oligonucleotides synthetically endowed with various cationic amines, guanidines and imidazoles–functionalities that are found at the active sites of e.g. RNase A (48,49).
Paralleling these elegant studies in rational RNase design, numerous reports have identified monomer nucleoside triphosphates (dXTPs where X is any given nucleobase) that are modified with protein-like functionalities that can be enzymatically polymerized for use in combinatorial selection to potentially enhance the catalytic repertoire of nucleic acids (50–63). For example, Joyce et al. (64) selected a Zn2+-dependent DNAzyme with RNase activity by incorporating a C5-imidazole functionalized deoxyuridine in lieu of its natural counterpart. Although this DNAzyme with three essential imidazoles operated with multiple turnover, it required 1–10 μM concentrations of Zn2+. Thus, it is unlikely that this DNAzyme will be active in cells where the free Zn2+ concentration is nanomolar or lower (65,66).
There are only two reported cases of M2+-independent RNase A mimicking DNAzymes bearing both imidazole and amino groups obtained by in vitro selection. Sidorov reported self-cleavage of a 12-nt RNA target (kobs ∼ 0.07 min−1) although the requirement for modifications was not absolute (see Discussion section) (30). Our early efforts in discovering synthetic M2+-independent RNase A mimics that are ‘anatomically’ similar to M2+-dependent ribozymes and DNAzymes began with the simultaneous enzymatic polymerization of 8-histaminyl-dATP and 5-aminoallyl-dUTP (Figure 1A) (67) and culminated in the discovery of 925-11, which was selected for self-cleavage of a single-embedded ribophosphodiester linkage where modified nucleobases 1 and 2 (Figure 1A) were obligately required for activity (68). Whereas at 37°C, kobs. for self-cleavage was found to be ∼0.04 min−1, detailed kinetic investigations identified a high rate constant for M2+-independent self-cleavage (kobs ∼ 0.2–0.3 min−1) and a temperature optimum of 13°C. The self-cleaving species was then converted to a trans-cleaving species that was synthesized using solid phase synthesis (69). This species cleaved the ribonucleoside linkage both with multiple turnover (kcat ∼ 0.03 min−1, 25°C) and high sequence specificity (36,37). Attempts to improve activity by reselection with either 20 or 40 degenerate positions failed and instead the core sequence of 925-11 along with closely related sequence variants populated subsequent selections (data not shown). This result suggested that few viable solutions to M2+-independent RNA cleavage exist when nucleotides 1 and 2 are used in the selection. ‘Tyranny of the small motif’ (16), a phenomenon attributed to the repeated discovery of the M2+-dependent unmodified 8-17 motif (70) appears to extend modified selections as well.
Figure 1.

(A) Chemical structure of the modified triphosphates dAimTP 1, dUaaTP 2, dUgaTP 3 and dCaaTP 4. (B) Synthesis of the modified C5-guanidinium deoxyuridine analog.
The modest catalytic properties of Sidorov's self-cleaving DNAzyme and of 925-11, along with our inability to reselect superior catalysts with the same two modified dXTPs led us to hypothesize that two modified bases are not sufficient to improve cleavage rates, and that a third functionality, a guanidinium, would augment catalytic activity as well as provide for catalysts that would be both chemically and sequentially different from 925-11. We based this hypothesis in part on the fact that the guanidinium group exhibits cationic character over a wide pH range (pKa ∼ 12–13) and concomitantly stabilizes both duplex and triplex structures (71). More recently, the guanidinium functionality was incorporated into aptamers that recognize glutamate, further highlighting its potential for anion recognition (61). Herein, we present the discovery of a new M2+-independent, RNase A-like self-cleaving DNAzyme with three different chemical functional groups: an imidazole, a potentially basic cationic amine, and a fully cationic guanidine group. This is the first example of a high-density functionalized DNA displaying any form of catalytic activity.
MATERIALS AND METHODS
Chemicals and reagents
dAimTP was synthesized according to a literature procedure (67), dUaaTP was purchased from Sigma-Aldrich and dCaaTP was obtained from TriLink. Starting materials and all buffer and metal salts were obtained from Sigma-Aldrich, save for Fe(NO3)3·9H2O, UO2(OAc)2·2H2O, CoCl2·7H2O and CuSO4 which were purchased from Fisher. LiClO4 was purchased from J.T. Baker. Flash chromatography was carried out using Silica Gel (230–400 mesh) from Silicycle. Thin layer chromatography (TLC) and preparative TLC (prep-TLC) were performed on precoated glass-backed plates containing Silica Gel 60 F254 from EMD Chemicals. HPLC purification was performed on an Agilent 1100 system using a Phenomenex Jupiter 10μ C4 300A column. All oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA, USA) and were purified by 10–15% 7 M urea denaturing PAGE. Ultrapure dNTPs were obtained from Fermentas. The nucleoside triphosphate dGTP α-[32P] was purchased from Perkin Elmer.
Enzymes
Sequenase Version 2.0 and single-stranded DNA-binding protein were purchased from GE Healthcare. Lambda exonuclease, Taq DNA polymerase and Vent exo(-) DNA polymerase were obtained from New England Biolabs. Streptavidin magnetic particles were purchased from Roche. pGEM-T-Easy Vector Systems kit was obtained from Promega.
Oligonucleotides (shown 5′ to 3′)
Biotin-T20GCGTGCCrCGTCTGTTGGGCCCTACCAACA 1, GAGCTCGCGGGGCGTGCN20CTGTTGGTAGGGCCCAACAGACG 2, phosphate-CGTCTGTTGGGCCCTACCA 3, GAGCTCGCGGGGCGTGC 4, phosphate-ACGACACAGAGCGTGCCCGTCTGTTGGGCCCTACCA 5, TTTTTTTTTTTTTTTTTTTTGAGCTCGCGGGGCGTGC 6, phosphate-TAATACGACTCACTATAGGGAGCTCGCGGGGCGTGC 7, GGGGCGTGCGACACTACGCGCTGCATGATGTTGGTAGGGCCCAACAGACGGGCACGCTCGTGTCGT 8, TTTTCTTTTCCCCCCTGACCTTCCCGATTA 9, GCAGCTGTAGATCTTAGCCAGGCCTTAAAAGAAAAGGGGGGACTGGAAGGGCTAA 10 (Oligos 9 and 10 were used to assess incorporations of the modified dNTPs, see Supplementary Material). Biotin-T40r(GCGUGCCCGUCU)GTTGGGCCCTACCAACA 11. Biotin-T40m(GCGUGC)r(CCG)m(UCU)GTTGGGCCCTACCAACA 12. Bold-faced letters indicate the position of the embedded rC in oligonucleotide 1 and the corresponding site in oligonucleotides 11 and 12, whereas ‘r’ designates a stretch of RNA bases and ‘m’ a stretch of 2′OMe bases.
Nucleoside synthesis
Synthesis of 5-(3-N,N′-di-Boc-guanidinoallyl)-2′-deoxyuridine triphosphate (5)
The triethylammonium salt of 2 (0.5 μmol) was suspended in a mixture of H2O (1.7 μl) and dioxane (5.1 μl). N,N′-di-Boc-N″-trifluoromethanesulfonylguanidine (72) (0.6 mg, 1.5 μmol) was added to this solution, and the mixture was thoroughly stirred until the bis-Boc protected trifluoromethanesulfonylguanidine was fully dissolved. The reaction was left at room temperature and stirred occasionally. After standing at room temperature overnight, the reaction was dried down and resuspended in H2O. Triphosphate 2 (0.125 μmol, 25%) and triphosphate 5 (0.375 μmol, 75%) were isolated by prep-TLC (dioxane/H2O/NH4OH 6:4:1). MS (MALDI−): 764.6 (M–H)−. λmax = 289 nm.
