

Abstract
Mice lacking histone deacetylase 9 (HDAC9) and its truncated variant, HDRP, exhibit post-axial polydactyly that manifests as an extra big toe on the right hind foot. Polydactyly in HDAC9/HDRP knockout mice occurs with incomplete penetrance and affects both genders similarly. Because polydactyly can result from overactivity of sonic hedgehog (Shh) signaling, we investigated whether HDRP acted as a negative regulator of the Shh pathway. We find that Gli1, a transcription factor and downstream mediator of Shh signaling, is expressed at substantially higher levels in the feet of perinatal HDAC9/HDRP-/- mice as compared with wild-type littermates. To more directly examine whether HDRP negatively-regulates Shh signaling we utilized cell lines that express components of the Shh pathway and that respond to the Shh agonist purmorphamine. We find that purmorphamine-mediated stimulation of Gli1 in the NIH 3T3 and HT22 cell lines is inhibited by the expression of HDRP. In HT22 cells, purmorphamine treatment leads to an increase in the rate of cell proliferation, which is also inhibited by HDRP. This inhibitory effect of HDRP on purmorphamine-mediated cell proliferation was also observed in primary cultures of glial cells. Although the mechanism by which it inhibits Gli1 induction and cell proliferation by purmorphamine is not clear, HDRP localizes to the nucleus suggesting it acts just upstream of Gli3 activation in the signaling cascade activated by Shh. Taken together our results suggest that HDRP acts as a negative regulator of the Shh pathway and that the absence of HDRP results in hyper-activation of this pathway resulting in polydactyly.
Keywords: HDRP, Shh, polydactyly, Gli1, purmorphamine
Introduction
Histone deacetylases (HDACs) are a class of enzymes originally identified on the basis of their ability to remove acetyl groups of lysine residues on histones thereby compacting chromatin in a transcriptionally-repressed conformation. Over the past few years, a large number of other non-histone nuclear and cytoplasmic proteins have been identified as substrates of HDACs. Through their actions on histones and other non-histone proteins HDACs serve as important regulators of a variety of cellular processes including proliferation, differentiation, cell survival, and cell death (1-4). Moreover, deregulated functioning of HDACs has been implicated in serious pathologies such as cancer and neurodegenerative disease (5, 6).
Eighteen individual HDACs have been identified in mammalian cells and these are divided into four classes depending primarily on their structural homology to yeast deacetylases. Class I HDACs (HDACs 1, 2, 3 and 8) are composed mainly on the HDAC catalytic domain, localized to the nucleus, and are expressed in most tissues (7). Class II HDACs (HDACs 4, 5, 6, 7, 9 and 10), on the other hand, are expressed more selectively and can generally translocate between the nucleus and cytoplasm in a phosphorylation-regulated manner (7). Within this group, HDACs 4, 5, 7, and 9 are subclassified as Class IIA HDACs whereas HDACs 6 and 10 represent Class IIB. Class III HDACs are called sirtuins. The seven members of this sub-family (Sirt1-7) are structurally and functionally distinct from all the other HDACs. Some of these proteins are nuclear, while others are cytoplasmic or localized to the mitochondria. The fourth class of HDACs contains a single member, HDAC11, which localizes predominantly to the nucleus (8).
We have previously focused on HDAC9 and a truncated form of this protein called HDAC-related protein (HDRP). HDRP is produced as the result of an RNA splicing event that truncates full length HDAC9 (human HDAC9, 1011 aa) into an isoform lacking the C-terminal deacetylase domain (human HDRP, 590 aa) (9). We described that the expression of both HDAC9 and HDRP was reduced in cultured neurons from the central nervous system primed to undergo cell death (10). Forced suppression of HDRP expression (but not HDAC9 expression) induces death of otherwise healthy neurons while the overexpression of HDRP prevents neuronal death. These results indicate that HDRP plays a protective role in the nervous system and at least in this neuronal model HDRP appears to have a more prominent role than HDAC9 (10). Mice lacking both HDAC9 and HDRP have been generated and found to be fertile and survive a normal lifespan (11). Although no overt anatomical anomalies, brain abnormalities, or neurological deficits are observed in the mutant (Wang and D’Mello, unpublished observation), HDAC9/HDRP knockout mice develop cardiac hypertrophy with age and in response to pressure overload (11). In addition, HDAC9/HDRP null mice are more sensitive to skeletal muscle denervation-induced hyperacetylation of muscle chromatin than wild-type counterparts (12). However, no phenotype was reported to be associated with this finding.
In this report we describe that HDAC9/HDRP knockout mice also exhibit post-axial polydactyly. Polydactyly is a frequently inherited condition in humans and other mammals. Several different polydactylous mutations have been identified in the mouse. Analysis of these mutant mice along with other experimental manipulations including limb bud transplants has identified the morphogenic signaling protein, sonic hedgehog (Shh) as a pivotal regulator of normal digit formation (13-15). Shh activates a signaling pathway in which a transmembrane protein Patched1 (Ptch1) inhibits the activity of another protein, Smoothened (Smo). Binding of Shh to Ptch1 relieves this inhibition, allowing Smo to transduce a signal to the nucleus via glioma (Gli) transcription factors, resulting in increased expression of target genes. Although the importance of Shh to proper digit formation is well accepted, the intracellular mechanisms involved are poorly understood. We provide evidence that HDRP acts as a negative regulator of Shh signaling providing an explanation for polydactyly in HDAC9/HDRP-deficient mice.
Materials and Methods
Reagents
Reagents and cell culture media were acquired from Invitrogen (Carlsbad, CA). Unless otherwise specified, all chemicals used were purchased from Sigma (St. Louis, MO). The following antibodies were used: Gli1, Gli3, and α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA); HDRP and Brd-U (Sigma, St. Louis, MO); glial fibrillary acidic protein (GFAP) (Cell Signaling Technology, Danvers, MA). For Western blotting, primary antibodies were used at a 1:1,000 dilution. Peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) used in this study were goat anti-rabbit IgG (1:10,000) and goat anti-mouse IgG (1:10,000). For Immunocytochemistry, fluorochrome-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Labs (West Grove, PA). Purmorphamine was purchased from Calbiochem (San Diego, CA).
