

Abstract
Lupus nephritis is a major contributor to morbidity and mortality in systemic lupus erythematosus, but little is known about the pathogenic processes that underlie the progressive decay in renal function. A common finding in lupus nephritis is thickening of glomerular basement membranes associated with immune complex deposition. It has been speculated that alterations in the synthesis or degradation of membrane components might contribute to such changes, and thereby to initiation and progression of nephritis through facilitation of immune complex deposition. Matrix metalloproteinases (MMPs) are enzymes that are intimately involved in the turnover of major glomerular basement membrane constituents, including collagen IV and laminins. Alterations in the expression and activity of MMPs have been described in a number of renal diseases, suggesting their relevance to the pathogenesis of various glomerulopathies. The same is true for their natural inhibitors, the tissue inhibitor of metalloproteinase family. Recent data from our group have identified an increase in proteolytic activity within the glomerulus coinciding with the development of proteinuria in the mouse model of systemic lupus erythematosus. (NXB × NZW)F1 Here we review current understanding of MMP/tissue inhibitor of metalloproteinase function within the kidney, and discuss their possible involvement in the development and progression of lupus nephritis.
Introduction
Systemic lupus erythematosus (SLE) is a complex auto-immune disease that is characterized by chronic inflammatory processes involving autoimmunity against multiple organ-specific and ubiquitous self-antigens. One commonly affected organ is the kidney, with the appearance of lupus nephritis ranging in severity from mild proteinuria to overt nephrotic syndrome progressing to end-stage renal disease. Although the molecular mechanisms that underlie the pathogenesis of nephritis remain largely obscure, disturbances in apoptotic signalling, phagocytosis and complement function have all been proposed as factors involved in initiation of auto-immunity and progression of the disease [1,2].
Expansion and/or disruption of the intraglomerular extra-cellular matrix is a well recognized phenomenon occurring during the development of lupus nephritis that may have an impact on renal immune complex deposition. Little is known, however, about the structure and composition of the expanded regions or the mediators of such changes. Increased or altered synthesis of extracellular matrix (ECM) constituents and/or their decreased breakdown could potentially play a role, although the contribution made by each of these factors remains unknown.
Another common finding in lupus nephropathy is the appearance of electron dense structures (EDSs) within mesangium or intimately linked to the glomerular capillary membranes, as seen on electron micrographs. These structures contain immune complexes with autoantibodies and chromatin fragments [3,4], and a recent study [5] has demonstrated a considerable affinity of nucleosomes toward the major matrix constituents laminin and collagen IV. It is therefore possible that alterations in the composition of the glomerular ECM may affect its interaction with immune complexes, thus facilitating their deposition and subsequent damage to glomerular structures. Indeed, qualitative as well as quantitative alterations in the makeup of the extracellular membranes of the glomerulus in lupus nephritis have already been described [6,7]. Candidate mediators of such changes include enzymes and signalling substances involved in maintaining the delicate balance between synthesis and breakdown of the proteins and proteoglycans that make up the ECM.
Although some studies have provided evidence of increased levels of expression of collagens and laminins, less is known about the kinetics of breakdown of these proteins. Turnover of ECM proteins is largely achieved through the action of matrix metalloproteinases (MMPs), which represent a major class of matrix-degrading proteinases. Thus, from its effect on capillary membranes and mesangial matrix composition, a putative role emerges for altered glomerular MMP activity in lupus nephritis. Exploring this possibility, however, is complicated by the many levels of regulation of proteinase activity. Also, there is an emerging appreciation of considerable functional divergence of both MMPs and their regulators, particularly the tissue inhibitors of metalloproteinase (TIMPs).
In this review we outline some of the current knowledge on MMP expression and regulation within the kidney in lupus nephritis, including clues gained from studies in other renal inflammatory diseases.
Matrix metalloproteinases
MMPs are a group of Zn2+-dependent proteins that are found in the extracellular milieu of various tissues. Based on sequence homology and substrate specificities, the MMPs can be classified into several subgroups including collagenases, gelatinases, stromelysins, matrilysins and the membrane-type metalloproteinases. There is considerable overlap in substrate specificities, and the MMPs appear to be involved in degradation of abundant ECM components, including laminins, collagens and fibronectin, but also in the release and turnover of cytokines and cell surface receptors of adjacent cells [8].
MMP-2 (gelatinase A) and MMP-9 (gelatinase B) constitute the gelatinases (Figure 1). On account of their propensity to cleave the major glomerular basement membrane component collagen IV, they have been particularly implicated in a variety of acute and chronic kidney diseases, including both immune and non-immune glomerulopathies, and are therefore the main focus of this review.
Figure 1.

Schematic structure of MMP-2 and MMP-9. The catalytic site contains three essential zinc ion binding sites. At the zymogen stage, a cysteine residue within the prodomain interacts with zinc to prevent substrate binding. The haemopexin domain mediates interaction with enzyme substrates. Specific to the gelatinases is the fibronectin-like domain, which further facilitates substrate binding. MMP, matrix metalloproteinase.
The gelatinases cleave a number of substrates, including native forms of collagens I, IV, V, VII, X and XI, elastin, laminin, fibronectin, myelin and the core protein of proteoglycans. (A comprehensive list of substrates for the various MMPs can be found in the Overall Lab Web Site [9].) Another metalloproteinase that is notable for its affinity for collagen IV is MMP-7 (matrilysin 1) [10]. Produced in both the tubular and glomerular compartment, it was recently described to be involved in several types of renal diseases with glomerular involvement, including diabetic nephropathy and X-linked Alport syndrome [11,12]. In addition to collagen IV, MMP-7 is a major factor in the turnover of tenascin (an oligomeric glyco-protein that is important for the functioning of the glomerular filtration barrier) [13] and other basement membrane components, such as laminin, entactin and proteoglycans, as well as in activation of several proinflammatory mediators, including MMP-2 and MMP-9 [14,15]. The relevance of MMP-7 in SLE has not yet been evaluated, but it remains an interesting candidate mediator of changes in membrane composition in lupus nephritis.
