

Abstract
Purpose
Type 4 cAMP phosphodiesterase (PDE4) inhibitors, compounds that activate cAMP-mediated signaling by inhibiting cAMP catabolism, potentiate glucocorticoid-mediated apoptosis in chronic lymphocytic leukemia (CLL) cells but the mechanism by which this occurs is unknown. In this study, we sought to address whether PDE4 inhibitors alter expression of glucocorticoid receptor (GRα) in CLL cells.
Experimental Design
CLL cells or normal hematopoietic cells were treated with PDE4 inhibitors followed by analysis of GRα transcript and protein by real-time PCR and Western analysis.
Results
PDE4 inhibitors up-regulate glucocorticoid receptor transcript levels in CLL cells but not normal circulating T cells, B cells, monocytes or neutrophils. As GRα transcript half-life does not vary in CLL cells treated with the prototypic PDE4 inhibitor rolipram, the four-fold increase in GRα mRNA levels observed within four hours of rolipram treatment appears to result from an increase in transcription. Rolipram treatment increases levels of transcripts derived from the 1A3 promoter to a greater extent than the 1B promoter. Treatment of CLL cells with cilomilast and roflumilast, two PDE4 inhibitors previously studied in clinical trials also augments GRα transcript levels and glucocorticoid-mediated apoptosis. Washout studies demonstrate that simultaneous treatment with both drug classes irreversibly augments apoptosis over the same time frame that glucocorticoid receptor up-regulation occurs. While treatment of CLL cells with glucocorticoids reduces basal GRα transcript levels in a dose-related manner, co-treatment with rolipram maintained GRα transcript levels above baseline.
Conclusion
Our results suggest that PDE4 inhibitors may sensitize CLL cells to glucocorticoid-induced apoptosis by augmenting GRα expression.
Keywords: CLL, PDE4 inhibitor
Introduction
Glucocorticoids are an important component of standard therapy for several lymphoid malignancies, including multiple myeloma, acute lymphoblastic leukemia and diffuse large B cell lymphoma. As early studies in patients with B cell chronic lymphocytic leukemia (B-CLL) demonstrated that addition of prednisone to chlorambucil augmented response rate but not median survival, glucocorticoids are not generally a standard component of initial therapy for patients with B-CLL (1, 2). Nonetheless, two studies of high dose glucocorticoid therapy have suggested that glucocorticoids can be of clinical benefit to a subset of patients with treatment-refractory B-CLL (3, 4).
Despite frequent responses to glucocorticoid treatment, monotherapy with glucocorticoids is not curative in any lymphoid malignancy, but the mechanisms underlying clinical glucocorticoid resistance remain controversial. Structural alterations in the GR are commonly identified in lymphoid cell lines that have been selected for glucocorticoid resistance by prolonged culture in dexamethasone, but comparable alterations in primary malignant lymphoid cells have been only infrequently reported (5-9). A detailed analysis of treated B-CLL patients failed to identify abnormalities in either the DNA or steroid binding domains of leukemic GRs (10). Non-structural modifications of glucocorticoid signaling pathways are likely to be important in clinical glucocorticoid resistance and efforts to identify and reverse these modifications may be therapeutically useful (8, 9). Several clinical studies in patients with acute and chronic lymphoid leukemias have reported a correlation between low leukemic cell GRα expression levels and poor response to treatment (11-13). However, numerous exceptions to such correlative studies have also been reported, leading to the suggestion that clinical resistance to GCs may also result from unrelated “downstream” signaling alterations (reviewed in (14)).
cAMP-mediated signaling can favorably alter the apoptotic response to glucocorticoids in specific lymphoid subsets, although the precise molecular explanation for this relationship remains unclear. Seminal early work done by Suzanne Bourgeois and colleagues demonstrated that isolation of Wehi-7 cells, a murine T cell lymphoma line, that were resistant to cAMP-mediated apoptosis due to alterations in protein kinase A made subsequent development of spontaneous glucocorticoid-resistant cells occur at higher frequencies (10-7) than in wildtype cells (> 10-10) (15). Gruol and Altschmied subsequently determined that RU486, ordinarily a GR antagonist for GC-induced lymphoid cytolysis, becomes an agonist in the setting of co-treatment with a cAMP analogue (16). Conversely, McConkey and colleagues reported that glucocorticoid receptor (GR) deficient ICR.27 cells, a variant of the CEM T cell lymphoma line, are insensitive to cAMP-induced apoptosis. Transfection of ICR.27 cells with the glucocorticoid receptor restored sensitivity to cAMP-mediated apoptosis (17). Finally, the catalytic subunit of PKA has been shown to associate with the glucocorticoid receptor (18).
One critical factor that regulates lymphoid sensitivity to glucocorticoids is the level of GR expression. Gruol et al determined that treatment of Wehi-7 cells with cAMP analogues raised glucocorticoid receptor transcript and protein levels (19). Several mechanisms have been proposed to explain why GR transcript levels rise following treatment of specific cell subsets with agents that augment cAMP signaling. In studies of rat hepatoma cells, Dong et al reported that treatment with 8-bromo-cAMP increases GR mRNA half-life from 4 hours to 10 hours (20). As treatment of such cell cultures with inhibitors of protein or mRNA synthesis had no effect on the ability of 8-bromo-cAMP to increase GR transcript levels, Dong et al argue that a principal mechanism by which cAMP signaling augments GR transcript levels must be through GR mRNA stabilization. In contrast, utilizing transfection of GR promoter luciferase constructs into HeLa cells, Penuelas et al determined that treatment with the adenylate cyclase activator forskolin doubled the transcriptional activity of the human GR promoter (21). After mapping and testing five putative CRE binding sites, the authors demonstrated loss of forskolin inducibility in promoter constructs shorter than 1 kB and the presence of a CRE element which bound CRE in vitro in shift assays. Thus, it appears that in some cell lineages, cAMP-induced augmentation of GR transcript levels is due to augmented transcription rather than mRNA stabilization.