Adenoviruses
A recombinant adenovirus expressing GFP was a kind gift from Kim A. Heidenreich (University of Colorado Health Sciences Center, Denver, CO). The HDRP (c-Myc tag) adenoviral vector was provided by Eric N. Olson (University of Texas Southwestern Medical Center, Dallas, TX).
Cell Culture
All cell cultures were maintained at 37°C and 5% CO2 in a humidified incubator. 293T (human embryonic kidney cells), NIH 3T3 (mouse fibroblasts), and HeLa (human cervical) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS. HT22 (mouse hippocampal) cells were grown in DMEM with 10% FBS but without sodium pyruvate. CHO (hamster ovarian) cells were cultured in DMEM/F-12 medium (ATCC, Manassas, VA) with 10% FBS. Cerebellar granule neurons and glia were extracted from 7-day-old Wistar rats cerebella as previously described (16). In brief, cells were cultured in basal Eagle’s medium with Earle’s salts (BME) containing 10% fetal bovine serum, 25 mM KCl, 2 mM glutamine (Gibco-BRL), and 100 μg/ml gentamicin on dishes coated with poly-L-lysine at a density of 1 × 106 cells/well in 24-well dishes. All animals used in experiments were treated in accordance with both on-site Institutional Animal Care and Use Committee policies in addition to those prescribed by the National Institutes of Health.
Adenovirus Infection
Adenoviral expression vectors were used to infect 1-day-old glial cultures, or cell lines one day after plating at a multiplicity of infection of 100. In brief, medium containing serum was removed, saved, and replaced with serum-free medium containing 4 μg/ml of hexadimethrin bromide (Sigma) and virus. Infections were carried out for 1 h with frequent mixing and then the cells were washed once and original medium was added back. Following infection, treatments were performed after 24 h.
RT-PCR
Trizol reagent (Invitrogen) was used to extract RNA from cells according to the manufacturer’s instructions and cDNA was synthesized using the Thermoscript RT-PCR system (Invitrogen) according to the manufacturer’s instructions. A PCR master mix (Promega, Madison, WI) was used to amplify the gene of interest. The gene-specific primers used for PCR amplification are listed below in 5′ to 3′ orientation and are as follows: Gli1 Forward: CTCAATGCCCACCCCAGT; Gli1 Reverse: GCCACAAAGTCCAGCTGAGTG; Gli3 Forward: ATCAGCTCTGCCTACCTGAGCA; Gli3 Reverse: ATGGAGGCAAATTTGGAGGATT; Ptch1 Forward: GTCTCAGGGTAGCTCTCATA; Ptch1 Reverse: GCATTCTGGCCCTAGCAATA; Smo Forward: CCCTGCTGTGTGCTGTCTAC; Smo Reverse: CAGGGTAGCGATTGGAGTTC; SHH Forward: GAACGATTTAAGGAACTCACC; SHH Reverse: GTCCAGGAAGGTGAGGAAGTC; HDRP Forward: CCTGGAGCTAAATCTTTGCCTA; HDRP Reverse: GGCTGGTGGAGTTCCATTAC; eHAND Forward: GAGCGGCCTTACTTCCAGAG; eHAND Reverse: ACGTCCATCAAGTAGGCGATGT; GFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC; GFP Reverse: GCGGATCTTGAAGTTCACCTTGATGCC; β-Actin Forward: AGGACTCCTATGTGGGTGACGA; and β-Actin Reverse: CGTTGCCAATAGTGATGACCTG. The HDAC9 knockout mice genotyping primers were used as previously described (11) and are: Neo: AGGCATGCTGGGGATGCGGTGGGC; MITR Forward: GCAATTGACTATGCGGCTCTGGGC; MITR Reverse: ATCAGCACTATAGAACAGCCACAGC.
Western Blotting
Cells were washed once with ice-cold phosphate-buffered saline (PBS) and lysed using a lysis buffer (1% Triton, 20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM sodium EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3- VO4,1 μg/ml leupeptin, and one protease inhibitor tablet). Cellular protein was measured using Bradford protein assay reagent (Bio-Rad, Hercules, CA). Protein concentration was normalized to 40 μg and subjected to Western blotting. Protein levels were then assayed using enhanced chemiluminescence (Amersham Bioscience, Piscataway, NJ).
Brd-U Immunocytochemistry
Cells were cultured in 4- or 24-well plates on coverslips coated with poly-L-lysine. Brd-U was added to cultures to a final concentration of 20 μM for 2 hours prior to fixation. Cells were fixed in 4% paraformaldehyde in PBS on ice for 30 min. The cells were washed in PBS for 5 min three times on ice followed by three 5-min washes with 0.5% triton × 100 on ice. Next, cells received one 10-min 1M HCl in H2O wash on ice and then a 30-min incubation with 2M HCl at 37°C. A wash with 0.1 M sodium borate, pH 8.5, for 12 min at room temperature (RT) followed. Next, three 5-min PBS washes at RT were done and cells blocked for 15 min at 37°C in antibody diluent (1% BSA, 0.5% tween 20, 0.1% sodium azide, 5% goat serum). Primary antibodies were added at 1:200 in antibody diluent at RT overnight. Coverslips were washed with PBS three times and fluorochrome-conjugated secondary antibodies added at 1:100 in antibody diluent. Three more washes with PBS followed. The cells were then counterstained with 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI) for 10 min at 25°C to visualize the nucleus. Coverslips were washed two more times with PBS and mounted onto slides. Images were obtained with a Nikon Eclipse 80i fluorescence microscope using a x60 objective for detailing HDRP expression in Glia and a x40 objective for all other images.