Synthesis of 5-(3-guanidinoallyl)-2′-deoxyuridine triphosphate (3)
Trifluoroacetic acid (17 μl) was added to triphosphate 5 (0.375 μmol), and the resulting mixture was stirred for 2 min. The solution was cooled down to –78°C and the reaction was quenched by addition of Et2O (170 μl). The mixture was then centrifuged, and the supernatant was removed. The pellet was washed with three portions of Et2O (85 μl) and dried under vacuum. The crude product was dissolved in H2O and purified by prep-TLC (dioxane/H2O/NH4OH 6:4:1). TLC showed that the reaction was not quite complete. Triphosphate 3 was further purified on HPLC using a linear gradient of 0–2% acetonitrile in H2O (over 20 min) containing triethylammonium bicarbonate (0.05 M, pH 7.5) yielding 85 nmol (23%) of product. Since the absorptions of the triphosphate modifications did not affect the absorption of 2, the triphosphate products were quantified using εmax = 7100 M−1 cm−1. Retention time: 4.7 min. MS (MALDI−): 564.0 (M–H)−. λmax = 289 nm.
Buffers
1 (cleavage buffer): 50 mM sodium cacodylate (pH 7.4), 200 mM NaCl, 1 mM EDTA. 2 (elution buffer): 1% LiClO4/Tris–HCl 10 mM (pH 8) in water. 3 (metals): 50 mM sodium cacodylate (pH 7.4), 200 mM NaCl and 500 μM M2+/3+ [M2+ = Co2+, Cu2+, Mn2+, Ba2+, Zn2+, Ca2+, Hg2+, (UO2)2+, Ni2+, Cd2+, Mn2+, Mg2+ and M3+ = Yb3+, Co3+, Ce3+, Fe3+, Eu3+, Sm3+] or 5 mM Mg2+. 4 (pH variance): 50 mM Tris–HCl, 200 mM NaCl, 1 mM EDTA. The pH of all buffers was adjusted to 5.97, 6.50, 6.94, 7.49, 7.94, 8.46 or 8.90. 5 (ionic strength): 50 mM sodium cacodylate (pH 7.4), 1 mM EDTA and NaCl (0 mM, 50 mM, 100 mM, 500 mM, 750 mM and 1 M). 6 (monovalent salts): 50 mM sodium cacodylate (pH 7.4), 1 mM EDTA and 200 mM MCl (M+ = K+ and Li+).
In Vitro selection
Thirty picomoles of oligonucleotide 1 (5′-biotin-T20GCGTGCCrCGTCTGTTGGGCCCTACCAACA-3′) were annealed to 30 pmol of template DNA (T20GAGCTCGCGGGGCGTGCN20CTGTTGGTAGGGCCCAACAGACG prepared by nested polymerase chain reaction (PCR) using primers 5 and 6), then enzymatically polymerized at 37°C for 3 h using 9.1 U of Sequenase in a mixture containing single-stranded binding protein (SSB, 5 U), 5 mM DTT, 50 μM dAimTP, 10 μM of each dUgaTP, dCaaTP, dGTP and 5–15 μCi of dGTP α-[32P]. The reaction was quenched by adding EDTA (25 mM final). The extension product was immobilized on 50 μl of prewashed magnetic streptavidin particles by incubating at room temperature for 30 min. After two short washes with 100 μl TEN buffer, the template strand was removed by five washes of 100 μl NaOH 0.1 M and EDTA 1 mM, followed by a neutralization wash of 200 μl cacodylate 25 mM (pH 6) and one 100 μl water wash. The particles were then suspended in 100 μl buffer 1 at room temperature for 60 min. The reaction time was decreased from 60 min down to 1 min over the selection (rounds 1 to 4: 60 min; rounds 5 and 6: 5 min; rounds 7 to 9: 1 min). From round 4 onwards, self-cleavage activity was measured at 1 min, 5 min and 60 min. Following magnetization, the supernatant was precipitated (1% LiClO4 in acetone), washed (EtOH), resuspended and resolved by 7% 7 M urea denaturing PAGE. The species corresponding to the cleaved product was eluted using buffer 2, precipitated and desalted. The PCR amplification of the resulting modified DNA followed the nested double PCR amplification method outlined in the selection of 925-11 (68). In the first amplification step, the modified DNA was PCR amplified using primers 3 and 4 and an internal label (10 μCi dGTP α-[32P]) for 30 cycles (15 s at 54°C, 40 s at 75°C and 15 s at 95°C). The reaction buffer included 0.07 U/μl Vent(exo-) DNA polymerase, 20 mM Tris–HCl (pH 8.8 at 25°C), 10 mM (NH4)2SO4, 10 mM KCl, 3 mM MgSO4, 0.1% gelatin, 7 μM oligonucleotides and 0.3 mM of each natural dNTP. Prior to purification by 10% 7 M urea denaturing PAGE, the amplicon was treated with lambda exonuclease. An aliquot was then further amplified using primers 5 and 6 with 0.1 U/μl Vent(exo-) DNA polymerase over 30 PCR cycles (using the same program as for the first amplification). The resulting product was precipitated (phenol–chloroform followed by an EtOH wash) and the phosphorylated strand was digested using lambda exonuclease. The single-stranded DNA product was then purified by 10% 7 M urea denaturing mini-PAGE and identified by UV-shadowing. The resulting DNA was used in the ensuing round of selection. A total of nine rounds of selection were performed.
Cloning of cDNAs
The nineth generation was amplified using Taq DNA polymerase with primers 3 and 7 to produce PCR products with 3′-A overhangs. These amplicons were then TA-cloned using the pGEM-T-Easy Vector Systems kit and were used to transform Escherichia coli DH10B via electroporation. The transformation was plated on LB Agar containing 100 mg/l ampicillin. White colonies were picked and used to inoculate 1 ml of TB containing Plasmid Miniprep Kit and were subjected to restriction digest using EcoRI. Plasmids containing a single insert of the correct size (as controlled by 2% agarose gels), were submitted for sequencing. The Nucleic Acid Protein Service Unit of UBC carried out the sequencing of the most active clones using an SP6 sequencing primer. From the 100 random clones, 34 contained single inserts. Synthetic oligonucleotides corresponding to the various clones were used as templates to synthesize modified DNA as described previously. Consequently, 5 pmol of each individual synthetic oligonucleotide were then immobilized on streptavidin magnetic particles. Following two short washes with 100 μl TEN buffer, the template strands were removed by three washes with 100 μl NaOH 0.1 M and EDTA 1 mM, followed by a neutralization wash of 200 μl cacodylate 25 mM (pH 6) and one 100 μl water wash. The modified DNAs were then incubated at room temperature in 40 μl of buffer 1. Eight time points were taken (2, 5, 10, 15, 30, 60, 120 and 1000 min) and resolved by 7% 7 M urea denaturing PAGE. Clone #86 showed the highest activity and was thus, fully characterized as reported herein.
Kinetic analysis of intramolecular cleavage
Thirty-two picomoles of primer 1 containing the embedded ribose, rC, (5′-biotin-T20GCGTGCCrCGTCTGTTGGGCCCTACCAACA-3′) was annealed to 30 pmol of synthetic template DNAzyme 9-86, then enzymatically polymerized at 37°C for 3 h using Sequenase 2.0 in a mixture containing SSB protein (5 U), 50 μM dAimTP, 10 μM of each dUgaTP, dCaaTP, dGTP and 5–15 μCi of dGTP α-[32P]. The reaction was quenched by adding EDTA (25 mM final). The extension product was immobilized on 50 μl of prewashed magnetic streptavidin particles by incubating at room temperature for 30 min. The template strand was then removed by five washes of 100 μl NaOH 0.1 M and EDTA 1 mM, followed by a neutralization wash of 200 μl cacodylate 25 mM (pH 6) and one 100 μl water wash. The slurry of streptavidin particles in water was then divided in an appropriate amount of tubes and decanted. The avidin-bound modified DNA was incubated in 90 μl of buffer 1. Five microliter of the slurries were quenched in 15 μl formamide [containing biotin (1 mM), EDTA (25 mM), 0.01% bromophenol blue and 0.01% xylene cyanole]. Samples were then heated (95°C, 5 min), cooled (0°C), magnetized and resolved by 7% 7 M urea denaturing PAGE. Visualization was carried out by means of a phosphorimager (Amersham Typhoon 9200) and polygons were drawn around the bands corresponding to the cleaved and uncleaved species. The data of the cleavage reactions were then fitted to first-order reactions with Sigmaplot 2001 (version 7.101) using equation (1):
| 1 |
Where Pt and P∞ are the fractions cleaved at time t and the end point of the reaction, respectively and k is the observed first-order rate constant. At least three independent sets of data were collected. In some cases, truncated bands were observed, even when using unmodified dNTPs. These unexplained truncates, were all found to be catalytically inactive as evidenced by their constant presence throughout the time course of the reaction. Indeed, because polygons were drawn around only the bands corresponding to the cleaved and uncleaved species, these truncated bands do not complicate the kinetics or interfere with a calculation of the rate constant.