Type 4 cAMP phosphodiesterase (PDE4) inhibitors offer a therapeutically plausible means by which to take advantage of the phenomenon of cAMP-mediated augmentation of glucocorticoid sensitivity in malignant lymphoid cells. The PDE4 family play a key role in catabolizing cAMP in a variety of human hematopoietic cells and PDE4 inhibitors are in late stage clinical studies for a variety of inflammatory illnesses, including asthma and chronic obstructive disease (22). In prior work, we have determined that inhibition of PDE4, in the absence of addition of exogenous adenylate cyclase activators such as forskolin or beta adrenergic agonists, increases cAMP levels, activates protein kinase A as judged by CREB phosphorylation, and induces apoptosis in primary B-CLL cells, albeit in well less than 100% of cells (23). Treatment with the prototypic PDE4 inhibitor rolipram induces mitochondrial release of cytochrome c, activation of caspase 9 and 3, and cleavage of PARP in CLL cells (24). PDE4 inhibitors also activate Rap1 in B-CLL cells due to cAMP-mediated activation of the Rap1 GDP exchange factor EPAC1, but EPAC activation appears to be anti-apoptotic (25). PDE4 inhibitors thus induce both PKA-mediated pro-apoptotic and EPAC-mediated anti-apoptotic signaling pathways in B-CLL cells, with the PKA-mediated pro-apoptotic pathway having a dominant effect.
PDE4 inhibitors such as rolipram augment hydrocortisone or dexamethasone-induced apoptosis in primary B-CLL cells, as well as transactivation of glucocorticoid response element (GRE)-containing reporter constructs (26). Both of these effects are reversed by the type I PKA antagonist Rp-8Br-cAMPS. The specific mechanism or mechanisms by which PDE4 inhibitors increase glucocorticoid sensitivity in B-CLL cells remain unknown. In this study, we sought to determine whether PDE4 inhibitors alter leukemic expression of glucocorticoid receptor. We find that PDE4 inhibitors augment expression of GRα at a transcriptional level and that among human primary hematopoietic cells, this effect is quite specific to B-CLL.
Methods and Materials
Materials
The following reagents were obtained from commercial sources: Rolipram (Biomol, Plymouth Meeting PA), forskolin (Sigma, St. Louis MO), actinomycin D (Calbiochem, San Diego CA), Rp-8Br-cAMPS (Biolog, Bremen, Germany). Cilomilast and roflumilast were obtained from Memory Pharmaceuticals (Montvale, NJ).
Cell Culture and Isolation
Blood samples were obtained in heparinized tubes with IRB-approved consent from flow cytometry-confirmed B-CLL patients that were either untreated or for whom at least 1 month had elapsed since chemotherapy. Patients with active infections or other serious medical conditions were not included in this study. Patients with white blood cell counts of less than 15,000/μl by automated analysis were excluded from this study. Whole blood was layered on Ficoll-Hystopaque (Sigma) and peripheral blood mononuclear cells (PBMC) isolated after centrification. PBMC were washed and resuspended at 1×107 cells/ml in complete media [RPMI-1640 (Mediatech) supplemented with 10% fetal bovine serum (Sigma), 20mM L-glutamine, 100 IU/ml penicillin and 100 μg/ml streptomycin (Mediatech)]. PBMC was found to contain >90% B-CLL by FACS without additional purification. Normal B cells, T cells, and monocytes were obtained from anonymous healthy donors (New York Biologics, Southampton NY) and isolated via magnetic negative depletion per the manufacturer's protocol (Miltenyi, Bergisch-Gladbach Germany) from PBMC. Polymorphonuclear cells (PMNs) were obtained by erythrocyte depletion of whole blood via dextran sedimentation followed by removal of PBMC using Ficoll seperation. With the exception of PMNs, which were used immediately after purification, all other primary normal and malignant cell populations were rested overnight at 37°C prior to use.
Western analysis
Following cell culture, cells were collected by centrifugation (400×g; 10 min.), washed once with phosphate buffered saline and lysed in ice-cold 10 mM HEPES-NaOH buffer (pH 7.4) containing 1%TritonX-100, 10% glycerol, 25 mM β-glycerophosphate, 100 mM NaCl, 2mM EDTA, 2mM EGTA, 1mM dithiothriotol, 1 mM vanadate, 1mM phenylmethanesulfonyl fluoride, and 1mM benzamidine. Cell lysates were transferred to 1.5 mL tubes and centrifuged at 14,000 rpm for 30 minutes (5°C) in a microcentrifuge to clarify samples of insoluble cellular debris. Concentrations of soluble proteins in samples of clarified supernatants were determined using Bradford assays. Samples were heat denatured at 100°C for 5 minutes in protein denaturing sample buffer. Levels of GR protein expression was examined in 50 μg aliquots of denatured protein samples that were subjected to electrophoretic separation through 8% SDS-polyacrylamide gels followed by electrotransfer onto Immobilon-P membrane (Millipore) in 10 mM 3-(cyclohexylamino)-1-propanesulfonic acid buffer (pH 11) containing 10% methanol. Primary glucocorticoid receptor antibodies (Santa Cruz Biotechnology, sc-1002) and secondary goat anti-rabbit IgG conjugated to horse radish peroxidase (HRP) (Santa Cruz Biotechnology, sc-2004) were diluted at 1:500 and 1:5000, respectively, in Tris buffered saline (20 mM Tris-HCl, pH 7.6; 137 mM NaCl; 0.1% Triton X-100) containing 5% non-fat milk (w/v) to immunoblot proteins on western membranes. Immunocomplexes with HRP activity on membranes were developed using enhanced chemiluminescent reagent (Pierce) as substrate and visualized by exposure to x-ray film. Membranes were blotted with primary monoclonal anti-α-tubulin (Sigma, T5168) to control for equal loading of protein samples on gels and transfer onto membranes.
Real-time PCR for GR exon 1A3-2, 1B-2, 1C-2, 8-9α and 8-9β transcripts
Cells were plated at a concentration of 1×107 cells/ml in complete media with or without drug treatment as indicated and incubated at 37°C for four hours. Total RNA was obtained using the RiboPure isolation kit (Ambion, Austin TX) per the manufacturer's protocol. Reverse transcription of 2-5 micrograms of total RNA was carried out with the Superscript III reverse transcription kit (Invitrogen, Carlsbad California) primed with random hexomers. An assay for the GR exon 8-9α transcript was devised using primer3 software available at http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi. The forward and reverse flanking oligonucleotide primers were 5′-AGCCATTGTCAAGAGGGAAG-3′ and 5′-TGATTGGTGATGATTTCAGCTA-3′, respectively. The FAM/TAMRA Taqman probe located in exon 8 was 5′-TCCAGCCAGAACTGGCAGCG-3′. Each reaction contained 900 nM forward and reverse primer, 50 nM probe, 1X Universal PCR Master Mix (Applied Biosystems, Foster City CA), and 2 μl of cDNA diluted 1:50. Flanking primers and internal FAM/TAMRA-labeled probe oligonucleotides and reaction conditions for the splice sites at GR exons 1A3-2, 1B-2 and 1C-2, as well as GR exon 8-9β were those reported by Pedersen and Vedeckis, with the exception that the Taqman probe for the exon 1A3-2 site was (27) 5′-TCAGTGAATATCAACTTCCTTCTCAGACACTTTAATGAA-3′ and the reverse primer for the exon 8-9β splice site was TGTGAGATGTGCTTTCTGGTTTTAA (28). cDNA was diluted 1:50 for measurement of exon 1A3-2, 1B-2, and 1C-2, and diluted 1:5 for measurement of exon 8-9β Predeveloped Taqman assay reagents (PDAR) for measuring 18S rRNA (Applied Biosystems) was used for normalization. Realtime PCR was carried out on a MX300P instrument (Stratagene, La Jolla,CA) Transcript abundance relative to control samples was calculated using the 2-ΔΔCt method. We established that the slopes of the curves for the amplification of GRα and rRNA did not vary by more than 10%. All oligonucleotides were purchased from IDT.