Staining of Mouse Bone/Limbs
The staining of mouse limbs was done as previously described (17). The mouse limb was eviscerated and skinned. Next, the mouse limb was placed into 70% ethanol for 5 days to fix the tissue. Staining was then performed by decanting the 70% ethanol and incubating in a staining solution (Alcian Blue 0.15% in 70% ethanol, Alizarin Red S 0.1% in 95% ethanol, and 2% potassium hydrogen phthalate in 70% ethanol mixed together to make a final solution of 1:1:18 respectively) for 3 days. The tissue was then gently washed with dH2O and a macerating solution added (0.75% KOH) and incubated for 3 days. After this time, the tissue was again gently washed in dH2O and a clearing solution added (70% ethanol, glycerol, benzyl alcohol solution, 2:2:1 ratio respectively) for 24 h. The next day, the clearing solution was decanted and the tissue was placed in a holding solution for 7 days. On the eighth day, the limbs were removed, gently dried using kimwipes, and digitally photographed.
Statistics
Graphs show mean values for data collected from three or more separate experiments. Error bars represent standard deviation and were also accrued from three or more separate experiments. Statistical significance was determined by using the unpaired t test, where P values of < 0.05 were scored as significant.
Results
Mice Lacking HDAC9/ HDRP Display Polydactyly
An analyses of over 200 pups from breedings of HDAC9+/- mice revealed the presence of polydactyly in about 25% of the HDAC9-/- pups. Polydactyly was not detected in wild-type or heterozygote littermates. When it was detected, polydactyly was observed on the right hind limb and involved the formation of an extra big toe (Fig. 1). Rarely, (∼2% of HDAC9-/- pups), an extra toe was also observed in the left hind limb. In this case also, the extra digit was the big toe. Both males and females displayed the defect. Thus, HDAC9/HDRP is a new autosomal mouse mutation leading to polydactyly with incomplete penetrance. Despite being part of the same subgroup of HDACs, HDAC4 and HDAC5 knockout mice did not display any type of limb abnormality (Morrison and D’Mello, unpublished observation).
Figure 1.
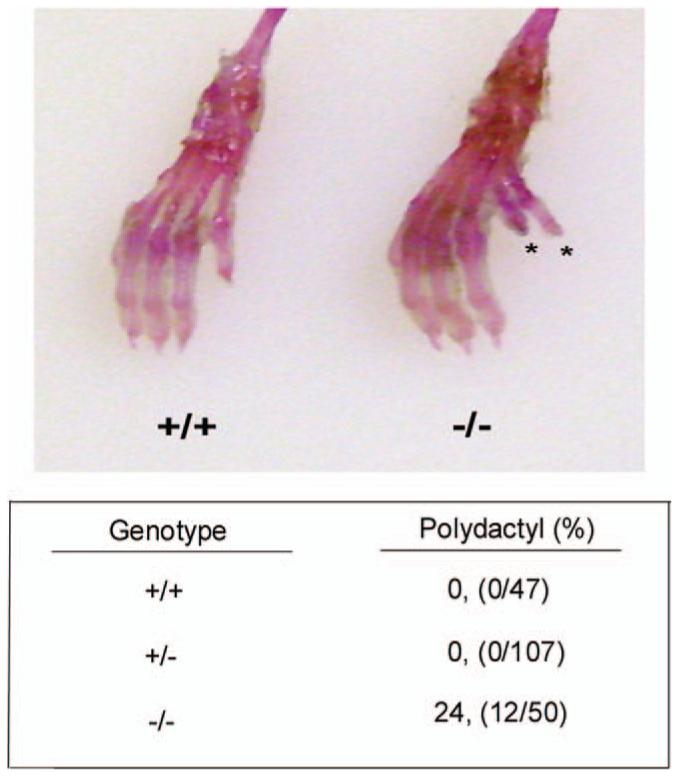
Polydactyly in HDAC9/HDRP null mice. Mouse limbs were skinned and stained with Alcian Blue and Alizarin Red to enhance bone and connective tissue features (representative image above). The lower panel shows the incidence of polydactyly. Polydactylous mice were scored and categorized by their corresponding HDAC9/HDRP genotype. The observed polydactyly manifested almost exclusively as an extra big toe on the right hind foot. We did note one instance out of 50 HDAC9 knockout mice examined that presented with an extra big toe on the left hind foot. A color version of the figure is available in the online journal.
HDAC9/HDRP Suppresses Gli1 Expression
Reports from a number of labs have demonstrated that increased or ectopic Shh signaling in the developing limb bud results in polydactyly (18, 19). Because HDAC9/HDRP-deficient mice display polydactyly we hypothesized that HDAC9/HDRP might negatively regulate the Shh signaling pathway. To examine this issue we analyzed the expression of important mediators of Shh signaling in organs isolated from wild-type and HDRP knockout mice. As shown in Figure 2, the expression level of the Shh pathway downstream target, Gli1, is substantially higher in HDAC9/HDRP-deficient polydactylous feet versus those from wild-type littermates (20). Interestingly, the difference in Gli1 expression diminishes by one month of age, a time at which the feet and digits have neared full development. Gli3, in comparison, shows no remarkable change as is the case for the Shh signal transducer, Smo. We did however observe an increase in the Shh receptor and Smo inhibitor, Ptch1, in both HDAC9/HDRP knockout limb and brain compared to wild-type but only for samples taken from neonates. Although lying upstream of Gli1 in the Shh signaling cascade, once activated Gli1 activates Ptch1 transcription suggesting that the increase in Ptch1 expression is possibly the result of a positive feedback mechanism (21). Indeed it has been shown that Shh signaling activation leads to GLI-dependent transcriptional activation of itself as well as Ptch1 (21). Subsequent data presented in this manuscript shows that Ptch1 induction does indeed occur following activation of the Shh pathway (Fig. 3) and this Ptch1 expression increase is independent of HDAC9/HDRP expression (Fig. 4A and 4C). As shown in Figure 2, there is no substantial difference between wild-type and knockout organ expression of Shh. Taken together, these findings suggest that HDAC9/HDRP acts at a point within the target cell’s Shh response machinery and not in a pathway regulating Shh production.
