

Abstract
We have identified an intergenic transcriptional activity that is located between the human HOXA1 and HOXA2 genes, shows myeloid-specific expression, and is up-regulated during granulocytic differentiation. The novel gene, termed HOTAIRM1 (HOX antisense intergenic RNA myeloid 1), is transcribed antisense to the HOXA genes and originates from the same CpG island that embeds the start site of HOXA1. The transcript appears to be a noncoding RNA containing no long open-reading frame; sucrose gradient analysis shows no association with polyribosomal fractions. HOTAIRM1 is the most prominent intergenic transcript expressed and up-regulated during induced granulocytic differentiation of NB4 promyelocytic leukemia and normal human hematopoietic cells; its expression is specific to the myeloid lineage. Its induction during retinoic acid (RA)–driven granulocytic differentiation is through RA receptor and may depend on the expression of myeloid cell development factors targeted by RA signaling. Knockdown of HOTAIRM1 quantitatively blunted RA-induced expression of HOXA1 and HOXA4 during the myeloid differentiation of NB4 cells, and selectively attenuated induction of transcripts for the myeloid differentiation genes CD11b and CD18, but did not noticeably impact the more distal HOXA genes. These findings suggest that HOTAIRM1 plays a role in the myelopoiesis through modulation of gene expression in the HOXA cluster.
Introduction
Human HOX gene clusters are known for the prevalence of intergenic transcription between coding genic members.1 Similar activity has also been observed in other developmentally important or tissue-specific gene loci, such as those containing the human beta globin genes, cardiac myosin heavy chain genes, and the interleukin-4 (IL-4)/IL-13 gene cluster.2–4 Extensive HOX gene cluster intergenic transcripts have been described largely as noncoding RNAs (ncRNAs), including both short microRNA (miRNA) species and long ncRNAs that are antisense to their canonical HOX neighbors. Well-defined HOX region ncRNAs include the mir-10 and mir-196 paralogs, bithoraxoid ncRNAs of the Drosophila bithorax complex, and human HOX antisense intergenic RNA (HOTAIR).5–7
Intergenic regions have been proposed as locations for novel radiational and reorganizing changes that have occurred in the evolution of HOX gene clusters, which are relatively constrained in structure in the higher vertebrates.5,8 Several recent studies have focused on expression of intergenic ncRNAs in the human HOX regions, especially the HOXA cluster, in tumor cell lines, tissues, and fibroblasts from different anatomic origins. All reported unusually active transcription within the intergenic regions, occurring in patterns coordinated with their HOX neighbors.7,9,10 Intergenic ncRNAs in the HOXA gene cluster were usually associated with CpG islands and their expression accompanied changes in either polycomb group repressive complex binding or methylation of histones, suggesting a pattern of cis modulation of the intergenic transcripts before the activation of adjacent HOX genes. However, the HOTAIR transcript, located between HOXC11 and HOXC12, was found to function in trans to repress a distal group of homologous HOXD genes by demarcating an extended silenced domain through interaction with the polycomb group complex PRC2 histone methyltransferase7,10,11
De novo genomic transcription mapping has revealed that intergenic ncRNA is possibly the most abundant form of transcriptional output from the genomes of humans and other higher eukaryotic organisms.12,13 Within the human genome, the majority of intergenic ncRNA are not highly conserved at the sequence level, with long ncRNAs generally less conserved than short miRNAs. Nevertheless, their expression patterns may be conserved among tissues or along developmental axes.14,15 More importantly, ncRNA function in gene regulation has emerged as an important mechanism in the control of many biologic processes in development and carcinogenesis.16
In the present study, we have identified intergenic transcription of a unique long ncRNA, here termed HOTAIRM1 (HOTAIR myeloid 1) after the nomenclature of Rinn et al,7 located between the human HOXA1 and HOXA2 genes. Transcript levels are associated with retinoic acid (RA)–mediated myeloid lineage differentiation and maturation. During differentiation induced by all trans retinoic acid (ATRA), shRNA-mediated knockdown of HOTAIRM1 attenuated the transcriptional induction of HOXA genes from the 3′ end of the cluster. The knockdown also interfered with the transcriptional induction of genes encoding β2 integrins CD11b and CD18. These findings indicate that HOTAIRM1 is a myeloid lineage-specific ncRNA that may play a regulatory role in myelopoiesis.