Temperature dependence
The single-stranded modified DNAs were incubated in 90 μl of buffer 1 (which was preheated at the appropriate temperature for 30 min prior to the experiment) at various temperatures. Buffer 1 was prepared at 24°C and cacodylate was chosen on account of the fact that the pH of a cacodylate solution remains relatively constant with temperature (ΔpH/ΔT = −0.0015 pH U/°C) (73). All experiments were carried out under mineral oil to prevent evaporation. Temperature varied <0.5°C in a VWR temperature controlled water bath. First-order rate constants (kobs) were then obtained by fitting the fraction cleaved to equation (1). The results shown are the average of two independent data sets and each reaction was carried out in triplicate. Data were then fit to the Arrhenius equation (2):
| 2 |
where Ea is the activation energy and A is the pre-exponential factor. Activation parameters were obtained by application of transition state theory using the Eyring equation (3) (74):
| 3 |
pH-rate profile and effect of ionic strength
The dependence of the rate of self-cleavage of DNAzyme 9-86 on pH was measured by incubating single-stranded modified DNAs obtained as described above in 90 μl buffer 4. The pH range was 5.94–8.90. The data were fitted to equation (4) (75):
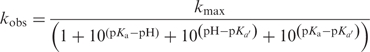 |
4 |
where kmax is the limit of the catalytic rate when [H+] reaches zero and Ka and Ka′ are the ionization constants of the catalytically essential groups involved. The dependence on ionic strength was measured by using a buffer containing varying concentrations of NaCl (buffer 5) at room temperature. Buffer alone (without addition of salt) contained <50 mM of monovalent cation (Na+). Again, the results shown are the average of two independent data sets and each reaction was carried out in triplicate.
Determination of the rate constant with the all-RNA and 2′OMe/RNA substrates
Thirty-two picomoles of primer 11 containing 12 RNA nucleotides (5′-biotin-T40r(GCGUGCCCGUCU)GTTGGGCCCTACCAACA-3′) or primer 12 containing three ribonucleotides denoted as ‘r’ flanked by 2′OMe-nucleotides denoted as ‘m’ (5′-biotin-T40m(GCGUGC)r(CCG)m(UCU)GTTGGGCCCTACCAACA-3′) were annealed to 30 pmol of synthetic template coding for DNAzyme 9-86, then enzymatically polymerized at 37°C for 3 h using Sequenase 2.0 in a mixture containing SSB protein (5 U), 50 μM dAimTP, 10 μM of each dUgaTP, dCaaTP, dGTP and 5–15 μCi of dGTP α-[32P]. The reaction was quenched by adding EDTA (25 mM final). The extension product was immobilized on 50 μl of prewashed magnetic streptavidin particles by incubating at room temperature for 30 min. The template strand was then removed by five washes of 100 μl NaOH and 0.1 M, EDTA 1 mM, followed by a neutralization wash of 200 μl cacodylate 25 mM (pH 6) and one 100 μl water wash. Ten units of SUPERase-Inhibitor (Ambion) were then added and the reactions were initiated by the addition of 90 μl of buffer 1. The kinetic analyses were then carried out in a similar manner to what was used with primer 1.
RESULTS
Synthesis of dUgaTP 3 and in vitro selection
The modified deoxynucleoside triphosphate analog dUgaTP (5-guanidinoallyl-dU) 3 was easily obtained by guanidinylation of the commercially available triphosphate dUaaTP 2 with N,N′-di-Boc-N″-trifluoromethanesulfonylguanidine under mild conditions, followed by removal of the Boc-protecting groups (Figure 1B) (52,72). In preliminary experiments, dUgaTP was examined for its compatibility with in vitro selection methods, which require a modified triphosphate to be a substrate for polymerases in the template-directed primer extension reaction which affords modified DNAs that must be competent templates in a PCR. The thermostable polymerases Vent (exo-) and Sequenase V 2.0 were both shown to incorporate dUgaTP 3 opposite dA in a primer extension reaction and yield full length products (Supplementary Material). Moreover, the modified DNA could be recopied into natural DNA by PCR: Vent (exo-) proved useful for this, whereas Taq was found to be a less potent enzyme to amplify such modification-rich strands (Supplementary Material). For compatibility with dAimTP 1 (67), we chose to employ Sequenase V 2.0 for the synthesis of modified DNA in the primer extension reaction and Vent (exo-) for amplification by PCR.
Consequently, an initial population was generated by polymerizing the three modified nucleoside triphosphates dAimTP 1, dCaaTP 2 and dUgaTP 3 (Figure 1A) along with dGTP on a template comprising 20 degenerate positions (≤1012 sequences) by application of a protocol applied in other in vitro selection experiments (45,68,76). The 5′-biotinylated primer used in the selection contained an embedded ribonucleoside (rC) flanked at both sides by target guide DNA sequences derived from a corresponding HIV-LTR mRNA sequence. Following removal of the template strand and neutralization, the single-stranded modified DNA was incubated in 200 mM NaCl, 50 mM cacodylate (pH 7.4) and 1 mM EDTA at room temperature for 60 min. The stringency was gradually increased over the course of the selection by gradually decreasing the reaction time from 60 min (G1–G4), to 5 min (G5–G6), and finally to 1 min (G7–G9). The selection progress is depicted in Figure 2A, where activity was gauged at several time points for each generation. Activity was observed at a relatively early stage of the selection (2% cleavage after 60 min for round 3) and the reaction times were decreased when the population achieved a relatively strong activity (>5% cleavage). While the fraction of sequences that cleaved after 60 min dropped in the final generations, the fraction of those cleaving within 5 min increased in generations 6–8 suggesting that increased stringency eliminated slow-to-cleave species in favor of both fast-to-cleave and inactive species. An explanation for this bifurcation is not immediately forthcoming. After nine generations, cloning and sequencing resulted in 34 distinct sequences (Supplementary Material).
Figure 2.

(A) Progress of the selection: the fraction is shown for each generation (round of selection). For the first four rounds a selection time of 60 min was used. In round 5, the selection time was reduced to 5 min. At round 7, the selection time was again decreased to 1 min and maintained until round 9. Starting at round 4, self-cleavage activity was measured at 1, 5 and 60 min. (B) Sequence and hypothetical 2D structure of the selected DNAzyme 9-86. (bold-face A, C and U indicate the position of the modified nucleosides 1, 2 and 3, respectively).
The randomized regions of the sequences obtained from the selection varied substantially in length (between 18 nt and 26 nt) and often contained mutations in the target sequence at the 5′-end. Synthetic oligonucleotides corresponding to the cloned sequences were then used in an initial kinetic survey to assess their activity. Clone 86 showed the highest catalytic activity and was characterized further. The sequence and hypothetical 2D structure of DNAzyme 9-86 is shown in Figure 2B. The degenerate region is slightly shorter than the initial library (19 nt instead of 20 nt) and consists of two putative hairpin loops. In addition, the AT and GC contents are roughly equal (42% and 58%, respectively) and only three imidazole-containing residues are present.
Kinetic analysis of self-cleavage
In order to investigate self-cleavage, DNAzyme 9-86 was prepared as described above for the selection. Generally, a single major product was observed along with a slightly shorter truncated band that was catalytically inactive and thus did not interfere with kinetic analysis (see Materials and Methods section). Others have also observed catalytically inactive truncated products (30,45). The full length transcript DNAzyme 9-86 self-cleaves with an average rate constant kobs of (0.134 ± 0.026) min−1 (error based on 11 different reactions carried out on different days with different modified DNA preparations) in 1 mM EDTA, 200 mM NaCl, 50 mM cacodylate pH 7.4 at 24°C (Figure 3). Cleavage is high yielding (>90%) and kinetics are best fit to a monophasic single-exponential equation with fit errors that are often as low as 1% suggesting a high degree of conformational homogeneity.
Figure 3.