Figure 2.
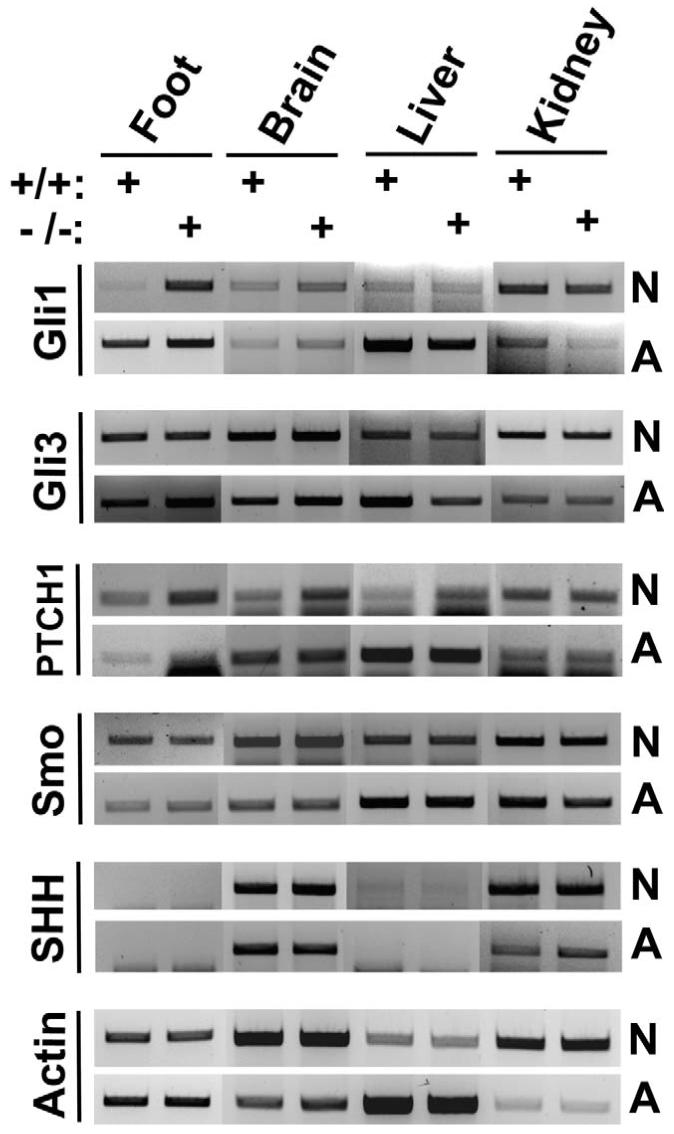
Polydactylous HDAC9/HDRP null mice exhibit increased SHH signaling. The indicated tissue samples were obtained from polydactylous HDAC9/HDRP null mice (-/-) and wild-type littermates (+/+) at either 4 days of age (N = neonatal) or 4 weeks of age (A = adolescent), RNA was extracted from whole organs and normalized between knockout and wild-type samples, cDNA was then made, and PCR performed using gene-specific primers.
Figure 3.
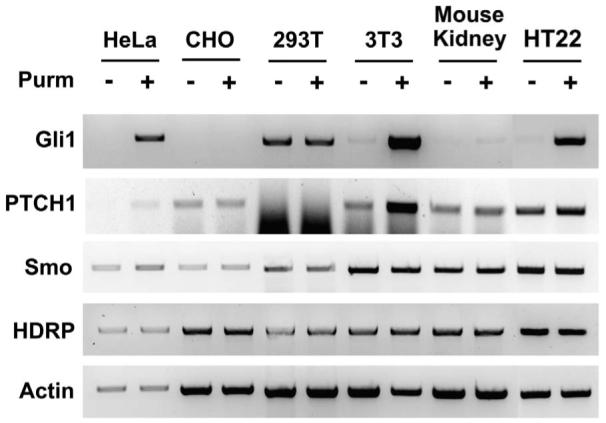
Characterization of SHH-responsive cell cultures. Cells were grown in medium with serum and upon reaching approximately 50% confluence the medium was switched to full medium without serum and with or without 2 μM purmorphamine (Purm). RNA was extracted 24 hours later, cDNA made, and PCR performed. Actin was amplified to ensure an equal amount of cDNA was used.
Figure 4.
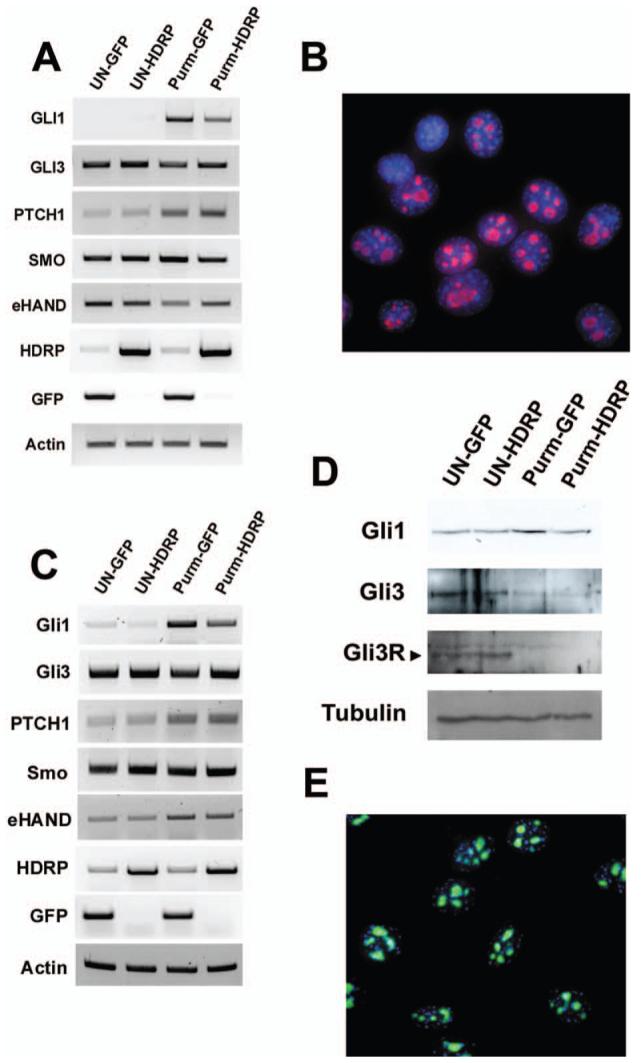
HDRP represses SHH signaling in NIH 3T3 and HT22 cell lines. Cells were grown in the presence of serum until 50% confluence when the cells were infected with HDRP or GFP containing adenovirus constructs. After 24 hours the medium was then changed to medium without serum and with or without (UN) 2 μM purmorphamine (Purm). RNA was extracted 24 hours after treatment, cDNA made, and PCR performed. Actin was amplified to ensure an equal amount of cDNA was used. (A) RT-PCR results for NIH 3T3 cell line. (C) RT-PCR results for HT22 cell line. (D) HT22 cells were treated as described above except that cells were treated with or without purmorphamine for 48 h following infection. Cells were then lysed, cellular protein was measured, normalized, and subjected to Western blotting. The resulting protein blot was sequentially probed with antibodies against Gli1 and Gli3 protein. To ensure equal protein loading α-tubulin levels were examined. Endogenous HDRP was detected in NIH 3T3 cells (B, red) and HT22 cells (E, green) by immunocytochemistry using an antibody against HDRP. DNA was counterstained with DAPI (blue) to visualize the nucleus. A color version of this figure is available in the online journal.