Apoptosis assays
One million cells were incubated in duplicate in 48 well plates with or without drug treatment as indicated for 48 hours in 1 mL of complete media. Cell were transferred to polypropylene Falcon FACS tubes, incubated for 10 minutes at 37°C with Hoechst 33342 (Molecular Probes, Eugene OR) at a final concentration of 0.25 μg/mL. Cells were then stored on ice until analysis on a MoFlo cytometer using a 450 nm bandpass filter. In some cases, apoptosis was detected as membrane depolarization with dihexyloxacarbocyanine (DiOC6(3)) from Molecular Probes (Eugene, OR) at a concentration of 400 nM. DiOC6(3) stained samples were incubated for 30 minutes at 37°C and stored on ice until analysis on a FACScan (Becton Dickenson, Franklin Lakes, NJ). Results obtained from Hoechst 33342 and DiOC6(3) staining were validated with Annexin-V/PI staining per the manufacturer's protocol (BD, Franklin Lakes, NJ). FACS data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR) by gating for the apoptotic population. The level of apoptosis detected in B-CLL cultures did not differ by more than 10% when measured by Hoechst 33342 or DiOC6(3).
Statistical Analysis
Statistical analysis and plotting was performed using Prism version 4 (GraphPad Software, San Diego CA) and SPSS version 12 (SPSS Inc., Chicago, Illinois). The significance of main and interaction effects was determined via repeated measures analysis of variance (ANOVA) tests with significance of subsequent pair-wise comparisons via Bonferroni post-hoc tests. In some cases multivariate analysis of variance (MANOVA) were used as indicated when the data violated the sphericity assumption for ANOVA.
Results
Rolipram augments GRα transcript and protein levels in B-CLL cells in a time and dose-dependent manner
Given prior reports that cAMP analogues augment GR levels in a subset of cell types, we used comparative quantification real-time RT-PCR to determine whether treatment of B-CLL cells with a PDE4 inhibitor altered expression of GRα transcript. In leukemic cells from eight patients, treatment of B-CLL cells with the PDE4 inhibitor rolipram (20 μM) augmented GRα transcript levels in a time and dose-dependent manner. The effect of rolipram exposure time on GRα transcript level was found to be significant by ANOVA (P = 0.017). GRα transcript levels rose significantly over the first six hours to a mean of 4.8±0.2 fold above baseline (P = 0.028) and maintained such a fourfold increase for at least 24 hours (Figure 1A). While comparable augmentation of GR transcript levels was observed at rolipram doses ranging from 1 to 20 μM, significant augmentation was not observed at 0.1 μM rolipram, a concentration at or below the EC50 of rolipram for inhibition of TNF secretion (Figure 1B) (29). Addition of the adenylate cyclase stimulator forskolin did not significantly augment GRα transcript in B-CLL cells, either when used alone or in combination with rolipram, a finding in keeping with prior studies demonstrating that rolipram activates PKA in B-CLL in the absence of exogenous adenylate cyclase activation (data not shown). Western analysis of rolipram-treated B-CLL cells from four patients demonstrated that PDE4-inhibitor-induced GR transcript up-regulation was associated with an increase in GR protein at four to six hours (Figure 1C).
Figure 1. GRα expression is up-regulated in B-CLL cells following treatment with the PDE4 inhibitor rolipram.
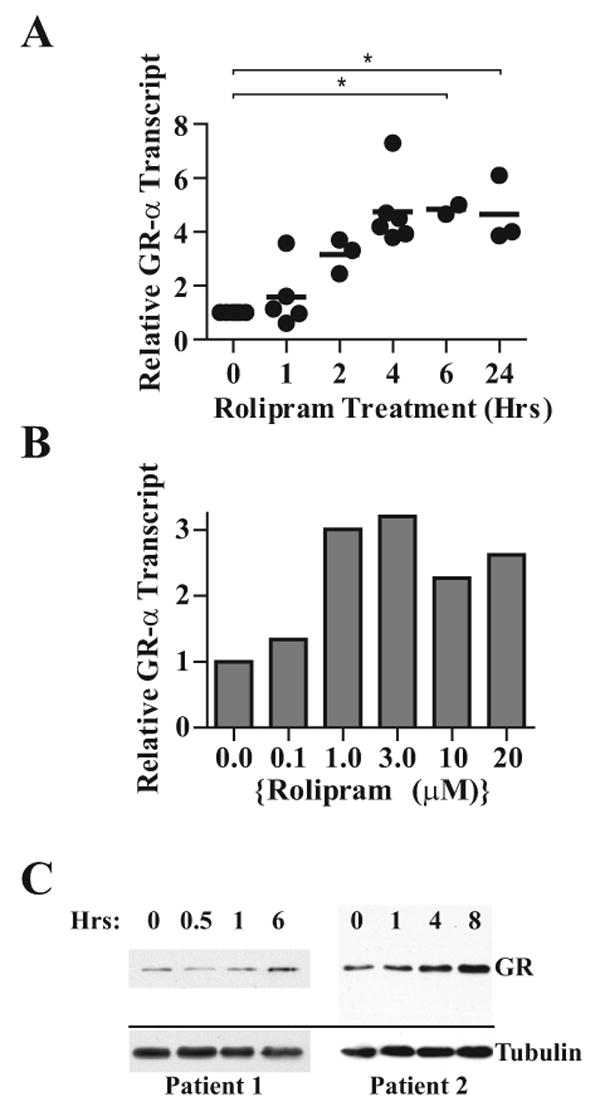
(A) B-CLL cells were treated for the indicated lengths of time with rolipram (20 μM), followed by RNA isolation, cDNA synthesis and real-time PCR for GRα using oligonucleotides that spanned exons 8 and 9α. Each point represents the fold increase in GRα transcript levels of an individual patient sample relative to the same patient's CLL cells treated with vehicle (DMSO) alone. The mean fold increase in transcript level is denoted with a horizontal line. Asterisks denote significant main effect for time at P < 0.05 (ANOVA). (B) B-CLL cells from an individual patient were treated for four hours with DMSO or rolipram at the indicated dosage (μM), followed by RNA isolation and real-time RT-PCR for GRα transcript levels relative to vehicle (DMSO) control. The data are representative of one of two similar experiments. (C) B-CLL cells were treated with DMSO alone (0 hr time point) or rolipram (20 μM) for the indicated amount of time, followed by lysis, protein quantification and immunoblot analysis for GRα protein expression (GR). Alpha-tubulin was also assessed by immunoblot analysis as an internal loading control. Results from two patients are shown and are representative of four patients tested.