MMP-2 is constitutively expressed in mesangial cells, with some contribution made by the podocytes and little or no expression in glomerular endothelial cells [16,17]. The expression is dramatically increased in various glomerulopathies, probably as a result of proinflammatory signalling [18,19]. MMP-9 is present at negligible levels in normal kidney glomeruli, but it is induced during the course of several renal inflammatory diseases, with mesangial cells and infiltrating neutrophils being the main sources [20]. Recent data from our laboratory [21] indicate an increase in glomerular proteolytic activity at around the onset of proteinuria in a model of lupus nephritis in (NZB × NZW)F1 mice, with MMP-9 being a major contributor.
Tissue inhibitors of metalloproteinases
The many roles of MMPs in vivo require a complex network of modulation of enzymatic activity. Key regulators are the TIMPs, of which four subtypes are currently known. The TIMPs form 1:1 complexes with metalloproteinases, with only modest variations in affinity toward the different MMPs [22]. The central role played by TIMPs in the regulation of MMP activity has led to the hypothesis that shifts in the balance between these two families of proteins could distort the kinetics of membrane turnover and cause pathological changes in membrane composition [23,24].
When considering the relevance of TIMP expression in renal disease, it is important to note that several members of the TIMP family appear to have functions of pathophysiological significance that is not directly related to ECM homeostasis/MMP regulation. One such facet is the apparent involvement in both pro-apoptotic and anti-apoptotic signalling pathways [25-27]. Various disturbances in apoptosis and the clearance of dead cells have been proposed to form a source of autoreactivity in SLE, which raises the possibility that TIMPs are involved in regulating apoptotic cell death in the context of autoimmune disorders.
A full account of the field of TIMP biology is beyond the scope of this review, and we limit the discussion to apoptosis-related aspects of their functioning. Whereas the molecular basis for TIMP-mediated signalling is still poorly understood, an emerging view is that they interact extensively with cell surface proteins, thus imposing modulation of various downstream signalling pathways.
Cell culture studies have reported anti-apoptotic effects of TIMP-1, some of which rely on its MMP-inhibitory function, whereas others appear independent of interaction with MMPs [28-30]. A recent in vitro study conducted in HeLa cervical cancer cells identified promyelocytic leukaemia zinc finger protein (PLZF) – a well known transcriptional repressor – to be a potential binding partner for TIMP-1 [31]. It was shown that the addition/over-expression of TIMP-1 reduced the percentage of apoptotic cells in this system in a PLZF-dependant manner. PLZF is expressed in myeloid cells, ovaries and, at low level, in kidney and lung tissues. The interaction between TIMP-1 and PLZF is reported to occur by direct interaction between the two proteins within the nucleus [31].
The concept of TIMP-1 translocating into the nucleus remains controversial [32,33], and the functions of TIMP in this location in vivo remain to be identified. However, as discussed below, several recent studies have reported that MMPs are present within the nucleus [34], offering new perspectives on the biological roles played MMPs and possibly TIMPs as well. For TIMP-2 and TIMP-4, reports have been partly contradictory, with both pro-apoptotic and anti-apoptotic effects being described, and further studies are awaited to characterize their putative roles in apoptosis and cell viability. TIMP-3 appears to have pro-apoptotic properties, attributed to the inhibition of both the MMP and ADAM (a disintegrin and metalloproteinase) families of matrix metalloproteinases [35]. One possible basis for these functions is the fact that certain ADAMs and MMPs are involved in the shedding of a number of cell surface receptors that are involved in pro-apoptotic and anti-apoptotic signalling pathways, including tumour necrosis factor receptors [36] and Fas receptor [37]. Studies conducted in tumor cells have shown that over-expression of TIMP-3 causes stimulation of Fas/Fas ligand signalling [38] and results in increased apoptotic activity. Involvement of TIMPs in the regulation of death pathways including Fas/Fas ligand is an interesting finding, because alterations in Fas function appear to be relevant to autoantibody production and possibly to the development of nephritis [39].
Challenges and pitfalls in assays of matrix metalloproteinase activity
Studies of tissue MMPs are complicated by the complexity of the regulatory network that governs their activity in vivo. A MMP-9 knockout mouse exhibited no or modest structural/functional abnormalities, both on a healthy background and in a model of Alport syndrome [40], which could be explained by redundancy of the system based on observations of compensatory upregulation of other MMPs, including MMP-2 [41]. Caution is therefore required when interpreting studies that are limited to one or a few MMPs, and such findings also suggest that therapeutic utilization of broad-spectrum inhibitors of MMP activity might be a more desirable strategy than more targeted ones, at least in some settings.
Increased gene expression and protein levels of the MMPs are often found to be accompanied by increases in the levels of one or more of the TIMPs. It is therefore not obvious what can be the net result of these opposing stimuli in terms of ECM turnover. In situ zymography is a technique that allows localization of active proteinases (including MMPs) within the tissues, providing valuable information about the net result of MMP regulation. It is done by incubating tissue sections with a fluorescence-marked substrate, which gives a direct visual impression of local proteinase activity [42].