HDRP Inhibits Shh-Induced Signaling
In order to avoid complicating any experimental results by introducing copious amounts of enzymatically active HDAC9 that could produce substantial off-target effects, we decided to utilize the naturally occurring HDAC9 isoform that lacks the deacetylase domain, HDRP. In fact, our previous research has indicated that in a rodent model of neurodegeneration HDRP plays a more prominent role in neuronal survival than HDAC9 (10). To test the hypothesis that HDRP inhibits cellular response to Shh we set out to identify pure cell populations that could respond to Shh so that subsequent more detailed molecular analysis could be performed. Five cell lines were assayed as well as a primary culture of kidney fibroblasts for their ability to respond to the pharmacological agonist of Smo and activator of Shh signaling, purmorphamine (Fig. 3). However, only the human cervical Hela cell line, the mouse fibroblast NIH 3T3 cell line, and the HT22 neuroblastoma cell line responded robustly to purmorphamine treatment as evidenced by increased Gli1 expression, a commonly used marker of Shh signaling. Not unexpectedly given that it acts by stimulating the activity of existing Smo protein, purmophamine treatment did not affect the expression of Smo (22).
Based on its responsiveness to purmorphamine in our hands as well as in previous reports we initially chose the NIH 3T3 cell line to investigate whether HDRP regulated the Shh signaling pathway (22-24). This cell line was infected with adenovirus expressing either GFP or HDRP and then treated with purmorphamine for 24 hrs. The expression of Smo was not altered by HDRP overexpression (Fig. 4A). In contrast, the induction of Gli1 by purmorphamine was substantially reduced by HDRP. Another downstream target of Shh signaling is eHAND (HAND1), a basic helix-loop-helix transcription factor. In the developing limb bud, the administration of Shh leads to a suppression of eHAND expression (25). A similar downregulation of eHAND is observed in purmorphamine-treated NIH 3T3 cells. This downregulation of eHAND is partially reversed by HDRP (Fig. 4A). As observed in most other cell types, HDRP localizes to the nucleus of NIH 3T3 cells under normal conditions (Fig. 4B). We performed these experiments in HT22 cells as well and obtained similar results (Fig. 4C, E). In addition, we observed that increasing HDRP expression within HT22 cells results in inhibition of purmorphamine-associated Gli1 induction at the protein level (Fig. 4D). Gli3 has been shown to be an activator of Gli1 transcription. However, in the absence of Shh signaling, Gli3 protein is cleaved into a repressor form known as Gli3R, which appears to actively inhibit Gli1 transcription (26-28). Interestingly, cleavage of full-length Gli3 protein into the Gli3R isoform was not affected by HDRP in the presence of purmorphamine (Fig. 4D). This finding indicates that HDRP-mediated suppression of Shh signaling occurs at both the RNA and protein levels and is independent of Gli3R production.
HDRP Inhibits Purmorphamine-Stimulated Cell Proliferation
Although HDRP inhibits the stimulation of Gli1 expression by purmorphamine it was unclear whether this action has any significance at the cellular level. We focused our attention on HT22. Treatment of HT22 cells with purmorphamine increases the rate of cell proliferation (Fig 5A). This effect of purmorphamine was significantly reduced when HDRP expression was enhanced (Fig 5A). Next, we wanted to determine if HDRP-mediated inhibition of the Shh pathway occurred in primary cultured cells. Since Gli1 was originally identified by its high expression in malignant gliomas we examined this phenomenon in primary glial cells (29). As with HT22 cells, we observed a reduction in purmorphamine-induced proliferation in glial cells with enhanced HDRP expression (Fig. 5B).
Figure 5.