cAMP-mediated augmentation of GR transcript levels has been variably attributed to increased GR half-life (in rat hepatoma cells) or GR transcription (in HeLa cells) (20, 21) To establish whether the increased levels of GR transcript observed in rolipram-treated B-CLL cells were the result of altered transcript half-life, we treated B-CLL cells with vehicle alone (DMSO) or rolipram (20 μM) for four hours, followed by treatment with the RNA polymerase inhibitor actinomycin D (10 μg/mL) for varying periods of time. Assessment of GRα transcript levels following such actinomycin D treatment revealed that the half-life of GRα transcript was not altered by rolipram treatment (P = 0.88, Figure 2), suggesting that in B-CLL cells, cAMP-mediated augmentation of GRα transcript occurs by a transcriptional mechanism.
Figure 2. PDE4 inhibitor-induced GRα transcript up-regulation is not due to altered transcript half-life.

B-CLL cells were treated for four hours with vehicle control alone (DMSO) or rolipram (20 μM), followed by treatment with the RNA polymerase inhibitor actinomycin D (10 μg/mL) for the indicated period of time. After RNA isolation and cDNA synthesis, samples were analyzed for relative GRα transcript levels by real-time PCR. All GR transcript levels were normalized to that observed in DMSO-treated cells at time 0. A non-linear curve fit for one phase exponential decay revealed no significant difference in the rates of reduction in GRα transcript levels (P = 0.88). The experiment is representative of two experiments performed.
Rolipram-mediated GRα transcript up-regulation is not observed in a range of normal hematopoietic cell types
In order to establish the specificity of PDE4 inhibitor-mediated GR transcript up-regulation, we carried out real-time RT-PCR analyses in a range of normal hematopoietic cells. Rolipram treatment did not augment GRα transcript levels in unpurified human mononuclear cells or in purified populations of human T cells, B cells, neutrophils and monocytes (Figure 3). In the absence of basal adenylate cyclase activity, PDE4 inhibitors may be relatively ineffective in activating cAMP-mediated signal transduction. However, forskolin (40 μM), either alone or in combination with rolipram, did not induce GR transcript up-regulation in these normal hematopoietic cell populations (data not shown). As it possible that a PDE family other than PDE4 might regulate GR levels in these cell populations, we examined whether addition of IBMX (50 μg/mL), a competitive inhibitor of nine of the eleven currently known PDE families, led to an increase in GR transcript. No augmentation of GR transcript levels was observed in unpurified human mononuclear cells (a predominantly T cell preparation) or in purified populations of human monocytes (data not shown). To establish whether the observed effect of PDE4 inhibitors on B-CLL GRα transcript levels was simply a function of lymphoid transformation, we examined primary leukemic cells from a patient with T-CLL and a patient with Sezary syndrome. In neither case was rolipram-induced augmentation of GRα transcript levels observed (Figure 3).
Figure 3. Among circulating hematopoietic lineage cells tested, PDE4-inhibitor-induced augmentation of GRα transcript levels is specific to B-CLL.

B-CLL cells, T-CLL cells, Sezary cells, human peripheral blood mononuclear cells (PBMC), T cells, B cells, neutrophils (PMNs) or monocytes were incubated for four hours in vehicle control alone (DMSO) or rolipram (20 μM) as indicated. RNA was isolated, cDNA synthesized, and levels of GRα transcript relative to vehicle-treated cells of each cell type was assessed by real-time RT-PCR. The data are representative of at least two samples tested for all cell populations except T-CLL, where only one patient sample was available.
Roflumilast and cilomilast augment glucocorticoid-mediated apoptosis and GRα transcript levels
To determine whether the alterations in GRα transcript observed following rolipram treatment of B-CLL cells are shared by other structurally distinct PDE4 inhibitors, we examined cilomilast and roflumilast, two PDE4 inhibitors that have been utilized in clinical trials testing the activity of PDE4 inhibitors in asthma and chronic obstructive pulmonary disease (COPD) (Figure 4A) (30) (31). Consistent with the hypothesis that PDE4 is in fact the rolipram target that results in augmented GRα transcript levels, cilomilast and roflumilast increased GRα transcript levels (P = 0.012; ANOVA) in B-CLL cells (Figure 4B). As previously observed with rolipram, both cilomilast (50 μM) and roflumilast (1.0 μM) augmented the efficacy with which glucocorticoids induce apoptosis in B-CLL (Figure 4C). In pooled data from ten B-CLL patients, combining PDE4 inhibitor and glucocorticoid treatment significantly augmented apoptosis relative to either agent alone (P < 0.01: MANOVA). Despite this statistically significant effect, however, it is important to point out that among these ten leukemic cell samples, several did not in fact show PDE4-inhibitor-mediated augmentation of glucocorticoid apoptosis. Such heterogeneity is similar to results we previously obtained in studies with rolipram. One patient whose leukemic cells were very sensitive to glucocorticoid-mediated apoptosis showed no further augmentation with the addition of cilomilast or roflumilast. Another leukemic cell sample which had relatively high basal apoptosis (39%) showed little or no sensitivity to any of the drug treatments. It is possible that this heterogeneity in apoptotic responses to combined glucocorticoid/PDE4 inhibitor treatment is due to genetic heterogeneity of the leukemias in this patient population.
Figure 4. Effect of several structurally distinct PDE4 inhibitors on glucocorticoid-mediated apoptosis and GRα transcript levels.