Matrix metalloproteinase activity in lupus nephritis and related diseases
Data on metalloproteinase activity in lupus nephritis is limited to few reports of altered gene expression patterns in murine and human kidneys [19,20,43]. There have also been reports of increased circulating levels of several MMPs, notably MMP-9, in sera from lupus patients [44-47]. Of note, circulating MMP-9 levels have been found to be inversely correlated to levels of antibodies against double-stranded DNA, which is commonly used as a marker of SLE disease activity [48]. No such correlations were observed for MMP-2 or MMP-3 [44,48,49]. The source(s) of serum MMPs probably includes circulating leucocytes, especially neutrophils and monocytes, whereas the contribution from the tissues is uncertain. Serum MMP measurements thus may be of limited value in elucidating their potential roles in end organ disease, and the organ-focused studies suggest different roles for the MMPs within the various tissue spaces. Nevertheless, increased circulating levels of MMP-9 have been described in SLE patients with evidence of neuropsychiatric manifestations [50]. Also, a recent study [51] identified increased MMP-9 activity in cerebrospinal fluid from SLE patients, with significantly higher levels in patients with evidence of central nervous system involvement. There are reports indicating a central role for MMPs in increasing permeability of the blood-brain barrier in inflammatory settings [52], which could be of relevance in SLE.
Matrix metalloproteinases and glomerulopathies
Owing to overlapping morphological and clinical presentations of various kinds of inflammatory diseases of the glomerulus, the results of studies of the involvement of MMP activity in other renal pathologies should provide valuable guidance in elucidating the role of these enzymes in lupus nephritis.
Although one might speculate that increased MMP activity is not detrimental, but rather represents a favourable compensatory response to aberrant matrix synthesis, this appears unlikely considering the favourable outcome of MMP inhibition/knockout strategies in other glomerulopathies [53]. Much of the data currently available come from work in models of antibody-induced nephropathies, such as anti-Thy 1.1 nephritis [54] and passive Heymann's nephritis [55], and from non-immune models such as ischemia/reperfusion renal scarring induced by ureteral ligation [56,57], all of which trigger inflammatory responses [58] that lead to progressive tubulointerstitial fibrosis and glomerulosclerosis. A common theme in these studies is a marked increase in either one or both of the gelatinases (MMP-2 or MMP-9) [18,19,59,60]. Often an increase in TIMP-1 is observed within the glomeruli, which (as mentioned above) complicates the interpretation of results. The finding that gelatinase levels are increased in a situation of ECM accumulation might appear paradoxical, because this would be expected to increase collagen breakdown. A simple explanation is that the increased expression is a compensatory response to an increase in the synthesis of matrix components. Although there are reports indicating that collagen IV increases early in glomerulonephritis [6], others have found collagen IV expression to appear relatively stable [61].
Preliminary data from our laboratory support the latter in the case of (NZB × NZW)F1 and MRL/lpr lupus-prone mice (Tveita A, unpublished data). As stated above, the relative contribution of increased synthesis and decreased degradation of collagen IV to ECM accumulation remains undetermined. Studies showing that MMP inhibition attenuates ECM accumulation in rat allograft nephropathy [62], anti-Thy 1.1 nephritis [63] and other experimental inflammatory renal diseases would suggest that matrix degradation plays at least some role in this process. As discussed below, there are also indications that the MMPs confer proliferative stimuli upon mesangial cells, providing another factor that might explain an increase in MMP activity in the face of nephritis and matrix proliferation [64].
Metalloproteinases and mesangial cell proliferation
The mesangium appears to play a central role in the development of glomerulonephritis. The contribution made by mesangial cells to inflammatory diseases of the kidneys is thought to be twofold: increased proliferation and proinflammatory response; and synthesis of matrix components, causing ECM accumulation. Mesangial cell culture experiments have implicated MMP-2 as a possible regulator of both of the above factors. Indeed, inhibition by both pharmacological and ribozyme-mediated approaches have shown reduction in MMP-2 activity to be associated with transformation of actively proliferating mesangial cells to a state of quiescence by induction of G0/G1 cell cycle arrest [65].
Treatment of cultured rat mesangial cell with a relatively unspecific MMP inhibitor caused a decrease in proliferative activity of up to 75% and evidence of decreased levels of activation [63]. In addition, MMP inhibitor experiments also demonstrated a significant increase in the number of apoptotic cells both in anti-Thy 1.1 nephritis and in cultured mesangial cells, probably mediated through a caspase-independent pathway [27]. A new aspect of function of MMPs has emerged from the reports of MMPs being present within mammalian cell nuclei [66,67]. It was recently shown that MMP-3 can translocate to the nucleus in vitro, where it was reported to exert pro-apoptotic functions mediated through its catalytic domain [34]. The same report showed evidence of intranuclear MMP-2 and MMP-3 on human liver sections.
Cryptic epitopes and immune complex deposition
Studies in multiple sclerosis and rheumatoid arthritis have demonstrated that cleavage of particular collagen fragments by MMPs leads to the exposure of highly immunoreactive epitopes [68,69]. These findings led to the proposition of a model for the generation of autoantibodies, termed the 'remnant epitope generate autoimmunity' (REGA) model [69,70]. Briefly, the underlying concept is that in an inflammatory context, a local increase in proteolytic activity generates a large number of substrate fragments for presentation by activated antigen-presenting cells, including exposed cryptic antigen epitopes. This leads to both quantitative and qualitative changes in the local antigen repertoire. Although highly speculative, one could envision a situation in which dysregulation of MMP activity leads to a quantitative increase in the exposure of such cryptic epitopes. Furthermore, qualitative alterations in ECM composition could lead to cleavage of substrates not normally found in this location, causing the appearance of novel epitopes within the matrix. In the face of a persisting inflammatory process, such as evolving lupus nephritis, quantitative and qualitative changes in antigen repertoire might conceivably increase production of autoantibodies against matrix structures. Alternatively, alterations in glomerular membrane composition could favour the deposition of immune complex-associated structures such as nucleosomes, thus accelerating the formation of EDS-like structures within the membranes.
In light of our recent findings of increased MMP activity and qualitative changes in collagen IV expression within glomeruli of lupus-prone mice during the development of nephritis, this scenario provides an attractive model to explain the relationship between immune complex deposits and renal dysfunction. In this framework, immune complexes propagate proinflammatory stimuli to resident and infiltrating cells, either directly or through complement activation, triggering an increase in MMP production and activity (Figure 2). In turn, MMPs mediate changes in glomerular basement membrane structure, favouring immune complex deposition and compromising the physical integrity of the membrane.