Methods
Human cell lines
NB4, HL-60, K-562, THP1, Jurkat, SuDHL6, HEK293, HeLa, HepG2, and Saos2 cell lines were obtained from ATCC (Manassas, VA). For granulocytic differentiation, NB4 cells were seeded at 2 × 105/mL in medium supplemented with 10% fetal bovine serum and 1 μM ATRA or 9-cis-retinoic acid (9-cis-RA) and cultured for up to 96 hours. For monocytic differentiation, NB4 cells seeded at 2 × 105/mL were primed with 100 μM vitamin D3 for 8 hours, washed, and then cultured in 1 μM tetradecanoyl phorbol acetate (TPA) for 72 hours. Total RNA was isolated with Tri-Reagent (Molecular Research Center, Cincinnati, OH), and purified with a MegaClear spin column (Ambion, Foster City, CA) followed by DNaseI digestion. RNA quality was checked by agarose gel electrophoresis.
Human leukocyte isolation
Human circulating neutrophils, monocytes, and mononuclear cells were isolated from venous blood of healthy volunteers as previously described.17 Human hematopoietic stem and progenitor cell populations were isolated from peripheral blood stem cells (mobilized by granulocyte colony-stimulating factor), and monocytes were prepared from peripheral blood, as previously described.18–20 The University of Massachusetts Medical School and Yale University human subjects committees approved procedures and consent forms at their respective sites. Volunteers' informed consent was obtained in accordance with the Declaration of Helsinki. All reagents and containers were certified as pyrogen-free by the manufacturers.
Polyribosome fractionation
From 5 × 107 to 1 × 108 NB4 cells differentiated in 1 μM ATRA for 24 to 48 hours were used for each gradient. Before harvesting, cells were incubated in media containing 100 μg/mL of cycloheximide for 5 minutes and washed twice with Hanks balanced salt solution containing 100 μg/mL cycloheximide. Cell pellets were resuspended in 1 mL of lysis buffer containing 0.5% Nonidet P40, 1 mM dithiothreitol, and 10 mM ribonucleoside-vanadyl complex (New England Biolabs, Ipswich, MA), and incubated on ice for 10 minutes. After centrifugation at 10 000g for 10 minutes at 4°C, the lysate was layered on top of a chilled 11 mL 15% to 50% (wt/vol) sucrose gradient and centrifuged at 150 000g in a prechilled Beckman SW41Ti rotor for 180 minutes at 4°C with slow deceleration. Gradients were collected in 22 equal fractions, monitored for absorbance at 254/280 nm; total RNA was isolated with TriReagent.
Microarray analysis
Transcription mapping was performed as previously described, using fragmented biotinylated ddATP end-labeled double-stranded cDNA samples analyzed on GeneChip ENCODE01 1.0 Arrays (Affymetrix, Santa Clara, CA), as part of the pilot ENCODE (ENCyclopedia Of DNA Elements) project.12 Hybridization, scanning, and signal scoring procedures have been reported previously.21 For each target position, a mismatch probe was used in background corrections for a perfectly matched probe; intensity estimates and significance of signal captured for each target were calculated within a sliding window centered on each probe pair and size-optimized for average exon size of investigated targets. Signal significance was ranked by P value from upper-sided paired Wilcoxon signed rank sum tests, and corresponding signal intensity was computed as a pseudomedian within its window. To analyze across datasets from different sources, all samples were scaled to an array median intensity of 150 after a quantile normalization conducted within each sample. Results were mapped to genomic positions in human genome assembly (hg) 18 (NCBI Build 36.1) and subjected to visual inspection with a P value threshold of positive signals that visualized signal stretches associated with known target structures.
Expression profiling with fragmented biotinylated NTP-labeled cRNA samples using HG_U133 GeneChip high density oligonucleotide arrays (Affymetrix) was performed with minor modifications of Affymetrix protocols as previously described.22 To analyze across multiple datasets from different cell types, preprocessing by within-sample quantile normalization and between-sample scaling was performed as described in the paragraph immediately above.
Real-time and multiplex reverse transcription–polymerase chain reaction
All primers (Table S1, available on the Blood website; see the Supplemental Materials link at the top of the online article) were designed to span an exon-exon boundary when possible and searched by BLAST (National Center for Biotechnology Information [NCBI]: http://www.ncbi.nlm.nih.gov/BLAST/) against GenBank (NCBI: http://www.ncbi.nlm.nih.gov/Genbank/) or by In-Silico polymerase chain reaction (PCR) in the UCSC Genome Browser (http://genome.ucsc.edu/). Because of high homology among HOX genes, primers for HOXA genes were further checked by multialignment to all clustered HOX gene sequences to verify specificity. First-strand cDNAs were reverse transcribed from total RNA using SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA). Real-time quantitative PCR assays using SYBR Green 1 chemistry were performed on an Applied Biosystems 7300 thermal cycler (Foster City, CA). Quantitation of the results on the basis of the threshold cycle for amplification was performed by the relative standard curve method according to the manufacturer's protocol. Relative expression levels of target genes were calculated from the results of at least triplicate experiments, normalized to either geometric means of a panel of endogenous reference genes including α1b tubulin (TUBA1B), ribosomal protein S6 (RPS6), peptidylprolyl isomerase A (PPIA), and hydroxymethylbilane synthase (HMBS), or to single reference genes with appropriate expression levels. Multiplex PCR using the Multiplex PCR Kit (QIAGEN, Valencia, CA) was used for comparative analyses of expression in RNA from cell lines or from a multiple tissue cDNA panel (Human cDNA Panel II; Clontech, Mountain View, CA), and for analysis of pull-down DNA fractions in chromatin immunoprecipitation. The endpoint for multiplex PCR was optimized for the log phase of amplification, based on the available threshold cycle range of target genes established in real-time PCR.