(A) Molecular structures of rolipram, roflumilast, and cilomilast. (B) B-CLL cells were treated as indicated with vehicle control (DMSO), 50 μM cilomilast, 1.0 μM roflumilast or 20 μM rolipram for four hours followed by RNA isolation and RT-PCR analysis for GRα transcript. The mean fold increase in transcript level of five patients is denoted with a horizontal line. (C) B-CLL cells were treated for 48 hours with PDE4 inhibitors alone or in the presence of increasing dosages of dexamethasone, as indicated, followed by assessment for apoptosis by FACS analysis. The PDE4 inhibitors cilomilast, roflumilast, and rolipram were added at 50 μM, 1.0 μM, and 20 μM respectively. The data shown are the mean and SEM of ten patients tested. (D) B-CLL cells were treated for 0, 4, 8, 24 or 48 hours with vehicle alone, rolipram (20 μM), dexamethasone (1 μM), or the combination of rolipram and dexamethasone. The drugs were removed by washing at the indicated time, followed by completion of culture in media alone until 48 hours had elapsed.
The synergistic apoptotic effects of combined PDE4 inhibitor/glucocorticoid treatment can be observed following drug treatment for as little as two hours
If the potential therapeutic benefit of combined PDE4 inhibitor/glucocorticoid therapy is to be explored clinically, it will be important to determine the length of time leukemic cells must be exposed to both agents in order to augment glucocorticoid-mediated apoptosis. CLL cells were treated with vehicle alone, rolipram (20 μM), dexamethasone (1 μM) or the combination of rolipram and dexamethasone for varying periods of time, followed by washing and completion of cell culture for 48 hours. Combined rolipram/glucocorticoid treatment for as little as two hours augmented apoptosis relative to treatment with either drug alone (Figure 4D). Treatment for eight hours with the drug combination resulted in a level of apoptosis that approached that observed following a full 48 hours of combined drug treatment. Although our studies did not address how much of the compounds remain associated with the leukemic cells after washing, these results suggest that PDE4 inhibitors potentiate glucocorticoid-mediated apoptosis in a relatively short period of time.
PDE4 inhibitors variably augment different classes of GRα transcript
Transcription of the glucocorticoid receptor gene is regulated by three promoters, 1A, 1B, and 1C (32). Prior studies in the human B cell line IM-9 have demonstrated that under basal conditions, roughly 1%, 32% and 66% of GRα transcripts are derived from promoters 1A, 1B and 1C, respectively (28). Using previously validated real-time PCR assays that detect splicing of exons 1A3, 1B and 1C to exon 2, we examined leukemic cells from six B-CLL patients for the effect of rolipram treatment on levels of GRα transcripts derived from these three promoters. As illustrated in Figure 5A, rolipram (20 μM) augmented GR transcripts derived from each of the three promoters: exon 1A3 (22.2±7.4 fold), exon 1B (3.6±0.5 fold) and exon 1C (7.1±0.9 fold). The up-regulation observed for transcripts containing exon 1A3 was significantly higher than that observed for transcripts containing exon 1B (3.6±0.5 fold) (P < 0.05; ANOVA).
Figure 5. Regulation of GRα and GRβ transcription by PDE4 inhibitors.

(A). Treatment of B-CLL cells with the PDE4 inhibitor rolipram alters relative expression of GR transcripts containing exons 1A, 1B, 1C, 8/9α and 8/9β. B-CLL cells from six patients were treated with rolipram (20 μM) for four hours, followed by RNA isolation, cDNA synthesis and real-time PCR for GRα or GRβ transcripts using oligonucleotides that spanned exons 1A3/2 (1A), 1B/2 (1B), 1C/2 (1C), or 8/9α (alpha). Transcripts are shown as relative to vehicle control treated cells for each exon analyzed. Asterisks denote significant exon 1 difference at P < 0.05 (ANOVA). A comparable analysis was also carried out for transcripts containing exon 8/9β (beta) but here only three patient samples were analyzed. (B). Co-treatment with a PDE4 inhibitor and dexamethasone maintains GRα transcript levels at greater than basal levels despite dexamethasone-induced down-regulation of GRα expression. B-CLL cells were treated with varying dosages of dexamethasone as indicated for four hours in the presence or absence of rolipram (20 μM), followed by RNA isolation, cDNA synthesis and real-time PCR for GRα using oligonucleotides that spanned exons 8 and 9α. All GR transcript levels were normalized to that observed in DMSO-treated cells (N=2). (C) Co-treatment with the enantiomeric cAMP antagonist Rp-8Br-cAMPS abrogates the ability of PDE4 inhibitors to augment GRα transcript levels. B-CLL cells were treated for the indicated period of time with rolipram (20 μM) or rolipram and Rp-8Br-cAMPS (500 μM), followed by RNA isolation, cDNA synthesis and real-time PCR for GRα (N=2).
GRβ has been reported to suppress transactivation of GRα by synthetic glucocorticoids and high levels of GRβ may correlate with insensitivity to GC-induced apoptosis (33, 34). We therefore examined GRβ regulation by PDE4 inhibitors in B-CLL. Treatment with rolipram augmented GRβ transcript levels seven-fold relative to levels observed in untreated CLL cells (Figure 5). Basal levels of GRβ in B-CLL cells appear to be far lower than those of GRα, as the real-time PCR threshold cycle numbers we observed for GRβ were 10 cycles greater than those for GRα despite comparable efficiency of amplification. These results are similar to the 1000-fold lower GRβ level reported by Vedeckis and colleagues using the same oligonucleotide primers in a quantitative real-time RT-PCR study of basal and glucocorticoid-treated GRα and β transcript levels in the EBV-transformed B cell-line IM-9 (28).
PDE4 inhibitors abrogate the ability of dexamethasone to reduce B-CLL GRα transcript levels
Exposure to glucocorticoids regulates intracellular GR levels, with resultant down-regulation of GR in most cell lineages, including B cells and B lineage cell lines, but with up-regulation of GR in thymocytes and T-ALL-derived cell lines (28). Using a tetracycline-regulated GR promoter transfected into a cell-line lacking functional GR, the glucocorticoid-induced autoinduction of GR expression in human T cell lines has been linked to their increased sensitivity to glucocorticoid-mediated apoptosis (35). We therefore sought to determine whether in CLL cells, co-treatment with PDE4 inhibitors would abrogate glucocorticoid-mediated attenuation of GR transcript levels. As expected, dexamethasone reduced GR transcript levels in CLL cells in a dose-dependent manner such that following treatment for six hours with 1 μM dexamethasone, GR transcript levels were one-third of those observed in untreated cells (Figure 5B). In contrast, co-treatment of CLL cells for six hours with 20 μM rolipram and varying doses of dexamethasone resulted in GR transcript levels above basal levels, even at 1 μM dexamethasone (Figure 5B). These results suggest that PDE4 inhibitors may augment glucocorticoid-mediated apoptosis in B-CLL cells as a result of their ability to block the normal down-regulation of GR transcript levels in glucocorticoid-treated cells.