Figure 2.
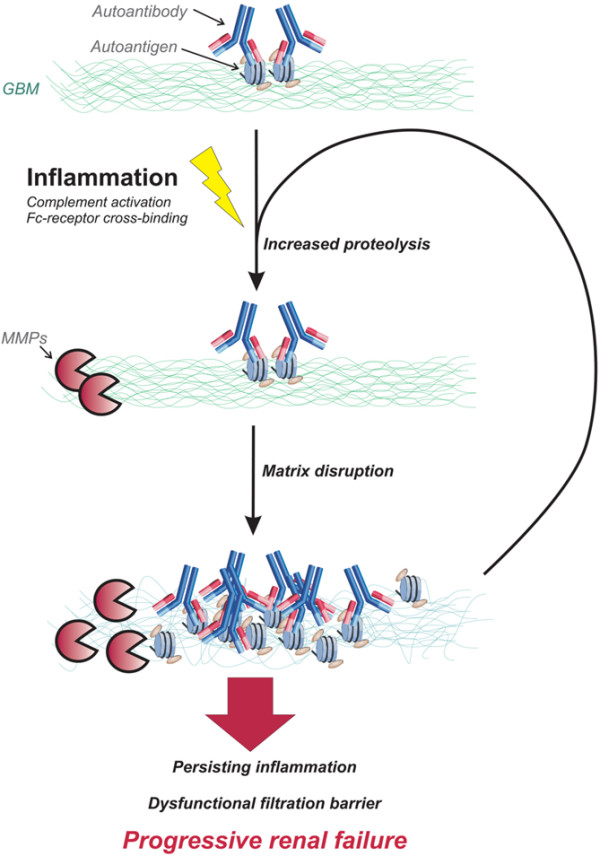
Conceptual framework for progression of lupus nephritis. An inflammatory reaction is brought about by complement- or Fc-mediated responses to autoantibodies in deposited immune complexes or locally exposed danger signals (such as necrotic chromatin; see text), triggering release of MMPs from intrinsic and infiltrating cells. Increased proteolytic degradation of the membrane exposes matrix components, facilitating binding of autoantibodies to capillary and mesangial antigens. This maintains the inflammatory reaction and continued stimulation of matrix degradation, leading to disruption of glomerular membrane barriers and progression toward end-stage renal failure. MMP, matrix metalloproteinase.
Central to the pathogenesis of lupus is continuous activation and proliferation of B-lymphocytes and T-lymphocytes with specificity for self-structures such as exposed chromatin fragments. Such structures may also serve as renal targets for the induced autoimmunity. Encounters with these antigens initiate proinflammatory signalling cascades, recruiting effector cells of the innate immune system, including monocytes/macrophages and neutrophils. As part of this inflammatory process, several MMPs and TIMPs are secreted by activated infiltrating cells and by cells intrinsic to the inflamed site, facilitating penetration into the tissue and structural remodeling as part of the healing process [71]. An inflammatory reaction invariably causes local cellular decay, serving as a potential reservoir for exposed self-structures, including nuclear antigens. Chromatin structures derived from dead cells are found deposited as EDSs in glomerular membranes in lupus kidneys, where they co-localize with deposited autoantibodies [4]. Ingestion of chromatin-containing immune complexes by infiltrating macrophages could conceivably upregulate MMP secretion through activation of the Toll-like receptor 9 signalling pathway [72,73]. Persistently increased glomerular MMP activity could therefore be the result of an inflammatory process that is maintained by retained necrotic or apoptotic cellular debris. In turn, excessive matrix degradation by the MMPs would facilitate the deposition of immune complexes by compromising the integrity of the glomerular membranes. In this manner, the combined presence of autoreactive lymphocytes and an inflammatory process that exposes the inciting autoantigens allows the translation of a latent systemic autoreactivity to a focused end organ inflammatory disease. Increased MMP activity forms part of a spectrum of changes at the site of inflammation that ensures continued engagement of the innate immune system and progression of local tissue damage.
Conclusion
MMP inhibitory strategies have been tested in animal models of a number of chronic inflammatory diseases, including chronic obstructive pulmonary diseases, inflammatory bowel disease, rheumatoid arthritis and atherosclerosis. The progress of various such trials was recently reviewed by Hu and coworkers [53]. For glomerulonephritis, MMP inhibition has exhibited promising results in rat anti-Thy 1.1 nephritis [63], but several key parameters in such a strategy remain ill defined, including the target MMP(s), timing and duration of intervention, specificity, dosage and delivery system. A more rigorous understanding of the spectrum of in vivo biochemical roles played by MMPs/TIMPs might be a prerequisite for the development and success of such targeted experimental and pharmacological interventions.
Our knowledge about the role played by MMPs within the context of lupus nephritis remains sparse and inconclusive. Studies in murine lupus-prone strains are underway and will hopefully shed light on this. The evidence that MMPs and TIMPs might be involved in the regulation of apoptosis provides further cause to look more closely into the matter of MMP activity, because disturbances in the clearance of apoptotic material is thought to be among the central elements in the development of lupus [74,75]. Also, by forming a part of the Toll-like receptor mediated response to danger signals such as necrotic chromatin, increased MMP activity could be an important factor in initiating end organ manifestations of an autoimmune response.
Identifying the signalling pathways that are involved in inducing the observed alterations in MMP expression may contribute to our understanding of the initiation of kidney damage in lupus nephritis. Hopefully, this might pave the road to therapeutic strategies directed at preventing the development of glomerulonephritis and kidney failure in lupus patients.