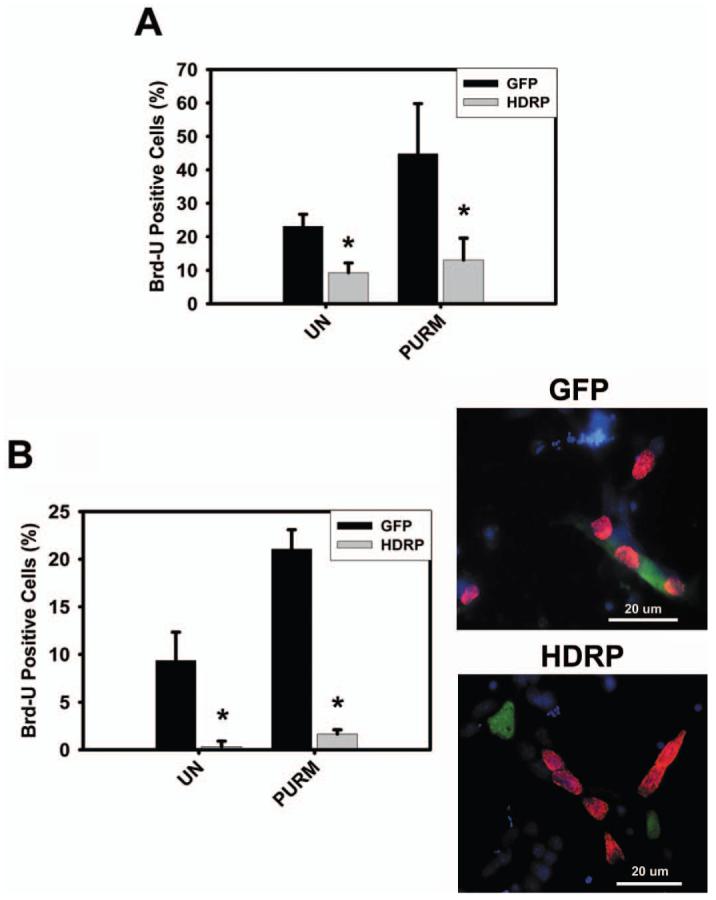
HDRP represses SHH-induced proliferation of HT22 and glial cells. (A) HT22 cells were grown in the presence of serum until 50% confluence when the cells were infected with HDRP or GFP containing adenovirus constructs. After 24 hours the medium was then changed to medium without serum and with or without (UN) 2 μM purmorphamine (Purm). Cells were incubated for an additional 48 hours, Brd-U was added to the medium for 2 hours, and immunocytochemistry performed. Brd-U and HDRP were detected using monoclonal antibodies against Brd-U and c-myc (HDRP contains a c-myc epitope tag) respectively. HDRP and GFP expressing cells were scored as Brd-U positive or negative and the results from three separate experiments were tabulated. (B) Glial cells were extracted from the cerebellum of rats and infected with HDRP or GFP containing adenovirus one day after plating. The following day the cells were treated with or without (UN) 2 μM purmorphamine (Purm). After 48 hours Brd-U was added for 2 hours and immunocytochemistry was performed. Cells expressing exogenous HDRP or GFP were scored as Brd-U positive or negative. The right panel shows a representative image of glial cells stained for GFP (green) or HDRP (green) and Brd-U (red). DAPI (blue) was used to visualize the nucleus and 20 μm scale bar shown for reference. * indicates statistical significance (P < 0.05) comparing GFP and HDRP groups for each treatment. A color version of this figure is available in the online journal.
We also examined how glial cultures from mice lacking HDRP/HDAC9 respond to purmorphamine. Interestingly, the rate of proliferation displayed by HDAC9/HDRP-deficient glial cells was similar to glia cultured from wild-type littermates (Fig. 6A). Since HDRP expression is detectable in wild-type glial cells (shown in Fig. 6B), a likely explanation for the lack of growth enhancement in knockout glial cultures is through compensation for HDAC9/HDRP loss by other HDAC family members.
Figure 6.
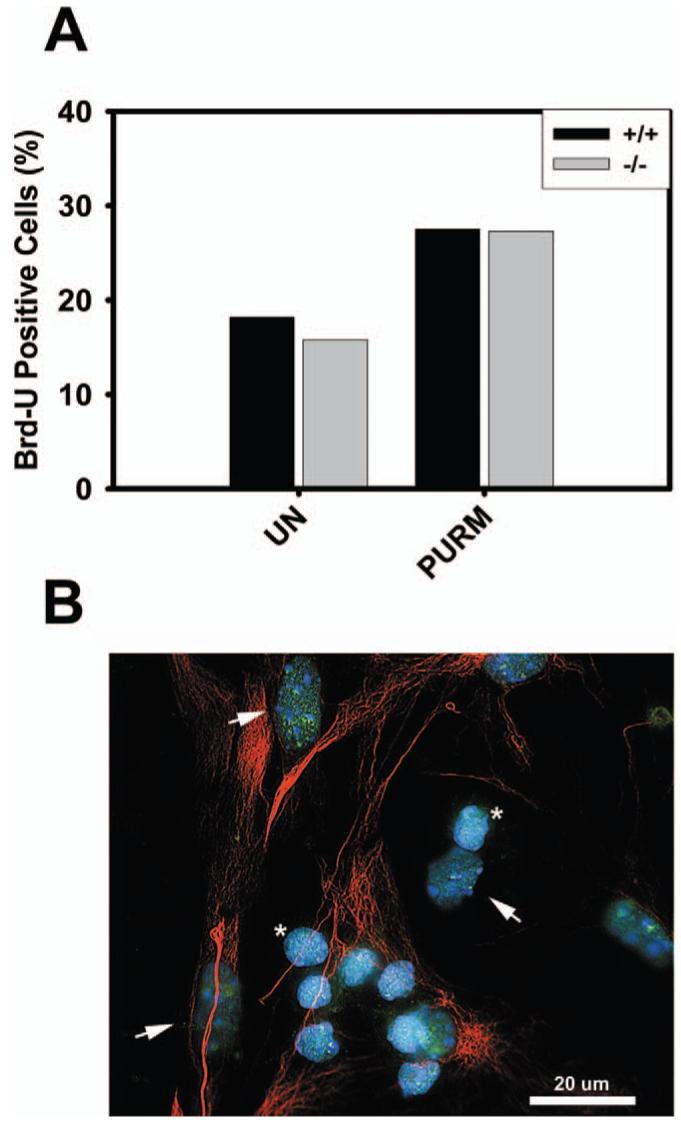
Proliferation of glial cells lacking HDAC9/HDRP. (A) Glial cells were extracted from HDAC9/HDRP null mice and wild-type littermates. The cells were treated with or without (UN) 2 μM purmorphamine (Purm) 48 hours after plating. Glial cells were then incubated for 48 hours, Brd-U was added to the medium for 2 hours, and immunocytochemistry subsequently performed. Brd-U was detected using monoclonal antibodies against Brd-U and glial cells were labeled using an antibody against GFAP. Glial cells were scored as Brd-U positive or negative and the results from two separate experiments were tabulated. (B) Endogenous HDRP (green) was detected in glial cells by immunocytochemistry using an antibody against HDRP. Glial cells (arrowheads) were positively identified using an antibody against GFAP (red). The smaller non-GFAP staining cells are CGNs (*). DNA was counterstained with DAPI (blue) to visualize the nucleus. A 20 μm scale bar is included for reference. A color version of this figure is available in the online journal.