Given our prior demonstration that inhibition of PKA signaling with the enantiomeric cAMP antagonist Rp-8-Br-cAMPS substantially or entirely blocked the ability of glucocorticoids to induce apoptosis in B-CLL cells as well as reduced transactivation of glucocorticoid response element (GRE)-containing reporter constructs, we sought to determine whether the same antagonist blocks PDE4 inhibitor-induced augmentation of GRα transcript levels (26). Co-treatment of CLL cells with Rp-8Br-cAMPS (500 μM) markedly reduced rolipram-induced augmentation of GRα at 4 hours (Figure 5C). These results are consistent with the hypothesis that PDE4 inhibitors regulate GRα transcript levels by a cAMP and PKA-mediated mechanism.
Discussion
This study demonstrates that treatment with several structurally distinct PDE4 inhibitors augments GR transcript levels in B-CLL cells but not in normal circulating hematopoietic cells. As co-treatment with PDE4 inhibitors and glucocorticoids also induces apoptosis in B-CLL cells to levels higher than that observed with either agent alone, these results suggest that the simultaneous use of these two classes of drug might be relatively selectively toxic to CLL cells (26). While it is experimentally difficult to determine whether the alterations in GR expression account for the augmented apoptosis observed when these drugs are combined, a variety of prior studies have demonstrated that levels of GR can play an important role in determining the outcome of glucocorticoid treatment. In cell lines expressing different levels of GR, the magnitude of transcriptional responses to glucocorticoids are roughly proportional to the number of receptors per cell (36). Thymocytes from transgenic mice carrying two extra copies of the GR show enhanced sensitivity to glucocorticoid-mediated apoptosis (37).
Consistent with actinomycin D experiments demonstrating that PDE4 inhibitors do not substantially alter GR transcript half-life, we find that rolipram augments GR transcripts derived from different promoters to varying degrees in CLL cells, suggesting a transcriptional mechanism for the observed increase in GR transcript levels. GR transcription in human lymphoid cells is regulated by at least three promoters (A-C), although the open reading frame of the GR gene, which begins in exon 2, is not altered by promoter usage (32). A quantitative analysis in the IM-9 human B cell line revealed that in such cells, promoters 1A, 1B and 1C accounted for 1%, 30% and 70% of all GRα transcripts, respectively (28). Alternative splicing of transcripts derived from the most 5′ promoter, 1A, results in three types of transcripts: 1A1, 1A2 and 1A3, the last of which was the most abundant (32). While a prior study in HeLa cells has suggested cAMP-mediated regulation of the most 3′ promoter, 1C, the effects of cAMP signaling on GR promoters 1A and 1B have not been reported (21). Transcripts containing 1B and 1C appear to be relatively ubiquitously expressed, while expression of exon 1A3-containing transcripts is particularly high in cell lines of hematopoietic lineage (32). In B-CLL cells, treatment with PDE4 inhibitors augments 1A3 transcripts to a greater degree than other GR transcripts (1A3 > 1B; p< 0.05). It is of interest that basal levels of GR transcripts containing exon 1A correlate with sensitivity to glucocorticoid-induced apoptosis, as expression of this form of GR transcript is particularly high in thymocytes and T lymphoblastoid cell lines (38). While the functional significance of the fact that PDE4 inhibitors preferentially induce expression of exon 1A3-containing GR transcripts in B-CLL cells remains unknown, it is possible that varying 5′ UTRs could alter either GR mRNA translation start site or translation efficiency. Two alternative translation initiation sites in exon 2 give rise to A and B forms of the GR. GRα B has twice the biologic activity of GRα A in gene expression studies, and different tissue types have differing ratios of GRα A and GRα B (39, 40).
Among all the primary hematopoietic cell populations tested, augmentation of GR transcript levels by PDE4 inhibitors is unique to B-CLL cells. This finding is in keeping with a prior study in which, among a variety of circulating hematopoietic cells examined, PDE4-inhibitor-induced EPAC activation was found only in B-CLL cells (25). The explanation for the unique impact of PDE4 inhibitor treatment on signaling in B-CLL cells remains unknown. Although we do not observe rolipram-induced up-regulation of GR transcript in primary circulating B cell samples, it is still possible that comparable responses might be observed in a normal B cell population that is not well represented among circulating B cells. As PDE4 inhibitors have potent effects on a variety of primary circulating hematopoietic cells, particularly T cells and monocytes, it is clearly not the case that the selective augmentation of GR transcript observed in B-CLL cells is due to the fact that PDE4 inhibitors initiate cAMP-mediated signaling only in B-CLL cells. Instead, the selectivity of the effects observed in CLL cells may be due to the magnitude or kinetics of the cAMP response, the effector proteins activated (i.e. type 1 vs. type 2 PKA, EPAC), or, perhaps most likely, cell-type-specific signaling induced by cAMP effector protein activation in B-CLL cells.
Glucocorticoids regulate expression of glucocorticoid receptor. In lymphoid cell subsets that are particularly sensitive to glucocorticoid-mediated apoptosis, glucocorticoids augment GR transcript levels (41). In other cell lineages, including B lineage cells, glucocorticoids reduce GRα transcript levels (28). Consistent with this literature, we find that treatment of B-CLL cells with dexamethasone reduces GRα transcript levels in a dose-dependent manner. In contrast, treatment with the combination of dexamethasone and a PDE4 inhibitor augmented GRα transcript levels. While treatment with both drugs resulted in augmentation of GRα transcript intermediate between those observed with treatment with either drug alone, even at the highest dose of dexamethasone used (1 μM), co-treatment resulted in an increment rather than a decrement in GRα transcript levels. These results suggest that PDE4 inhibitors may increase glucocorticoid-mediated apoptosis in B-CLL cells because they counteract the normal dampening of glucocorticoid-mediated signaling that occurs in B lineage cells as a result of glucocorticoid-induced down-regulation of GRα levels.