Abbreviations
ADAM: a disintegrin and metalloproteinase; ECM: extracellular matrix; EDS: electron dense structure; MMP: matrix metalloproteinase; PLZF: promyelocytic leukaemia zinc finger protein; SLE: systemic lupus erythematosus; TIMP: tissue inhibitor of metalloproteinase.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
We thank Dr Jan-Olof Winberg for critical review of the manuscript.
Contributor Information
Anders Tveita, Email: anders.aune.tveita@fagmed.uit.no.
Ole Petter Rekvig, Email: olepr@fagmed.uit.no.
Svetlana N Zykova, Email: svetlana@fagmed.uit.no.
References
- Mortensen ES, Fenton KA, Rekvig OP. Lupus nephritis: the central role of nucleosomes revealed. Am J Pathol. 2008;172:275–283. doi: 10.2353/ajpath.2008.070563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berden JH, Grootscholten C, Jurgen WC, Vlag J van der. Lupus nephritis: a nucleosome waste disposal defect? J Nephrol. 2002;15(suppl 6):S1–S10. [PubMed] [Google Scholar]
- Kalaaji M, Sturfelt G, Mjelle JE, Nossent H, Rekvig OP. Critical comparative analyses of anti-alpha-actinin and glomerulus-bound antibodies in human and murine lupus nephritis. Arthritis Rheum. 2006;54:914–926. doi: 10.1002/art.21622. [DOI] [PubMed] [Google Scholar]
- Kalaaji M, Fenton KA, Mortensen ES, Olsen R, Sturfelt G, Alm P, Rekvig OP. Glomerular apoptotic nucleosomes are central target structures for nephritogenic antibodies in human SLE nephritis. Kidney Int. 2007;71:664–672. doi: 10.1038/sj.ki.5002133. [DOI] [PubMed] [Google Scholar]
- Mjelle JE, Rekvig OP, Fenton KA. Nucleosomes possess high affinity for glomerular laminin and collagen IV and bind nephritogenic antibodies in murine lupus-like nephritis. Ann Rheum Dis. 2007;66:1661–1668. doi: 10.1136/ard.2007.070482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergijk EC, Van Alderwegen IE, Baelde HJ, de Heer E, Funabiki K, Miyai H, Killen PD, Kalluri RK, Bruijn JA. Differential expression of collagen IV isoforms in experimental glomerulosclerosis. J Pathol. 1998;184:307–315. doi: 10.1002/(SICI)1096-9896(199803)184:3<307::AID-PATH5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Peutz-Kootstra CJ, Hansen K, De Heer E, Abrass CK, Bruijn JA. Differential expression of laminin chains and anti-laminin autoantibodies in experimental lupus nephritis. J Pathol. 2000;192:404–412. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH707>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Somerville RP, Oblander SA, Apte SS. Matrix metalloproteinases: old dogs with new tricks. Genome Biol. 2003;4:216. doi: 10.1186/gb-2003-4-6-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Overall Lab Web Page http://www.clip.ubc.ca
- Imai K, Yokohama Y, Nakanishi I, Ohuchi E, Fujii Y, Nakai N, Okada Y. Matrix metalloproteinase 7 (matrilysin) from human rectal carcinoma cells. Activation of the precursor, interaction with other matrix metalloproteinases and enzymic properties. J Biol Chem. 1995;270:6691–6697. doi: 10.1074/jbc.270.12.6691. [DOI] [PubMed] [Google Scholar]
- McLennan SV, Kelly DJ, Schache M, Waltham M, Dy V, Langham RG, Yue DK, Gilbert RE. Advanced glycation end products decrease mesangial cell MMP-7: a role in matrix accumulation in diabetic nephropathy? Kidney Int. 2007;72:481–488. doi: 10.1038/sj.ki.5002357. [DOI] [PubMed] [Google Scholar]
- Rao VH, Lees GE, Kashtan CE, Delimont DC, Singh R, Meehan DT, Bhattacharya G, Berridge BR, Cosgrove D. Dysregulation of renal MMP-3 and MMP-7 in canine X-linked Alport syndrome. Pediatr Nephrol. 2005;20:732–739. doi: 10.1007/s00467-004-1805-5. [DOI] [PubMed] [Google Scholar]
- Mignatti P. Extracellular matrix remodeling by metalloproteinases and plasminogen activators. Kidney Int Suppl. 1995;49:S12–S14. [PubMed] [Google Scholar]
- Haro H, Crawford HC, Fingleton B, Shinomiya K, Spengler DM, Matrisian LM. Matrix metalloproteinase-7-dependent release of tumor necrosis factor-alpha in a model of herniated disc resorption. J Clin Invest. 2000;105:143–150. doi: 10.1172/JCI7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang FQ, So J, Reierstad S, Fishman DA. Matrilysin (MMP-7) promotes invasion of ovarian cancer cells by activation of progelatinase. Int J Cancer. 2005;114:19–31. doi: 10.1002/ijc.20697. [DOI] [PubMed] [Google Scholar]
- Martin J, Knowlden J, Davies M, Williams JD. Identification and independent regulation of human mesangial cell metalloproteinases. Kidney Int. 1994;46:877–885. doi: 10.1038/ki.1994.345. [DOI] [PubMed] [Google Scholar]
- Lenz O, Striker LJ, Jacot TA, Elliot SJ, Killen PD, Striker GE. Glomerular endothelial cells synthesize collagens but little gelatinase A and B. J Am Soc Nephrol. 1998;9:2040–2047. doi: 10.1681/ASN.V9112040. [DOI] [PubMed] [Google Scholar]
- Camp TM, Smiley LM, Hayden MR, Tyagi SC. Mechanism of matrix accumulation and glomerulosclerosis in spontaneously hypertensive rats. J Hypertens. 2003;21:1719–1727. doi: 10.1097/00004872-200309000-00022. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Liu XG, Chen GP, Zhang XR, Guo MY. Significance of MMP-2 and TIMP-2 mRNA expressions on glomerular cells in the development of glomerulosclerosis. Chin Med Sci J. 2004;19:84–88. [PubMed] [Google Scholar]