Kinetic analysis of self-cleavage. (A) Representative autoradiographic image of denaturing PAGE 7% showing the fraction cleaved over a period of 900 min. (B) Graphical analysis of this particular run: kobs = (0.167 ± 0.007) min−1 (R2 > 0.99); error is standard deviation from the exponential fit. Reaction conditions: 1 mM EDTA, 200 mM NaCl, 50 mM cacodylate pH 7.4, 24°C. A slightly truncated species is observed and remains relatively constant throughout the time course suggesting the enzymatic production of minor, catalytically inactive species. Rate constants were calculated using equation (1) based on the relative autoradiographic densities in both the uncleaved and cleaved bands. Time points were 1, 3, 6, 9, 12, 20, 30, 45, 60, 90, 120, 150, 180, 240 and 900 min.
The need for modifications
As with DNAzyme 925-11, the observed activity for DNAzyme 9-86 is strictly dependent on the presence of the modified bases; synthesis of strands where one, two or three modified dXTP(s) is/are replaced with an unmodified counterpart(s) results in species entirely devoid of catalytic activity (Figure 4). Indeed, the replacement of any one of the modified dNTPs by a natural counterpart led to a total suppression of activity (<10% cleavage over 1200 min, Figure 4A), as well as significantly higher amounts of truncated products that form even when using a complement of four unmodified dNTPs. The ablation of two of the modifications is even more deleterious (Figure 4B) and no modifications also gave inactive strands (data not shown). In addition, incorporation of aminoallyl-dUTP in lieu of dUgaTP, along with both dCaaTP and dAimTP, led to a complete suppression of activity suggesting that the guanidinium cation itself is required and cannot be readily replaced by an allylammonium ion (data not shown). Finally, exchanging dAimTP 1 for a closely related chemical homolog (4-imidazolylpropylamino-dA) dAhomTP (76,77), which contains but one additional methylene in the spacer linking the imidazole moiety to the adenine, led to catalytically inactive species (Figure 4B,C). These observations suggest that the incorporation of the three modified dNTPs leads to a finely tuned catalytic surface consistent with a veritable active site that is sensitive to rather minute alterations at the molecular level.
Figure 4.

Gel images (PAGE 7%) demonstrating the importance of the three modifications. (A) Modified DNAs with dAimTP, dCaaTP and dUgaTP single knockouts. (B) Modified DNAs synthesized with dAimTP as sole modification and with dAhomTP instead of dAimTP. As a control, the elongated strands were also cleaved with RNase A to identify anticipated cleavage products that are denoted with asterisks. (C) Structure of dAhomTP, which when added as a triphosphate in lieu the histaminyl dATP is incorporated into strands that manifest <10% activity after 1260 min. Time points were 3, 5, 10, 20, 30, 60 and 1260 min.
Effect of temperature and pH-rate profile
Because changes in temperature and pH often have a particularly drastic effect on the rate of M2+-independent DNAzymes, 9-86 self-cleavage was investigated as a function of changing these parameters. The dependence of the rate constant on temperature is shown in Figure 5A. Even though DNAzyme 9-86 was selected at room temperature (24°C), the rate constant increases nearly linearly with temperature and reaches a maximum at 37°C before decreasing at higher temperatures. A break in the linear increase is reproducibly observed between 20°C and 30°C, hinting at a possible dynamic rearrangement of the secondary and tertiary structures of the DNAzyme, reflecting a second, catalytically competent conformation (78,79). In addition, the linear part of the Arrhenius plot (Figure 5B) was fit to equation (2) and gave an activation energy of 15.9 kcal mol−1. By application of transition state theory [using the Eyring equation (3)], an enthalpy of activation of ΔH‡ = 15.3 kcal mol−1 and an entropy of activation of ΔS‡ = −11.9 eu were obtained (Δ G‡298K=18.8 kcal mol−1).
Figure 5.

(A) Temperature dependence of the intramolecular RNA cleavage, measured in 200 mM NaCl, 50 mM cacodylate pH 7.4, and 1 mM EDTA over a range of 4–53°C. Cacodylate was chosen for a relatively constant pH at variable temperature (ΔpH/ΔT = −0.0015 pH U/°C), and error bars indicate standard error on triplicate runs. (B) Arrhenius plot of the temperature dependence of DNAzyme 9-86 in the interval 4–32°C. Fitting the data to a linear regression gave Ea = 15.9 ± 2.3 kcal mol−1 (R2 = 0.94).
As might be expected, the pH-rate profile adopts a bell shape (Figure 6). The rate of RNA-cleavage steadily increases with the pH over the range of 6.0–7.5 and reaches a plateau between pH 7.5 and 8.0 before decreasing over the range of 8–9. At lower pH values (i.e. between 6 and 7) the logarithmic value of kobs increased linearly with pH with a slope close to unity (0.86) (data not shown), suggesting that a single deprotonation event is involved in the rate-limiting step at pH values less than neutral. Using equation (4), the pH-rate profile analysis revealed pKa's of 7.4 and 8.1, consistent with pKa's of two catalytically relevant groups, most likely two imidazoles, although we cannot exclude the possibility of other functionalities acting as acids or bases (see Discussion section).
Figure 6.

Cleavage rate dependence on pH, measured in (50 mM buffer, 200 mM NaCl, 1 mM EDTA) at room temperature, error bars indicate standard error on triplicate runs. Values for pKa were calculated using equation (4) (See Materials and methods section) to be 7.4 ± 0.1, 8.1 ± 0.1 and kmax = 0.29 ± 0.06 min−1 (R2 = 0.96).
Site of cleavage and comparison of the activity of DNAzyme 9-86 for DNA/RNA chimeric and all-RNA substrates
DNAzyme 9-86 was selected to catalyze self-cleavage of a single ribophosphodiester linkage embedded in a DNA strand. Not surprisingly, DNAzyme 9-86 was incapable of self-cleavage when the 2′OH was replaced with an all-DNA or 2′OMe target (data not shown), thus demonstrating that cleavage does not occur via direct hydrolysis of an RNA or DNA linkage. In addition, this also excludes the possibility of DNA scission in the vicinity of the target ribose that might have proceeded either oxidatively (80) or via a two-step mechanism involving first depurination (81) followed by an AP-lyase-like catalyzed fragmentation (82). Moreover, the 3′-cleavage product was competently phosphorylated by T4 polynucleotide kinase showing that cleavage leads to the generation of a free 5′OH in the 3′-product (Supplementary Material) (46). Although the 5′-cleavage product remains uncharacterized, it is likely that DNAzyme 9-86 generates a 2′,3′-cyclic phosphate; otherwise it would be unprecedented if 9-86, instead of exploiting the 2′OH for anchimeric attack, were to activate water for cleavage. The resistance of the analogous DNA and 2′OMe sequences further argues against activation of water for hydrolysis.
Interestingly, DNAzyme 9-86 was also able to cleave a 12-nt long RNA substrate at presumably one specific location (Table 1 and Supplementary Material). Moreover, DNAzyme 9-86 could even cleave a 3-nt long RNA substrate embedded within a 9-nt long sequence of 2′OMe-containing ribonucleotides to afford a product of identical electrophoretic mobility. However, exchanging the chimeric DNA/RNA substrate for either an all-RNA or a 2′OMe/RNA chimeric substrate resulted in a substantial (∼100-fold) loss of activity (Table 1), but it is nonetheless noteworthy that this DNAzyme can cleave an all-RNA sequence (see Discussion section).
Table 1.
Rate constants of deoxyribozyme 9-86 with different substrates
| Substrate | kobs (min−1) |
|---|---|
| DNA/RNA chimera | 0.134 ± 0.026 |
| All-RNA | 0.0014 ± 0.0001 |
| 2′OMe/RNA chimera | 0.0021 ± 0.0002 |
Effect of monovalent ionic strength
Because DNAzyme 9-86 was selected in the presence of 200 mM NaCl, the effect of ionic strength on the cleavage rate was also investigated. In contrast to 925-11, where the value of kobs fell linearly with decreasing ionic strength, in the concentration range tested (50 mM to 1 M) the observed rate of intramolecular cleavage is virtually indifferent to the ionic strength. Only when examined in 50 mM sodium cacodylate and no added NaCl was there a significant loss in rate of cleavage (Figure S5A, Supplementary Material). Finally, exchanging Na+ for either Li+ or K+ resulted in no inhibitory effect, suggesting that DNAzyme 9-86 operates independently of the nature of the monovalent salt employed (data not shown).