Discussion
A central issue in developmental biology is to understand how diverse cell populations assemble in their proper spatial pattern and proportions to generate functional organs or body parts. A popular model system that has been used to study the signaling mechanisms of cell specification and pattern formation is the vertebrate limb. During limb development a signaling center called the zone of polarizing activity (ZPA) develops along the posterior margin of the limb bud and directs the specification of the identity and the number of digits of the limb. The pattern formation activity of the ZPA is mediated by Shh. Grafts of the ZPA or application of beads coated with Shh to the anterior margin of the developing limb both result in mirror-image digits duplications (30-32). Additionally, analysis of a number of polydactylous mouse mutants reveals an ectopic Shh expression in the anterior part of the limb bud (13-15).
Here we report that absence of HDAC9/HDRP in mice leads to the formation of an extra big toe on the right hind paw in approximately one quarter of mice. The fact that fewer than 100% of HDAC9/HDRP knockout mice exhibited an extra digit suggests that polydactyly in these mice might arise from multiple factors and may have an environmental component. However, evidence presented here suggests that the Shh pathway is a key target for HDAC9/HDRP action as it pertains to polydactyly. Analysis of the Shh axis, the most-closely linked pathway involved in digit patterning, revealed that the downstream Shh effector, Gli1, RNA levels were upregulated in polydactylous neonatal mice lacking the HDAC9 gene. Not surprisingly, given that limb patterning and development was near complete, this disparity was reduced in this line of mice at one month of age. The expression pattern of Shh did not vary between samples from wild-type and HDAC9/HDRP null mice suggesting that Shh production was normal in both mice and that enhanced Gli1 RNA expression was likely due to an altered target cell response.
To further the test hypothesis that HDRP represses Shh signaling in target cells we first examined the ability of five cell lines and a primary kidney cell culture for their capacity to respond at the transcriptional level to purmorphamine. In our efforts to identify and characterize these cell lines we discovered two lines that respond to purmorphamine that were previously unreported to do so. Both HeLa and HT22 cell lines display a transcription-based response profile similar to the known purmorphamine and Shh sensitive NIH 3T3 mouse fibroblast cell line. In addition to expressing all of the major Shh signaling components NIH 3T3 and HT22 cells express HDRP that localizes to the nucleus, which is also the site of Gli1 and Gli3 function. Discovery of HT22 and HeLa cells’ responsiveness to purmorphamine may be useful tools in the future study of Shh activity throughout the body. We examined NIH 3T3 cells as well as HT22 cells following infection with HDRP or GFP encoding adenovirus vectors and found that increasing the level of HDRP in both of these cell types inhibited the purmorphamine-induced increase in Gli1 RNA levels. We also showed that Gli1 induction is inhibited at the protein level and that Gli3 protein cleavage to produce Gli3R does not play a role in HDRP-mediated Gli1 repression suggesting that HDRP works by another mechanism.
The cellular consequence of HDRP-mediated Gli1 inhibition was tested in HT22 cells. We showed that increasing HDRP using adenovirus prevented purmorphamine-induced cell proliferation and this stands to reason as previous work has shown overactive Shh pathway function to result in cell proliferation and cancer (33, 34). We report here that glial cells express HDRP and like HT22 and NIH 3T3 cells, HDRP is localized within the nucleus. Again we found that increasing the levels of HDRP resulted in a profound decrease in proliferation caused by purmorphamine. We then asked whether glia that lacked HDRP would proliferate to a greater extent than wild-type cells. Glial cells were cultured from wild-type and HDAC9/HDRP null mice and treated with purmorphamine; however, no discernable difference in proliferation rates was observed. We speculate that compensation by one or more other HDAC family members might explain this result.
In summary, our report demonstrates a role for HDAC9/HDRP in limb patterning and sheds light into the involvement of HDACs in the regulation of Shh signaling. Shh signaling is involved in a variety of normal processes and perturbation of this signaling pathway has been shown to result in pathologies such as cancer. Further studies are warranted to find out the extent to which HDAC9/HDRP is associated in these physiological and pathophysiological processes.
Acknowledgments
This research was supported by NIH grant NS40408 to S.R.D.