- Urushihara M, Kagami S, Kuhara T, Tamaki T, Kuroda Y. Glomerular distribution and gelatinolytic activity of matrix metalloproteinases in human glomerulonephritis. Nephrol Dial Transplant. 2002;17:1189–1196. doi: 10.1093/ndt/17.7.1189. [DOI] [PubMed] [Google Scholar]
- Tveita A, Rekvig OP, Zykova SN. Increased glomerular matrix metalloproteinase activity in murine lupus nephritis. Kidney Int. 2008;74:1150–1158. doi: 10.1038/ki.2008.308. [DOI] [PubMed] [Google Scholar]
- Murphy G, Willenbrock F. Tissue inhibitors of matrix metalloendopeptidases. Methods Enzymol. 1995;248:496–510. doi: 10.1016/0076-6879(95)48032-3. [DOI] [PubMed] [Google Scholar]
- Zaoui P, Barro C, Maynard C, Descotes JL, Maurizi-Balzan J, Cordonnier DJ, Morel F. Inter-regulated balance between gelatinases and tissue inhibitor (TIMP-1) in isolated human glomeruli. Ren Fail. 1998;20:201–209. doi: 10.3109/08860229809045103. [DOI] [PubMed] [Google Scholar]
- Han SY, Jee YH, Han KH, Kang YS, Kim HK, Han JY, Kim YS, Cha DR. An imbalance between matrix metalloproteinase-2 and tissue inhibitor of matrix metalloproteinase-2 contributes to the development of early diabetic nephropathy. Nephrol Dial Transplant. 2006;21:2406–2416. doi: 10.1093/ndt/gfl238. [DOI] [PubMed] [Google Scholar]
- Chromek M, Tullus K, Lundahl J, Brauner A. Tissue inhibitor of metalloproteinase 1 activates normal human granulocytes, protects them from apoptosis, and blocks their transmigration during inflammation. Infect Immun. 2004;72:82–88. doi: 10.1128/IAI.72.1.82-88.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirco R, Liu XW, Jung KK, Kim HR. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev. 2006;25:99–113. doi: 10.1007/s10555-006-7893-x. [DOI] [PubMed] [Google Scholar]
- Daniel C, Duffield J, Brunner T, Steinmann-Niggli K, Lods N, Marti HP. Matrix metalloproteinase inhibitors cause cell cycle arrest and apoptosis in glomerular mesangial cells. J Pharmacol Exp Ther. 2001;297:57–68. [PubMed] [Google Scholar]
- Liu XW, Taube ME, Jung KK, Dong Z, Lee YJ, Roshy S, Sloane BF, Fridman R, Kim HR. Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells from extrinsic cell death: a potential oncogenic activity of tissue inhibitor of metalloproteinase-1. Cancer Res. 2005;65:898–906. [PubMed] [Google Scholar]
- Liu XW, Bernardo MM, Fridman R, Kim HR. Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells against intrinsic apoptotic cell death via the focal adhesion kinase/phosphatidylinositol 3-kinase and MAPK signaling pathway. J Biol Chem. 2003;278:40364–40372. doi: 10.1074/jbc.M302999200. [DOI] [PubMed] [Google Scholar]
- Guedez L, Stetler-Stevenson WG, Wolff L, Wang J, Fukushima P, Mansoor A, Stetler-Stevenson M. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J Clin Invest. 1998;102:2002–2010. doi: 10.1172/JCI2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho SB, Chung BM, Lee JH. TIMP-1 regulates cell proliferation by interacting with the ninth zinc finger domain of PLZF. J Cell Biochem. 2007;101:57–67. doi: 10.1002/jcb.21127. [DOI] [PubMed] [Google Scholar]
- Ritter LM, Garfield SH, Thorgeirsson UP. Tissue inhibitor of metalloproteinases-1 (TIMP-1) binds to the cell surface and translocates to the nucleus of human MCF-7 breast carcinoma cells. Biochem Biophysic Res Commun. 1999;257:494–499. doi: 10.1006/bbrc.1999.0408. [DOI] [PubMed] [Google Scholar]
- Hockenbery DM. MMPs in unusual places. Am J Pathol. 2006;169:1101–1103. doi: 10.2353/ajpath.2006.060553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si-Tayeb K, Monvoisin A, Mazzocco C, Lepreux S, Decossas M, Cubel G, Taras D, Blanc JF, Robinson DR, Rosenbaum J. Matrix metalloproteinase 3 is present in the cell nucleus and is involved in apoptosis. Am J Pathol. 2006;169:1390–1401. doi: 10.2353/ajpath.2006.060005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AH, Zaltsman AB, George SJ, Newby AC. Divergent effects of tissue inhibitor of metalloproteinase-1, -2, or -3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. J Clin Invest. 1998;101:1478–1487. doi: 10.1172/JCI1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Kung H, Durum SK, Colburn NH, Sun Y. TIMP-3 induces cell death by stabilizing TNF-alpha receptors on the surface of human colon carcinoma cells. Cytokine. 1997;9:770–780. doi: 10.1006/cyto.1997.0233. [DOI] [PubMed] [Google Scholar]
- Ahonen M, Poukkula M, Baker AH, Kashiwagi M, Nagase H, Eriksson JE, Kahari VM. Tissue inhibitor of metalloproteinases-3 induces apoptosis in melanoma cells by stabilization of death receptors. Oncogene. 2003;22:2121–2134. doi: 10.1038/sj.onc.1206292. [DOI] [PubMed] [Google Scholar]