Effect of various M2+ ions
Our interest in exploring the advantages and limitations of using modified dXTPs to select for M2+-independent RNA-cleaving agents stems from the realization that besides Mg2+, the intracellular concentrations of other, free divalent metal cations are generally submicromlar if not much lower (65,66,83,84). Nevertheless, the significance of this work extends beyond the potential use as anti-mRNA agents; modifications, particularly imidazoles, specifically recognize certain divalent metal cations for sensing or for use in chemical catalysis at micromolar concentrations or higher. For instance, it was not surprising that 925-11 was strongly inhibited by mercury cations (85), a finding that prompted us to reselect for DNAzyme 10-13 that could be stimulated by Hg2+ and could thus act as a sensor (76).
In light of the potential for enhanced affinity for soft metal cations, we investigated to what extent the activity of DNAzyme 9-86 was impaired by the presence of 500 μM M2+/M3+, 200 mM NaCl and cacodylate 50 mM, pH 7.4 at room temperature (Table 2 and Supplementary Material). At 0.5 mM, physiologically relevant metal ions, such as Ca2+ and Mg2+ had little or no effect on the cleavage rate (even in the absence of NaCl in the cleavage buffer; data not shown). Other di- and trivalent metal cations, such as Mn2+, Co3+ and Fe3+ were found to be only mildly inhibitory. More aminophilic metals, such as Ni2+ and Zn2+, along with soft metal cations such as Cd2+, Cu2+ and Hg2+ having an exemplary affinity for soft imidazoles completely abolished cleavage. Unlike 925-11, DNAzyme 9-86 showed less selectivity for mercury since many other metal ions displayed a similar deleterious effect on its catalytic activity. Nevertheless, DNAzyme 9-86 still maintains a strong affinity for mercuric cations. Indeed, by application of the same kinetic analysis based on a steady-state inhibition model described in the metal survey of DNAzyme 925-11 (85), an apparent dissociation constant (KAPPd) of 80.9 ± 0.4 nM was calculated for Hg2+ (data not shown), which is similar to what had been measured for 925-11 (KAPPd = 110 ± 9 nM) (85). The significance of these findings is further addressed in the Discussion section.
Table 2.
Effect of metal cations on the cleavage rate of DNAzyme 9-86a
| Metal | kobs (min−1) | krel |
|---|---|---|
| Buffer only | 0.1041 | 1 |
| Mg2+ (5 mM) | 0.0580 | 0.56 |
| Mg2+ (0.5 mM) | 0.07752 | 0.75 |
| Mn2+ | 0.03807 | 0.37 |
| Ba2+ | 0.06509 | 0.63 |
| Co3+ | 0.04507 | 0.43 |
| Ce3+ | 0.005429 | 0.05 |
| Co2+ | 0.004388 | 0.04 |
| UO22+ | 0.07837 | 0.75 |
| Ca2+ | 0.07629 | 0.73 |
| Fe3+ | 0.05431 | 0.52 |
| Hg2+ | n.d.b | n.d. |
| Eu3+ | n.d. | n.d. |
| Sm3+ | n.d. | n.d. |
| Cu2+ | n.d. | n.d. |
| Yb3+ | n.d. | n.d. |
| Cd2+ | n.d. | n.d. |
| Ni2+ | n.d. | n.d. |
| Zn2+ | n.d. | n.d. |
aReaction conditions: 200 mM NaCl, 50 mM cacodylate pH 7.4, 500 μM M2+/3+, RT.
bNo cleavage detected.
DISCUSSION AND CONCLUSION
The highly functionalized DNAzyme 9-86 bears three different side chains mimicking the side chains of histidine, lysine and arginine, which populate the active sites of numerous protein enzymes. These functionalities allow this small 19-nt DNAzyme 9-86 to catalyze the cleavage of a single ribocytosine embedded in a DNA strand with a rate constant of 0.13 min−1. This catalytic activity occurs without the aid of any divalent metal cations, and thus proceeds under conditions wherein most catalytic nucleic acids are catalytically impaired, if not entirely inactive. The rate achieved by DNAzyme 9-86 is comparable with what had been observed for DNAzyme 925-11 at 13°C (68), yet is much more active at 37°C, and exceeds catalytic rates by a >100-fold over other unmodified DNAzymes (45,46) and at least 2-fold over similarly modified DNAzymes (30).
Factors such as the presence of metal cations, variation of pH and change of the temperature affect the rate of RNA-cleavage of DNAzyme 9-86. Because nucleic acids are negatively charged biopolymers, their folding strongly depends on the presence of cations such as Na+ to neutralize the negative charges on the backbones and attenuate the resulting repulsion (86). However, increasing the ionic strength (from 50 mM to 1 M) of the cleavage buffer had a minimal effect on the rate constant of DNAzyme 9-86. This might be attributed to the stabilizing effect of the guanidinium side chains, which are known to stabilize duplexes and triplexes (71). Although the absence of any added salt (NaCl), resulted in a loss of activity, DNAzyme 9-86 efficiently cleaves the embedded ribocytosine bond at salt concentrations even as low as 50 mM, which is quite noteworthy since i) most other M2+-independent DNAyzmes and ribozymes are only active at 1–4 M monovalent cations and ii) 925-11 displayed significant reduction in activity when the monovalent ionic strength was reduced from 200 mM to 50 mM.
As expected, DNAzyme 9-86 is most active in the total absence of divalent metal ions, which reflects the initial conditions used for the in vitro selection. In particular Mg2+, which is the only readily available intracellular divalent metal cation, had little effect on the rate constant of DNAzyme 9-86. Furthermore, DNAzyme 9-86 is strongly inhibited by a number of transition metal cations but only at concentrations that are much higher than physiological. This finding suggests that: (i) at physiological levels of such metal cations, DNAzyme 9-86 is active, and (ii) these three modified nucleosides may be useful for selecting interesting M2+-based catalysts for sensing and/or chemical transformations.
Interestingly, a ∼5-fold overall increase in rate constant was observed when the temperature was raised from 4°C to 37°C at which point the rate of self-cleavage reached an apparent maximum in contrast to 925-11 that diminished drastically at 37°C. A similar temperature dependence has been observed for DNAzyme 8-17, albeit with a more impressive rate increase (87). The activation parameters of 9-86 are similar to those observed for a high temperature Zn2+-dependent DNAzyme (74), but differ substantially from the values reported for DNAzyme 8-17 (87) and the hammerhead ribozyme (79).
The bell-shaped pH-rate profile of DNAzyme 9-86 is consistent with a two-step protonation–deprotonation mechanism: fitting the data to equation (4) revealed pKa's of 7.4 and 8.1, which are consistent with two imidazole groups involved in general base and general acid catalysis. Although we cannot exclude the possibility that a pKa-perturbed allylammonium or its conjugate base may act respectively as an acid or base, it is noteworthy that when the imidazoles are positioned on a slightly longer linker (propyl versus ethyl), no activity was observed. This suggests that at least two of the three imidazoles are intimately involved in some aspect of catalysis rather than structural positioning. Nevertheless, pH-dependent conformational changes along with the possibility of other pKa perturbed groups may account for a bell-shaped pH-rate profile. As such kinetic ambiguity is seen in naturally occurring ribozymes (88) and because many ionizable groups may influence folding in this case, we are cautious in concluding that the bell-shaped rate profile necessarily represents acid and base catalysis or that it is due to two specific imidazoles. Future studies including affinity labeling will help to identify critical groups needed for acid/base catalysis (89).
Most DNAzymes that have been selected for the cleavage of single ribonucleotide linkage within a chimeric DNA substrate are either inactive or suffer drastic (∼1000-fold) losses of activity in the presence of all-RNA substrates. In contrast, DNAzyme 17E cleaves an all-RNA substrate despite having been selected to cleave a DNA/RNA chimeric substrate (90). However, this property is not totally unexpected since 17E has a similar catalytic core to 8-17, which had been selected to target an all-RNA strand. Compared with the complete inactivity of 925-11 against an all-RNA target, it is thus significant that DNAzyme 9-86 shows a propensity to cleave an all-RNA substrate, further suggesting that the additional functionalities of DNAzyme 9-86 indeed enhance activity in terms of recognizing all-RNA substrates.