High dose glucocorticoid therapy can lead to clinical responses in a subset of patients with treatment refractory B-CLL (3) (4). The experiments described in this study suggest that addition of a PDE4 inhibitor to such therapy might fairly selectively augment apoptosis in B-CLL cells as a result of augmentation of GR transcript levels. Our observation that treatment with PDE4 inhibitors for as few as four hours augments glucocorticoid-induced killing of B-CLL cells augers well for the potential clinical applicability of PDE4 inhibitor/glucocorticoid therapy for B cell malignancies, as it is likely that therapeutically effective serum levels of PDE4 inhibitors could be safely maintained for such a period of time. Our future studies will focus on the mechanisms underlying the selectivity with which PDE4 inhibitors alter cAMP metabolism in CLL cells.
Acknowledgments
The authors wish to thank Lewis Weintraub M.D. for assistance in obtaining leukemic cell samples and Paul Epstein PhD for very helpful discussions.
This work was funded in part by National Cancer Institute grant CA106705 and by a National Heart, Lung, and Blood Institute-supported BUSM Hematology Training Grant HL007501 (J.A.M.).
References
- 1.Han T, Ezdinli EZ, Shimaoka K, Desai DV. Chlorambucil vs combined chlorambucil-corticosteroid therapy in chronic lymphocytic leukemia. Cancer. 1973;31:502–8. doi: 10.1002/1097-0142(197303)31:3<502::aid-cncr2820310303>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 2.Sawitsky A, Rai KR, Glidewell O, Silver RT. Comparison of daily versus intermittent chlorambucil and prednisone therapy in the treatment of patients with chronic lymphocytic leukemia. Blood. 1977;50:1049–59. [PubMed] [Google Scholar]
- 3.Molica S. High-dose dexamethasone in refractory B-cell chronic lymphocytic leukemia patients. Am J Hematol. 1994;47:334. doi: 10.1002/ajh.2830470421. [DOI] [PubMed] [Google Scholar]
- 4.Thornton PD, Hamblin M, Treleaven JG, Matutes E, Lakhani AK, Catovsky D. High dose methyl prednisolone in refractory chronic lymphocytic leukaemia. Leuk Lymphoma. 1999;34:167–70. doi: 10.3109/10428199909083393. [DOI] [PubMed] [Google Scholar]
- 5.Moalli PA, Pillay S, Weiner D, Leikin R, Rosen ST. A mechanism of resistance to glucocorticoids in multiple myeloma: transient expression of a truncated glucocorticoid receptor mRNA. Blood. 1992;79:213–22. [PubMed] [Google Scholar]
- 6.Irving JA, Minto L, Bailey S, Hall AG. Loss of heterozygosity and somatic mutations of the glucocorticoid receptor gene are rarely found at relapse in pediatric acute lymphoblastic leukemia but may occur in a subpopulation early in the disease course. Cancer Res. 2005;65:9712–8. doi: 10.1158/0008-5472.CAN-05-1227. [DOI] [PubMed] [Google Scholar]
- 7.de Lange P, Segeren CM, Koper JW, et al. Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor-mediated effects in transfected cells. Cancer Res. 2001;61:3937–41. [PubMed] [Google Scholar]
- 8.Rabindran SK, Danielsen M, Stallcup MR. Glucocorticoid-resistant lymphoma cell variants that contain functional glucocorticoid receptors. Mol Cell Biol. 1987;7:4211–7. doi: 10.1128/mcb.7.12.4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zawydiwski R, Harmon JM, Thompson EB. Glucocorticoid-resistant human acute lymphoblastic leukemic cell line with functional receptor. Cancer Res. 1983;43:3865–73. [PubMed] [Google Scholar]
- 10.Soufi M, Kaiser U, Schneider A, Beato M, Westphal HM. The DNA and steroid binding domains of the glucocorticoid receptor are not altered in mononuclear cells of treated CLL patients. Exp Clin Endocrinol Diabetes. 1995;103:175–83. doi: 10.1055/s-0029-1211347. [DOI] [PubMed] [Google Scholar]
- 11.Pui CH, Dahl GV, Rivera G, Murphy SB, Costlow ME. The relationship of blast cell glucocorticoid receptor levels to response to single-agent steroid trial and remission response in children with acute lymphoblastic leukemia. Leuk Res. 1984;8:579–85. doi: 10.1016/0145-2126(84)90006-7. [DOI] [PubMed] [Google Scholar]
- 12.Shahidi H, Vottero A, Stratakis CA, et al. Imbalanced expression of the glucocorticoid receptor isoforms in cultured lymphocytes from a patient with systemic glucocorticoid resistance and chronic lymphocytic leukemia. Biochem Biophys Res Commun. 1999;254:559–65. doi: 10.1006/bbrc.1998.9980. [DOI] [PubMed] [Google Scholar]
- 13.Kato GJ, Quddus FF, Shuster JJ, et al. High glucocorticoid receptor content of leukemic blasts is a favorable prognostic factor in childhood acute lymphoblastic leukemia. Blood. 1993;82:2304–9. [PubMed] [Google Scholar]
- 14.Haarman EG, Kaspers GJ, Veerman AJ. Glucocorticoid resistance in childhood leukaemia: mechanisms and modulation. Br J Haematol. 2003;120:919–29. doi: 10.1046/j.1365-2141.2003.04189.x. [DOI] [PubMed] [Google Scholar]
- 15.Gruol DJ, Campbell NF, Bourgeois S. Cyclic AMP-dependent protein kinase promotes glucocorticoid receptor function. J Biol Chem. 1986;261:4909–14. [PubMed] [Google Scholar]
- 16.Gruol DJ, Altschmied J. Synergistic induction of apoptosis with glucocorticoids and 3′,5′-cyclic adenosine monophosphate reveals agonist activity by RU 486. Mol Endocrinol. 1993;7:104–13. doi: 10.1210/mend.7.1.8383286. [DOI] [PubMed] [Google Scholar]
- 17.Kiefer J, Okret S, Jondal M, McConkey DJ. Functional glucocorticoid receptor expression is required for cAMP-mediated apoptosis in a human leukemic T cell line. J Immunol. 1995;155:4525–8. [PubMed] [Google Scholar]
- 18.Doucas V, Shi Y, Miyamoto S, West A, Verma I, Evans RM. Cytoplasmic catalytic subunit of protein kinase A mediates cross-repression by NF-kB and the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2000;97:11893–8. doi: 10.1073/pnas.220413297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gruol DJ, Rajah FM, Bourgeois S. Cyclic AMP-dependent protein kinase modulation of the glucocorticoid-induced cytolytic response in murine T-lymphoma cells. Mol Endocrinol. 1989;3:2119–27. doi: 10.1210/mend-3-12-2119. [DOI] [PubMed] [Google Scholar]