- Finan KM, Hodge G, Reynolds AM, Hodge S, Holmes MD, Baker AH, Reynolds PN. In vitro susceptibility to the pro-apoptotic effects of TIMP-3 gene delivery translates to greater in vivo efficacy versus gene delivery for TIMPs-1 or -2. Lung Cancer. 2006;53:273–284. doi: 10.1016/j.lungcan.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Nakajima A, Hirai H, Kayagaki N, Yoshino S, Hirose S, Yagita H, Okumura K. Treatment of lupus in NZB/W F1 mice with monoclonal antibody against Fas ligand. J Autoimmun. 2000;14:151–157. doi: 10.1006/jaut.1999.0356. [DOI] [PubMed] [Google Scholar]
- Andrews KL, Betsuyaku T, Rogers S, Shipley JM, Senior RM, Miner JH. Gelatinase B (MMP-9) is not essential in the normal kidney and does not influence progression of renal disease in a mouse model of Alport syndrome. Am J Pathol. 2000;157:303–311. doi: 10.1016/S0002-9440(10)64541-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Khurana M, Rao VH, Cosgrove D, Rougier JP, Werner MC, Shield CF, 3rd, Werb Z, Kalluri R. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med. 2006;3:e100. doi: 10.1371/journal.pmed.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galis ZS, Sukhova GK, Libby P. Microscopic localization of active proteases by in situ zymography: detection of matrix metalloproteinase activity in vascular tissue. FASEB J. 1995;9:974–980. doi: 10.1096/fasebj.9.10.7615167. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ebihara I, Osada S, Takahashi T, Yamamoto M, Tomino Y, Koide H. Gene expression of metalloproteinases and their inhibitor in renal tissue of New Zealand black/white F1 mice. Clin Sci (Lond) 1993;85:295–301. doi: 10.1042/cs0850295. [DOI] [PubMed] [Google Scholar]
- Faber-Elmann A, Sthoeger Z, Tcherniack A, Dayan M, Mozes E. Activity of matrix metalloproteinase-9 is elevated in sera of patients with systemic lupus erythematosus. Clin Exp Immunol. 2002;127:393–398. doi: 10.1046/j.1365-2249.2002.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyszer G, Lambiri I, Nagel R, Keysser C, Keysser M, Gromnica-Ihle E, Franz J, Burmester GR, Jung K. Circulating levels of matrix metalloproteinases MMP-3 and MMP-1, tissue inhibitor of metalloproteinases 1 (TIMP-1), and MMP-1/TIMP-1 complex in rheumatic disease. Correlation with clinical activity of rheumatoid arthritis versus other surrogate markers. J Rheumatol. 1999;26:251–258. [PubMed] [Google Scholar]
- Kotajima L, Aotsuka S, Fujimani M, Okawa-Takatsuji M, Kinoshita M, Sumiya M, Obata K. Increased levels of matrix metalloproteinase-3 in sera from patients with active lupus nephritis. Clin Exp Rheumatol. 1998;16:409–415. [PubMed] [Google Scholar]
- Zucker S, Hymowitz M, Conner C, Zarrabi HM, Hurewitz AN, Matrisian L, Boyd D, Nicolson G, Montana S. Measurement of matrix metalloproteinases and tissue inhibitors of metalloproteinases in blood and tissues. Clinical and experimental applications. Ann N Y Acad Sci. 1999;878:212–227. doi: 10.1111/j.1749-6632.1999.tb07687.x. [DOI] [PubMed] [Google Scholar]
- Makowski GS, Ramsby ML. Concentrations of circulating matrix metalloproteinase 9 inversely correlate with autoimmune antibodies to double stranded DNA: implications for monitoring disease activity in systemic lupus erythematosus. Mol Pathol. 2003;56:244–247. doi: 10.1136/mp.56.4.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker S, Mian N, Drews M, Conner C, Davidson A, Miller F, Birembaut P, Nawrocki B, Docherty AJ, Greenwald RA, Grimson R, Barland P. Increased serum stromelysin-1 levels in systemic lupus erythematosus: lack of correlation with disease activity. J Rheumatol. 1999;26:78–80. [PubMed] [Google Scholar]
- Ainiala H, Hietaharju A, Dastidar P, Loukkola J, Lehtimaki T, Peltola J, Korpela M, Heinonen T, Nikkari ST. Increased serum matrix metalloproteinase 9 levels in systemic lupus erythematosus patients with neuropsychiatric manifestations and brain magnetic resonance imaging abnormalities. Arthritis Rheum. 2004;50:858–865. doi: 10.1002/art.20045. [DOI] [PubMed] [Google Scholar]
- Trysberg E, Blennow K, Zachrisson O, Tarkowski A. Intrathecal levels of matrix metalloproteinases in systemic lupus erythematosus with central nervous system engagement. Arthritis Res Ther. 2004;6:R551–R556. doi: 10.1186/ar1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun-Bryce S, Rosenberg GA. Gelatinase B modulates selective opening of the blood-brain barrier during inflammation. Am J Physiol. 1998;274:R1203–R1211. doi: 10.1152/ajpregu.1998.274.5.R1203. [DOI] [PubMed] [Google Scholar]
- Hu J, Steen PE Van den, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- Bagchus WM, Hoedemaeker PJ, Rozing J, Bakker WW. Glomerulonephritis induced by monoclonal anti-Thy 1.1 antibodies. A sequential histological and ultrastructural study in the rat. Lab Invest. 1986;55:680–687. [PubMed] [Google Scholar]
- Heymann W, Hackel DB, Harwood S, Wilson SG, Hunter JL. Production of nephrotic syndrome in rats by Freund's adjuvants and rat kidney suspensions. Proc Soc Exp Biol Med. 1959;100:660–664. doi: 10.3181/00379727-100-24736. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Avila G, Iturria C, Vadillo-Ortega F, Ovalle C, Montano M. Changes in matrix metalloproteinases during the evolution of interstitial renal fibrosis in a rat experimental model. Pathobiology. 1998;66:196–204. doi: 10.1159/000028023. [DOI] [PubMed] [Google Scholar]