In contrast to the self-cleaving DNAzyme selected by Sidorov et al. where some residual activity was observed in the same sequence lacking both modifications, and approximately 10% or 30% activity was seen when either the amine or imidazole was respectively ablated, the functionalities adorning DNAzyme 9-86 are vital for the catalytic activity. Indeed, replacing all of any one modified nucleotide with its unmodified congener led to a total suppression of catalytic activity. Moreover, even replacing histaminyl-dA for imidazolylpropylamino-dA abolished virtually all activity, suggesting that the putative active site of DNAzyme 9-86 is intolerant to even the minutest alterations either in regards to chemical composition or the spatial orientation of the side chains that are involved for either folding or catalysis, or both.
In conclusion, the in vitro selection discovery of DNAzyme 9-86 demonstrates how incorporation of three different nucleotides can be used combinatorially to discover catalytically active DNA enzymes which could be used in practical applications ranging from the catalysis of chemically useful reactions to the elaboration of novel biomaterials. In this regard, the addition of a third modification, in this case a guanidine, quantitatively and qualitatively improves the M2+-independent RNA-cleaving activity when compared with two antecedent modified M2+-independent RNA-cleaving DNAzymes that presented only two protein-like functionalities and did not cleave as effectively at 37°C. This is particularly true with regards to activity at elevated temperatures and under reduced ionic strength. Due to its ability to function under physiological conditions (presence of metal cations such as Mg2+, Ca2+, cleavage of all-RNA substrates, low salt concentrations, pH 7.5 and 37°C), DNAzyme 9-86 could be considered for potential in vivo applications, especially provided that it can be engineered into a trans-cleaving catalyst. Such experiments, along with reselection for cleavage against all-RNA targets, are underway. Finally, in so far as DNAzymes can be regarded as aptamers to transition states, the findings suggest that modified dNTPs should afford DNA aptamers with enhanced chemical properties.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swiss National Science Foundation (grant PBBE2-108568 to M.H.); CIHR (to M.H.); Senior Scholar Award of from the Michael Smith Foundation for Health Research in British Columbia (to D.M.P.). Funding for open access charge: CIHR (Canadian Institutes for Health Research).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank David Dietrich for the synthesis of the dAimTP and Jason Thomas for fruitful discussions.
REFERENCES
- 1.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 2.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment - RNA ligands to bacteriophage -T4 DNA-polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 3.Breaker RR, Joyce GF. A DNA enzyme that cleaves RNA. Chem. Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 4.Cuenoud B, Szostak JW. A DNA metalloenzyme with DNA-ligase activity. Nature. 1995;375:611–614. doi: 10.1038/375611a0. [DOI] [PubMed] [Google Scholar]
- 5.Li YF, Liu Y, Breaker RR. Capping DNA with DNA. Biochemistry. 2000;39:3106–3114. doi: 10.1021/bi992710r. [DOI] [PubMed] [Google Scholar]
- 6.Purtha WR, Coppins RL, Smalley MK, Silverman SK. General deoxyribozyme-catalyzed synthesis of native 3′-5′ RNA linkages. J. Am. Chem. Soc. 2005;127:13124–13125. doi: 10.1021/ja0533702. [DOI] [PubMed] [Google Scholar]
- 7.Li YF, Breaker RR. Phosphorylating DNA with DNA. Proc. Natl Acad. Sci. USA. 1999;96:2746–2751. doi: 10.1073/pnas.96.6.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santoro SW, Joyce GF. A general purpose RNA-cleaving DNA enzyme. Proc. Natl Acad. Sci. USA. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emilsson GM, Breaker RR. Deoxyribozymes: new activities and new applications. Cell. Mol. Life Sci. 2002;59:596–607. doi: 10.1007/s00018-002-8452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chinnapen DJ-F, Sen D. A deoxyribozyme that harnesses light to repair thymine dimers in DNA. Proc. Natl Acad. Sci. USA. 2004;101:65–69. doi: 10.1073/pnas.0305943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pradeepkumar PI, Höbartner C, Baum DA, Silverman SK. DNA-catalyzed formation of nucleopeptide linkages. Angew. Chem. Int. Ed. 2008;47:1753–1757. doi: 10.1002/anie.200703676. [DOI] [PubMed] [Google Scholar]
- 12.Seelig B, Jaschke A. A small catalytic RNA motif with Diels-Alderase activity. Chem. Biol. 1999;6:167–176. doi: 10.1016/S1074-5521(99)89008-5. [DOI] [PubMed] [Google Scholar]
- 13.Fusz S, Eisenfuhr A, Srivatsan SG, Heckel A, Famulok M. A ribozyme for the aldol reaction. Chem. Biol. 2005;12:941–950. doi: 10.1016/j.chembiol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Breaker RR. Engineered allosteric ribozymes as biosensor components. Curr. Opin. Biotechnol. 2002;13:31–39. doi: 10.1016/s0958-1669(02)00281-1. [DOI] [PubMed] [Google Scholar]
- 15.Fiammengo R, Jaschke A. Nucleic acid enzymes. Curr. Opin. Biotechnol. 2005;16:614–621. doi: 10.1016/j.copbio.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Joyce GF. Directed evolution of nucleic acid enzymes. Annu. Rev. Biochem. 2004;73:791–836. doi: 10.1146/annurev.biochem.73.011303.073717. [DOI] [PubMed] [Google Scholar]
- 17.Peracchi A. DNA catalysis: potential, limitations, open questions. ChemBioChem. 2005;6:1316–1322. doi: 10.1002/cbic.200500098. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Brown AK, Meng X, Cropek DM, Istok JD, Watson DB, Lu Y. A catalytic beacon sensor for uranium with parts-per-trillion sensitivity and millionfold selectivity. Proc. Natl Acad. Sci. USA. 2007;96:2056–2061. doi: 10.1073/pnas.0607875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Lu Y. Rational design of ‘Turn-On’ allosteric DNAzyme catalytic beacons for aqueous mercury ions with ultrahigh sensitivity and selectivity. Angew. Chem. Int. Ed. 2007;46:7587–7590. doi: 10.1002/anie.200702006. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Lu Y. A DNAzyme catalytic beacon sensor for paramagnetic Cu2+ ions in aqueous solution with high sensitivity and selectivity. J. Am. Chem. Soc. 2007;129:9838–9839. doi: 10.1021/ja0717358. [DOI] [PubMed] [Google Scholar]
- 21.Seetharaman S, Zivarts M, Sudarsan N, Breaker RR. Immobilized RNA switches for the analysis of complex chemical and biological mixtures. Nat. Biotechnol. 2001;19:336–341. doi: 10.1038/86723. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Lu Y. A highly sensitive and selective catalytic DNA biosensor for lead ions. J. Am. Chem. Soc. 2000;122:10466–10467. [Google Scholar]
- 23.Lu Y. New transition-metal-dependent DNAzymes as efficient endonucleases and as selective metal biosensors. Chem. Eur. J. 2002;8:4588–4596. doi: 10.1002/1521-3765(20021018)8:20<4588::AID-CHEM4588>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 24.Schubert S, Kurreck J. Ribozyme- and deoxyribozyme-strategies for medical applications. Curr. Drug Targets. 2004;5:667–681. doi: 10.2174/1389450043345092. [DOI] [PubMed] [Google Scholar]