References
- 1.Marzio G, Wagener C, Gutierrez MI, Cartwright P, Helin K, Giacca M. E2F family members are differentially regulated by reversible acetylation. J Biol Chem. 2000;275(15):10887–10892. doi: 10.1074/jbc.275.15.10887. [DOI] [PubMed] [Google Scholar]
- 2.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92(15):1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 3.Morrison BE, Majdzadeh N, D’Mello SR. Histone deacetylases: focus on the nervous system. Cell Mol Life Sci. 2007;64(17):2258–2269. doi: 10.1007/s00018-007-7035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majdzadeh N, Morrison BE, D’Mello SR. Class IIA HDACs in the regulation of neurodegeneration. Front Biosci. 2008;13:1072–1082. doi: 10.2741/2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem. 1993;268(30):22429–22435. [PubMed] [Google Scholar]
- 6.Salminen A, Tapiola T, Korhonen P, Suuronen T. Neuronal apoptosis induced by histone deacetylase inhibitors. Brain Res Mol Brain Res. 1998;61(12):203–206. doi: 10.1016/s0169-328x(98)00210-1. [DOI] [PubMed] [Google Scholar]
- 7.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370(Pt 3):737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. 2002;277(28):25748–25755. doi: 10.1074/jbc.M111871200. [DOI] [PubMed] [Google Scholar]
- 9.Petrie K, Guidez F, Howell L, Healy L, Waxman S, Greaves M, Zelent A. The histone deacetylase 9 gene encodes multiple protein isoforms. J Biol Chem. 2003;278(18):16059–16072. doi: 10.1074/jbc.M212935200. [DOI] [PubMed] [Google Scholar]
- 10.Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D’Mello SR. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006;26(9):3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110(4):479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mejat A, Ramond F, Bassel-Duby R, Khochbin S, Olson EN, Schaeffer L. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat Neurosci. 2005;8(3):313–321. doi: 10.1038/nn1408. [DOI] [PubMed] [Google Scholar]
- 13.Chan DC, Laufer E, Tabin C, Leder P. Polydactylous limbs in Strong’s Luxoid mice result from ectopic polarizing activity. Development. 1995;121(7):1971–1978. doi: 10.1242/dev.121.7.1971. [DOI] [PubMed] [Google Scholar]
- 14.Masuya H, Sagai T, Moriwaki K, Shiroishi T. Multigenic control of the localization of the zone of polarizing activity in limb morphogenesis in the mouse. Dev Biol. 1997;182(1):42–51. doi: 10.1006/dbio.1996.8457. [DOI] [PubMed] [Google Scholar]
- 15.Buscher D, Ruther U. Expression profile of Gli family members and Shh in normal and mutant mouse limb development. Dev Dyn. 1998;211(1):88–96. doi: 10.1002/(SICI)1097-0177(199801)211:1<88::AID-AJA8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 16.D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A. 1993;90(23):10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young AD, Phipps DE, Astroff AB. Large-scale double-staining of rat fetal skeletons using Alizarin Red S and alcian blue. Teratology. 2000;61(4):273–276. doi: 10.1002/(SICI)1096-9926(200004)61:4<273::AID-TERA5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 18.Ming JE, Roessler E, Muenke M. Human developmental disorders and the Sonic hedgehog pathway. Mol Med Today. 1998;4(8):343–349. doi: 10.1016/s1357-4310(98)01299-4. [DOI] [PubMed] [Google Scholar]
- 19.Vargas FR, Roessler E, Gaudenz K, Belloni E, Whitehead AS, Kirke PN, Mills JL, Hooper G, Stevenson RE, Cordeiro I, Correia P, Felix T, Gereige R, Cunningham ML, Canun S, Antonarakis SE, Strachan T, Tsui LC, Scherer SW, Muenke M. Analysis of the human Sonic Hedgehog coding and promoter regions in sacral agenesis, triphalangeal thumb, and mirror polydactyly. Hum Genet. 1998;102(4):387–392. doi: 10.1007/s004390050709. [DOI] [PubMed] [Google Scholar]
- 20.Lee J, Platt KA, Censullo P, Ruiz i Altaba A. Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development. 1997;124(13):2537–2552. doi: 10.1242/dev.124.13.2537. [DOI] [PubMed] [Google Scholar]
- 21.Katoh Y, Katoh M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network (review) Int J Mol Med. 2006;18(6):1019–1023. [PubMed] [Google Scholar]
- 22.Sinha S, Chen JK. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat Chem Biol. 2006;2(1):29–30. doi: 10.1038/nchembio753. [DOI] [PubMed] [Google Scholar]
- 23.Riobo NA, Saucy B, Dilizio C, Manning DR. Activation of heterotrimeric G proteins by Smoothened. Proc Natl Acad Sci U S A. 2006;103(33):12607–12612. doi: 10.1073/pnas.0600880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gustafsson MK, Pan H, Pinney DF, Liu Y, Lewandowski A, Epstein DJ, Emerson CP., Jr. Myf5 is a direct target of long-range Shh signaling and Gli regulation for muscle specification. Genes Dev. 2002;16(1):114–126. doi: 10.1101/gad.940702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandez-Teran M, Piedra ME, Rodriguez-Rey JC, Talamillo A, Ros MA. Expression and regulation of eHAND during limb development. Dev Dyn. 2003;226(4):690–701. doi: 10.1002/dvdy.10271. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Zhang H, Choi SC, Litingtung Y, Chiang C. Sonic hedgehog signaling regulates Gli3 processing, mesenchymal proliferation, and differentiation during mouse lung organogenesis. Dev Biol. 2004;270(1):214–231. doi: 10.1016/j.ydbio.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 27.Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100(4):423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- 28.Meyer NP, Roelink H. The amino-terminal region of Gli3 antagonizes the Shh response and acts in dorsoventral fate specification in the developing spinal cord. Dev Biol. 2003;257(2):343–355. doi: 10.1016/s0012-1606(03)00065-4. [DOI] [PubMed] [Google Scholar]
- 29.Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O’Brien SJ, Wong AJ, Vogelstein B. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236(4797):70–73. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 30.Saunders JW, Gasseling MT. New insights into the problem of pattern regulation in the limb bud of the chick embryo. Prog Clin Biol Res. 1983;110(Pt A):67–76. [PubMed] [Google Scholar]
- 31.Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75(7):1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 32.Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75(7):1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 33.Fujita E, Khoroku Y, Urase K, Tsukahara T, Momoi MY, Kumagai H, Takemura T, Kuroki T, Momoi T. Involvement of Sonic hedgehog in the cell growth of LK-2 cells, human lung squamous carcinoma cells. Biochem Biophys Res Commun. 1997;238(2):658–664. doi: 10.1006/bbrc.1997.7262. [DOI] [PubMed] [Google Scholar]
- 34.Wetmore C. Sonic hedgehog in normal and neoplastic proliferation: insight gained from human tumors and animal models. Curr Opin Genet Dev. 2003;13(1):34–42. doi: 10.1016/s0959-437x(03)00002-9. [DOI] [PubMed] [Google Scholar]