- Iimura O, Takahashi H, Yashiro T, Madoiwa S, Sakata Y, Asano Y, Kusano E. Effect of ureteral obstruction on matrix metalloproteinase-2 in rat renal cortex. Clin Exp Nephrol. 2004;8:223–229. doi: 10.1007/s10157-004-0287-x. [DOI] [PubMed] [Google Scholar]
- Zoja C, Abbate M, Remuzzi G. Progression of chronic kidney disease: insights from animal models. Curr Opin Nephrol Hypertens. 2006;15:250–257. doi: 10.1097/01.mnh.0000222691.53970.83. [DOI] [PubMed] [Google Scholar]
- Harendza S, Schneider A, Helmchen U, Stahl RA. Extracellular matrix deposition and cell proliferation in a model of chronic glomerulonephritis in the rat. Nephrol Dial Transplant. 1999;14:2873–2879. doi: 10.1093/ndt/14.12.2873. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Horikoshi S, Osada S, Shofuda K, Shirato I, Tomino Y. Macrophage-derived MT1-MMP and increased MMP-2 activity are associated with glomerular damage in crescentic glomerulonephritis. J Pathol. 2000;191:299–305. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH637>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Tomita M, Koike H, Han GD, Shimizu F, Kawachi H. Decreased collagen-degrading activity could be a marker of prolonged mesangial matrix expansion. Clin Exp Nephrol. 2004;8:17–26. doi: 10.1007/s10157-003-0258-7. [DOI] [PubMed] [Google Scholar]
- Lutz J, Yao Y, Song E, Antus B, Hamar P, Liu S, Heemann U. Inhibition of matrix metalloproteinases during chronic allograft nephropathy in rats. Transplantation. 2005;79:655–661. doi: 10.1097/01.tp.0000151644.85832.b5. [DOI] [PubMed] [Google Scholar]
- Steinmann-Niggli K, Ziswiler R, Kung M, Marti HP. Inhibition of matrix metalloproteinases attenuates anti-Thy1.1 nephritis. J Am Soc Nephrol. 1998;9:397–407. doi: 10.1681/ASN.V93397. [DOI] [PubMed] [Google Scholar]
- Marti HP. The role of matrix metalloproteinases in the activation of mesangial cells. Transplant Immunol. 2002;9:97–100. doi: 10.1016/s0966-3274(02)00006-0. [DOI] [PubMed] [Google Scholar]
- Turck J, Pollock AS, Lee LK, Marti HP, Lovett DH. Matrix metalloproteinase 2 (gelatinase A) regulates glomerular mesangial cell proliferation and differentiation. J Biol Chem. 1996;271:15074–15083. doi: 10.1074/jbc.271.25.15074. [DOI] [PubMed] [Google Scholar]
- Kwan JA, Schulze CJ, Wang W, Leon H, Sariahmetoglu M, Sung M, Sawicka J, Sims DE, Sawicki G, Schulz R. Matrix metalloproteinase-2 (MMP-2) is present in the nucleus of cardiac myocytes and is capable of cleaving poly (ADP-ribose) polymerase (PARP) in vitro. FASEB J. 2004;18:690–692. doi: 10.1096/fj.02-1202fje. [DOI] [PubMed] [Google Scholar]
- Wang W, Schulze CJ, Suarez-Pinzon WL, Dyck JR, Sawicki G, Schulz R. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation. 2002;106:1543–1549. doi: 10.1161/01.cir.0000028818.33488.7b. [DOI] [PubMed] [Google Scholar]
- Nelissen I, Martens E, Steen PE Van den, Proost P, Ronsse I, Opdenakker G. Gelatinase B/matrix metalloproteinase-9 cleaves interferon-beta and is a target for immunotherapy. Brain. 2003;126:1371–1381. doi: 10.1093/brain/awg129. [DOI] [PubMed] [Google Scholar]
- Opdenakker G, Dillen C, Fiten P, Martens E, Van Aelst I, Steen PE Van den, Nelissen I, Starckx S, Descamps FJ, Hu J, Piccard H, Van Damme J, Wormald MR, Rudd PM, Dwek RA. Remnant epitopes, autoimmunity and glycosylation. Biochim Biophys Acta. 2006;1760:610–615. doi: 10.1016/j.bbagen.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Opdenakker G, Steen PE Van den, Van Damme J. Gelatinase B: a tuner and amplifier of immune functions. Trends Immunol. 2001;22:571–579. doi: 10.1016/s1471-4906(01)02023-3. [DOI] [PubMed] [Google Scholar]
- Gill SE, Parks WC. Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol. 2008;40:1334–1347. doi: 10.1016/j.biocel.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim EJ, Lee SH, Lee JG, Kim JR, Yun SS, Baek SH, Lee C. Toll-like receptor 9 dependent activation of MAPK and NF-kB is required for the CpG ODN-induced matrix metalloproteinase-9 expression. Exp Mol Med. 2007;39:239–245. doi: 10.1038/emm.2007.27. [DOI] [PubMed] [Google Scholar]
- Merrell MA, Ilvesaro JM, Lehtonen N, Sorsa T, Gehrs B, Rosenthal E, Chen D, Shackley B, Harris KW, Selander KS. Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol Cancer Res. 2006;4:437–447. doi: 10.1158/1541-7786.MCR-06-0007. [DOI] [PubMed] [Google Scholar]
- Gaipl US, Voll RE, Sheriff A, Franz S, Kalden JR, Herrmann M. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev. 2005;4:189–194. doi: 10.1016/j.autrev.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Lorenz HM, Herrmann M, Winkler T, Gaipl U, Kalden JR. Role of apoptosis in autoimmunity. Apoptosis. 2000;5:443–449. doi: 10.1023/a:1009692902805. [DOI] [PubMed] [Google Scholar]