- 25.Dass CR. Deoxyribozymes: cleaving a path to clinical trials. Trends Pharmacol. Sci. 2004;25:395–397. doi: 10.1016/j.tips.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Breaker RR, Joyce GF. A DNA enzyme with Mg2+-dependent RNA phosphoesterase activity. Chem. Biol. 1995;2:655–660. doi: 10.1016/1074-5521(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 27.Zaug AJ, Been MD, Cech TR. The tetrahymena ribozyme acts like an RNA restriction endonuclease. Nature. 1986;324:429–433. doi: 10.1038/324429a0. [DOI] [PubMed] [Google Scholar]
- 28.Zivarts M, Breaker RR. Engineered allosteric ribozymes that respond to specific divalent metal ions. Nucleic Acids Res. 2005;33:622–631. doi: 10.1093/nar/gki182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heidenreich O, Kang SH, Brown DA, Xu X, Swiderski P, Rossi JJ, Eckstein F, Nerenberg M. Ribozyme-mediated RNA degradation in nuclei suspension. Nucleic Acids Res. 1995;23:2223–2228. doi: 10.1093/nar/23.12.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sidorov AV, Grasby JA, Williams DM. Sequence-specific cleavage of RNA in the absence of divalent metal ions by a DNAzyme incorporating imidazolyl and amino functionalities. Nucleic Acids Res. 2004;32:1591–1601. doi: 10.1093/nar/gkh326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trepanier J, Tanner JE, Momparler RL, Le ONL, Alvarez F, Alfieri C. Cleavage of intracellular hepatitis C RNA in the virus core protein coding region by deoxyribozymes. J. Viral Hepat. 2006;13:131–138. doi: 10.1111/j.1365-2893.2005.00684.x. [DOI] [PubMed] [Google Scholar]
- 32.Mulquiney PJ, Kuchel PW. Free magnesium-ion concentration in erythrocytes by P-31 NMR: the effect of metabolite-haemoglobin interactions. NMR Biomed. 1997;10:129–137. doi: 10.1002/(sici)1099-1492(199705)10:3<129::aid-nbm459>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 33.Murphy E, Freudenrich CC, Levy LA, London RE, Lieberman M. Monitoring cytosolic free magnesium in cultured chicken heart-cells by use of the fluorescent indicator Furaptra. Proc. Natl Acad. Sci. USA. 1989;86:2981–2984. doi: 10.1073/pnas.86.8.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy E, Steenbergen C, Levy LA, Raju B, London RE. Cytosolic free magnesium levels in ischemic rat-heart. J. Biol. Chem. 1989;264:5622–5627. [PubMed] [Google Scholar]
- 35.Suzuki Y, Komatsu H, Ikeda T, Saito N, Araki S, Citterio D, Hisamoto H, Kitamura Y, Kubota T, Nakagawa J, et al. Design and synthesis of Mg2+-selective fluoroionophores based on a coumarin derivative and application for Mg2+ measurement in a living cell. Anal. Chem. 2002;74:1423–1428. doi: 10.1021/ac010914j. [DOI] [PubMed] [Google Scholar]
- 36.Ting R, Thomas JM, Lermer L, Perrin DM. Substrate specificity and kinetic framework of a DNAzyme with an expanded chemical repertoire: a putative RNaseA mimic that catalyzes RNA hydrolysis independent of a divalent metal cation. Nucleic Acids Res. 2004;32:6660–6672. doi: 10.1093/nar/gkh1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ting R, Thomas JM, Perrin DM. Kinetic characterization of a cis- and trans-acting M2+-independent DNAzyme that depends on synthetic RNaseA-like functionality - BURST-phase kinetics from the coalescence of two active DNAzyme folds. Can. J. Chem. 2007;85:313–329. [Google Scholar]
- 38.Peracchi A. Prospects for antiviral ribozymes and deoxyribozymes. Rev. Med. Virol. 2004;14:47–64. doi: 10.1002/rmv.415. [DOI] [PubMed] [Google Scholar]
- 39.Sun LQ, Cairns MJ, Saravolac EG, Baker A, Gerlach WL. Catalytic nucleic acids: from lab to applications. Pharmacol. Rev. 2000;52:325–347. [PubMed] [Google Scholar]
- 40.James HA, Gibson I. The therapeutic potential of ribozymes. Blood. 1998;91:371–382. [PubMed] [Google Scholar]
- 41.Roth A, Breaker RR. An amino acid as a cofactor for a catalytic polynucleotide. Proc. Natl Acad. Sci. USA. 1998;95:6027–6031. doi: 10.1073/pnas.95.11.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Mei SHJ, Brennan JD, Li Y. Assemblage of signaling DNA enzymes with intriguing metal-ion specificities and pH dependences. J. Am. Chem. Soc. 2003;125:7539–7545. doi: 10.1021/ja035208+. [DOI] [PubMed] [Google Scholar]
- 43.Nakano S, Chadalavada DM, Bevilacqua PC. General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 44.Jayasena VK, Gold L. In vitro selection of self-cleaving RNAs with a low pH optimum. Proc. Natl Acad. Sci. USA. 1997;94:10612–10617. doi: 10.1073/pnas.94.20.10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geyer CR, Sen D. Evidence for the metal-cofactor independence of an RNA phosphodiester-cleaving DNA enzyme. Chem. Biol. 1997;4:579–593. doi: 10.1016/s1074-5521(97)90244-1. [DOI] [PubMed] [Google Scholar]
- 46.Faulhammer D, Famulok M. Characterization and divalent metal-ion dependence of in vitro selected deoxyribozymes which cleave DNA/RNA chimeric oligonucleotides. J. Mol. Biol. 1997;269:188–202. doi: 10.1006/jmbi.1997.1036. [DOI] [PubMed] [Google Scholar]
- 47.Carrigan MA, Ricardo A, Ang DN, Benner SA. Quantitative analysis of a RNA-cleaving DNA catalyst obtained via in vitro selection. Biochemistry. 2004;43:11446–11459. doi: 10.1021/bi049898l. [DOI] [PubMed] [Google Scholar]
- 48.Niittymaki T, Lonnberg H. Artificial ribonucleases. Org. Biomol. Chem. 2006;4:15–25. doi: 10.1039/b509022a. [DOI] [PubMed] [Google Scholar]
- 49.Kuzuya A, Komiyama M. Site-selective artificial ribonucleases and their applications. Curr. Org. Chem. 2007;11:1450–1459. [Google Scholar]
- 50.Gourlain T, Sidorov A, Mignet N, Thorpe SJ, Lee SE, Grasby JA, Williams DM. Enhancing the catalytic repertoire of nucleic acids. II. Simultaneous incorporation of amino and imidazolyl functionalities by two modified triphosphates during PCR. Nucleic Acids Res. 2001;29:1898–1905. doi: 10.1093/nar/29.9.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SE, Sidorov A, Gourlain T, Mignet N, Thorpe SJ, Brazier JA, Dickman MJ, Hornby DP, Grasby JA, Williams DM. Enhancing the catalytic repertoire of nucleic acids: a systematic study of linker length and rigidity. Nucleic Acids Res. 2001;29:1565–1573. doi: 10.1093/nar/29.7.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jager S, Rasched G, Kornreich-Leshem H, Engeser M, Thum O, Famulok M. A versatile toolbox for variable DNA functionalization at high density. J. Am. Chem. Soc. 2005;127:15071–15082. doi: 10.1021/ja051725b. [DOI] [PubMed] [Google Scholar]
- 53.Hocek M, Fojta M. Cross-coupling reactions of nucleoside triphosphates followed by polymerase incorporation. Construction and applications of base-functionalized nucleic acids. Org. Biomol. Chem. 2008;6:2233–2241. doi: 10.1039/b803664k. [DOI] [PubMed] [Google Scholar]
- 54.Augustin MA, Ankenbauer W, Angerer B. Progress towards single-molecule sequencing: enzymatic synthesis of nucleotide-specifically labeled DNA. J. Biotechnol. 2001;86:289–301. doi: 10.1016/s0168-1656(00)00420-x. [DOI] [PubMed] [Google Scholar]