- 20.Dong Y, Aronsson M, Gustafsson JA, Okret S. The mechanism of cAMP-induced glucocorticoid receptor expression. Correlation to cellular glucocorticoid response. J Biol Chem. 1989;264:13679–83. [PubMed] [Google Scholar]
- 21.Penuelas I, Encio IJ, Lopez-Moratalla N, Santiago E. cAMP activates transcription of the human glucocorticoid receptor gene promoter. J Steroid Biochem Mol Biol. 1998;67:89–94. doi: 10.1016/s0960-0760(98)00097-1. [DOI] [PubMed] [Google Scholar]
- 22.Lerner A, Epstein PM. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem J. 2006;393:21–41. doi: 10.1042/BJ20051368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim DH, Lerner A. Type 4 cyclic adenosine monophosphate phosphodiesterase as a therapeutic target in chronic lymphocytic leukemia. Blood. 1998;92:2484–94. [PubMed] [Google Scholar]
- 24.Moon EY, Lerner A. PDE4 inhibitors activate a mitochondrial apoptotic pathway in chronic lymphocytic leukemia cells that is regulated by protein phosphatase 2A. Blood. 2003;101:4122–30. doi: 10.1182/blood-2002-10-3208. [DOI] [PubMed] [Google Scholar]
- 25.Tiwari S, Felekkis K, Moon EY, Flies A, Sherr DH, Lerner A. Among circulating hematopoietic cells, B-CLL uniquely expresses functional EPAC1, but EPAC1-mediated Rap1 activation does not account for PDE4 inhibitor-induced apoptosis. Blood. 2004;103:2661–7. doi: 10.1182/blood-2003-06-2154. [DOI] [PubMed] [Google Scholar]
- 26.Tiwari S, Dong H, Kim EJ, Weintraub L, Epstein PM, Lerner A. Type 4 cAMP phosphodiesterase (PDE4) inhibitors augment glucocorticoid-mediated apoptosis in B cell chronic lymphocytic leukemia (B-CLL) in the absence of exogenous adenylyl cyclase stimulation. Biochem Pharmacol. 2005;69:473–83. doi: 10.1016/j.bcp.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Fan WT, Koch CA, de Hoog CL, Fam NP, Moran MF. The exchange factor Ras-GRF2 activates Ras-dependent and Rac-dependent mitogen-activated protein kinase pathways. Curr Biol. 1998;8:935–8. doi: 10.1016/s0960-9822(07)00376-4. [DOI] [PubMed] [Google Scholar]
- 28.Pedersen KB, Vedeckis WV. Quantification and glucocorticoid regulation of glucocorticoid receptor transcripts in two human leukemic cell lines. Biochemistry. 2003;42:10978–90. doi: 10.1021/bi034651u. [DOI] [PubMed] [Google Scholar]
- 29.Seldon PM, Meja KK, Giembycz MA. Rolipram, salbutamol and prostaglandin E2 suppress TNFalpha release from human monocytes by activating Type II cAMP-dependent protein kinase. Pulm Pharmacol Ther. 2005;18:277–84. doi: 10.1016/j.pupt.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 30.Rennard SI, Schachter N, Strek M, Rickard K, Amit O. Cilomilast for COPD: results of a 6-month, placebo-controlled study of a potent, selective inhibitor of phosphodiesterase 4. Chest. 2006;129:56–66. doi: 10.1378/chest.129.1.56. [DOI] [PubMed] [Google Scholar]
- 31.Timmer W, Leclerc V, Birraux G, et al. The new phosphodiesterase 4 inhibitor roflumilast is efficacious in exercise-induced asthma and leads to suppression of LPS-stimulated TNF-alpha ex vivo. J Clin Pharmacol. 2002;42:297–303. doi: 10.1177/00912700222011328. [DOI] [PubMed] [Google Scholar]
- 32.Breslin MB, Geng CD, Vedeckis WV. Multiple promoters exist in the human GR gene, one of which is activated by glucocorticoids. Mol Endocrinol. 2001;15:1381–95. doi: 10.1210/mend.15.8.0696. [DOI] [PubMed] [Google Scholar]
- 33.Fruchter O, Kino T, Zoumakis E, et al. The human glucocorticoid receptor (GR) isoform {beta} differentially suppresses GR{alpha}-induced transactivation stimulated by synthetic glucocorticoids. J Clin Endocrinol Metab. 2005;90:3505–9. doi: 10.1210/jc.2004-1646. [DOI] [PubMed] [Google Scholar]
- 34.Koga Y, Matsuzaki A, Suminoe A, Hattori H, Kanemitsu S, Hara T. Differential mRNA expression of glucocorticoid receptor alpha and beta is associated with glucocorticoid sensitivity of acute lymphoblastic leukemia in children. Pediatr Blood Cancer. 2005;45:121–7. doi: 10.1002/pbc.20308. [DOI] [PubMed] [Google Scholar]
- 35.Ramdas J, Liu W, Harmon JM. Glucocorticoid-induced cell death requires autoinduction of glucocorticoid receptor expression in human leukemic T cells. Cancer Res. 1999;59:1378–85. [PubMed] [Google Scholar]
- 36.Vanderbilt JN, Miesfeld R, Maler BA, Yamamoto KR. Intracellular receptor concentration limits glucocorticoid-dependent enhancer activity. Mol Endocrinol. 1987;1:68–74. doi: 10.1210/mend-1-1-68. [DOI] [PubMed] [Google Scholar]
- 37.Reichardt HM, Umland T, Bauer A, Kretz O, Schutz G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol Cell Biol. 2000;20:9009–17. doi: 10.1128/mcb.20.23.9009-9017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purton JF, Monk JA, Liddicoat DR, et al. Expression of the glucocorticoid receptor from the 1A promoter correlates with T lymphocyte sensitivity to glucocorticoid-induced cell death. J Immunol. 2004;173:3816–24. doi: 10.4049/jimmunol.173.6.3816. [DOI] [PubMed] [Google Scholar]
- 39.Yudt MR, Cidlowski JA. Molecular identification and characterization of a and b forms of the glucocorticoid receptor. Mol Endocrinol. 2001;15:1093–103. doi: 10.1210/mend.15.7.0667. [DOI] [PubMed] [Google Scholar]
- 40.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–42. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 41.Pedersen KB, Geng CD, Vedeckis WV. Three mechanisms are involved in glucocorticoid receptor autoregulation in a human T-lymphoblast cell line. Biochemistry. 2004;43:10851–8. doi: 10.1021/bi049458u. [DOI] [PubMed] [Google Scholar]