- 55.Brakmann S, Nieckchen P. The large fragment of Escherichia coli DNA polymerase I can synthesize DNA exclusively from fluorescently labeled nucleotides. ChemBioChem. 2001;2:773–777. doi: 10.1002/1439-7633(20011001)2:10<773::AID-CBIC773>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 56.Thum O, Jager S, Famulok M. Functionalized DNA: a new replicable biopolymer. Angew. Chem. Int. Ed. 2001;40:3990–3993. doi: 10.1002/1521-3773(20011105)40:21<3990::AID-ANIE3990>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 57.Latham JA, Johnson R, Toole JJ. The application of a modified nucleotide in aptamer selection – novel thrombin aptamers containing 5-(1-pentynyl)-2′-deoxyuridine. Nucleic Acids Res. 1994;22:2817–2822. doi: 10.1093/nar/22.14.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Battersby TR, Ang DN, Burgstaller P, Jurczyk SC, Bowser MT, Buchanan DD, Kennedy RT, Benner SA. Quantitative analysis of receptors for adenosine nucleotides obtained via in vitro selection from a library incorporating a cationic nucleotide analog. J. Am. Chem. Soc. 1999;121:9781–9789. doi: 10.1021/ja9816436. [DOI] [PubMed] [Google Scholar]
- 59.Masud MM, Kuwahara M, Ozaki H, Sawai H. Sialyllactose-binding modified DNA aptamer bearing additional functionality by SELEX. Bioorg. Med. Chem. 2004;12:1111–1120. doi: 10.1016/j.bmc.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 60.Shoji A, Kuwahara M, Ozaki H, Sawai H. Modified DNA aptamer that binds the (R)-Isomer of a thalidomide derivative with high enantioselectivity. J. Am. Chem. Soc. 2007;129:1456–1464. doi: 10.1021/ja067098n. [DOI] [PubMed] [Google Scholar]
- 61.Ohsawa K, Kasamatsu T, Nagashima JI, Hanawa K, Kuwahara M, Ozaki H, Sawai H. Arginine-modified DNA aptamers that show enantioselective recognition of the dicarboxylic acid moiety of glutamic acid. Anal. Sci. 2008;24:167–172. doi: 10.2116/analsci.24.167. [DOI] [PubMed] [Google Scholar]
- 62.Tarasow TM, Tarasow SL, Eaton BE. RNA-catalysed carbon-carbon bond formation. Nature. 1997;389:54–57. doi: 10.1038/37950. [DOI] [PubMed] [Google Scholar]
- 63.Wiegand TW, Janssen RC, Eaton BE. Selection of RNA amide syntheses. Chem. Biol. 1997;4:675–683. doi: 10.1016/s1074-5521(97)90223-4. [DOI] [PubMed] [Google Scholar]
- 64.Santoro SW, Joyce GF, Sakthivel K, Gramatikova S, Barbas CF. RNA cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc. 2000;122:2433–2439. doi: 10.1021/ja993688s. [DOI] [PubMed] [Google Scholar]
- 65.Bozym RA, Thompson RB, Stoddard AK, Fierke CA. Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor. ACS Chem. Biol. 2006;1:103–111. doi: 10.1021/cb500043a. [DOI] [PubMed] [Google Scholar]
- 66.Colvin RA, Bush AI, Volitakis I, Fontaine CP, Thomas D, Kikuchi K, Holmes WR. Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. Am. J. Physiol. Cell Physiol. 2008;294:C726–C742. doi: 10.1152/ajpcell.00541.2007. [DOI] [PubMed] [Google Scholar]
- 67.Perrin DM, Garestier T, Helene C. Expanding the catalytic repertoire of nucleic acid catalysts: simultaneous incorporation of two modified deoxyribonucleoside triphosphates bearing ammonium and imidazolyl functionalities. Nucleosides Nucleotides. 1999;18:377–391. doi: 10.1080/15257779908043083. [DOI] [PubMed] [Google Scholar]
- 68.Perrin DM, Garestier T, Helene C. Bridging the gap between proteins and nucleic acids: a metal-independent RNAseA mimic with two protein-like functionalities. J. Am. Chem. Soc. 2001;123:1556–1563. doi: 10.1021/ja003290s. [DOI] [PubMed] [Google Scholar]
- 69.Lermer L, Roupioz Y, Ting R, Perrin DM. Toward an RNaseA mimic: a DNAzyme with imidazoles and cationic amines. J. Am. Chem. Soc. 2002;124:9960–9961. doi: 10.1021/ja0205075. [DOI] [PubMed] [Google Scholar]
- 70.Schlosser K, Gu J, Lam JCF, Li Y. In vitro selection of small RNA-cleaving deoxyribozymes that cleave pyrimidine-pyrimidine junctions. Nucleic Acids Res. 2008;36:4768–4777. doi: 10.1093/nar/gkn396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roig V, Asseline U. Oligo-2′-deoxyribonucleotides containing uracil modified at the 5-position with linkers ending with guanidinium groups. J. Am. Chem. Soc. 2003;125:4416–4417. doi: 10.1021/ja029467v. [DOI] [PubMed] [Google Scholar]
- 72.Feichtinger K, Zapf C, Sings HL, Goodman M. Diprotected triflylguanidines: a new class of guanidinylation reagents. J. Org. Chem. 1998;63:3804–3805. [Google Scholar]
- 73.Gore MG. Spectrophotometry and Spectrofluorimetry. UK: Oxford University Press; 2000. p. 339. [Google Scholar]
- 74.Nelson KE, Bruesehoff PJ, Lu Y. In vitro selection of high temperature Zn2+-dependent DNAzymes. J. Mol. Evol. 2005;61:216–225. doi: 10.1007/s00239-004-0374-3. [DOI] [PubMed] [Google Scholar]
- 75.Roychowdhury-Saha M, Burke DH. Extraordinary rates of transition metal ion-mediated ribozyme catalysis. Rna. 2006;12:1846–1852. doi: 10.1261/rna.128906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hollenstein M, Hipolito C, Lam C, Dietrich D, Perrin DM. A highly selective DNAzyme sensor for mercuric ions. Angew. Chem. Int. Ed. 2008;47:4346–4350. doi: 10.1002/anie.200800960. [DOI] [PubMed] [Google Scholar]
- 77.Lam C, Hipolito C, Perrin DM. Synthesis and enzymatic incorporation of modified deoxyadenosine triphosphates. Eur. J. Org. Chem. 2008;2008:4915–4923. [Google Scholar]
- 78.Jose AM, Soukup GA, Breaker RR. Cooperative binding of effecters by an allosteric ribozyme. Nucleic Acids Res. 2001;29:1631–1637. doi: 10.1093/nar/29.7.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peracchi A. Origins of the temperature dependence of hammerhead ribozyme catalysis. Nucleic Acids Res. 1999;27:2875–2882. doi: 10.1093/nar/27.14.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carmi N, Shultz LA, Breaker RR. In vitro selection of self-cleaving DNAs. Chem. Biol. 1996;3:1039–1046. doi: 10.1016/s1074-5521(96)90170-2. [DOI] [PubMed] [Google Scholar]
- 81.Sheppard TL, Ordoukhanian P, Joyce GF. A DNA enzyme with N-glycosylase activity. Proc. Natl Acad. Sci. USA. 2000;97:7802–7807. doi: 10.1073/pnas.97.14.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.May JP, Ting R, Lermer L, Thomas JM, Roupioz Y, Perrin DM. Covalent Schiff base catalysis and turnover by a DNAzyme: a M2+ independent AP-endonuclease mimic. J. Am. Chem. Soc. 2004;126:4145–4156. doi: 10.1021/ja037625s. [DOI] [PubMed] [Google Scholar]
- 83.Marban E, Kitakaze M, Kusuoka H, Porterfield JK, Yue DT, Chacko VP. Intracellular free calcium-concentration measured with F-19 Nmr-spectroscopy in intact ferret hearts. Proc. Natl Acad. Sci. USA. 1987;84:6005–6009. doi: 10.1073/pnas.84.16.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 85.Thomas JM, Ting R, Perrin DM. High affinity DNAzyme-based ligands for transition metal cations - a prototype sensor for Hg2+ Org. Biomol. Chem. 2004;2:307–311. doi: 10.1039/b310154a. [DOI] [PubMed] [Google Scholar]
- 86.Tan ZJ, Chen SJ. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys. J. 2006;90:1175–1190. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bonaccio M, Credali A, Peracchi A. Kinetic and thermodynamic characterization of the RNA-cleaving 8-17 deoxyribozyme. Nucleic Acids Res. 2004;32:916–925. doi: 10.1093/nar/gkh250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bevilacqua PC. Mechanistic considerations for general acid-base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry. 2003;42:2259–2265. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- 89.Thomas JM, Perrin DM. Active site labeling of G8 in the hairpin ribozyme: implications for structure and mechanism. J. Am. Chem. Soc. 2006;128:16540–16545. doi: 10.1021/ja063942y. [DOI] [PubMed] [Google Scholar]
- 90.Li J, Zheng W, Kwon AH, Lu Y. In vitro selection and characterization of a highly efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2000;28:481–488. doi: 10.1093/nar/28.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.