Rapid amplification of cDNA ends and Northern blot analysis
Rapid amplification of cDNA ends (RACE) primers were targeted to regions of interest identified on tiling and expression arrays (Table S1). 5′ and 3′ RACE were performed using the FirstChoice RLM-Race kit (Ambion), following the manufacturer's protocols. PCR products were sequenced and aligned by BLAT to the latest human genome assembly in the UCSC genome browser. Northern blots were performed by standard techniques on 25-μg samples of glyoxal-denatured total RNA, as previously described.23 Hybridization of radiolabeled probe (Table S1) was detected on a Molecular Dynamics PhosphorImager (GE Healthcare, Piscataway, NJ).
Chromatin immunoprecipitation
Procedures were modified from the methods of Wang et al.24 Briefly, 2 × 107 NB4 cells were grown with or without 1 μM ATRA. Formaldehyde was added directly to culture media to a final concentration of 1% and crosslinking proceeded at room temperature for 30 minutes before addition of glycine to a final concentration of 0.14 M. Cells were washed with ice-cold phosphate-buffered saline, then attached cells were scraped and collected in cold phosphate-buffered saline. Lysed and micrococcal nuclease-digested samples were sonicated to shear the cross-linked chromatin to 0.5- to 1-kb fragments, then immunoprecipitated overnight at 4°C with 2 μg mouse monoclonal anti-RNA polymerase II (RNAP II) antibody A-10 (Santa Cruz Biotechnology, Santa Cruz, CA). Subsequent steps of crosslinking reversal, proteinase K digestion and purification of immunoprecipitated DNA were performed as previously described.24 For multiplex PCR reactions, 2.5% to 10% of the immunoprecipitated DNA and 0.0125% of total DNA (input) from a 10-cm plate served as templates.
shRNA-expressing lentiviral vector construction and transduction
siRNA-targeting sites of HOTAIRM1 transcript were designed on the siRNA selection server at the Whitehead Institute for Biomedical Research25 and further tested by BLAST for off-target homology within the HOXA cluster. Oligonucleotides for shRNA hairpin expression (Figure S1) were inserted into the pLKO.1-TRC backbone plasmid (Addgene plasmid #10878), according to the vendor's protocol, to construct the lentiviral pLKO.1-HOTAIRM1 vectors. The pLKO.1-HOTAIRM1 and pLKO.1 scramble shRNA (Addgene plasmid #1864) lentiviral vectors were produced and pseudotyped in the HEK293T cell line by cotransfecting with packaging plasmids pCMV-VSVG (Addgene plasmid #8454) and pCMV-dR8.2 (Addgene plasmid #8455), and titers of viral supernatants determined in NB4 cells. For long-term silencing of HOTAIRM1, clones of stably transduced NB4 cells were generated; 2 × 105 cells were exposed for 24 hours to pseudo-lentiviral particles with multiplicities of infection ranging from 1 to 4, in the presence of 8 g/mL polybrene (Sigma-Aldrich, St Louis, MO), then washed and plated in soft agar medium containing 0.75 μg/mL puromycin for 6 to 9 days as previously described.26 Colonies were identified by phase-contrast microscope, then transferred to 48-well plates and expanded in fresh medium. Clones with more than 70% knockdown of HOTAIRM1 during ATRA induction were identified by reverse-transcription (RT)–PCR.
Results
Microarray analysis of intergenic transcription between human HOXA1 and HOXA2
To examine intergenic transcription in the HOXA cluster during myelopoiesis, we conducted a transcription mapping study of human peripheral blood neutrophils and in NB4 and HL-60 cells induced by ATRA to granulocytic differentiation, using data from the ENCODE project.12 The analysis included data deposited in Gene Expression Omnibus (GEO) database (GSE2678, GSE2679, GSE2802) from our laboratories and others. The data were generated on GeneChip (Affymetrix) oligonucleotide tiling arrays designed to detect all transcripts from the pilot ENCODE regions, including the HOXA cluster.12 Publicly available tiling array data on HeLa (GSE2800), GM06990 (GSE2800), and placental (GSE2671) transcription mapping with polyA+ RNA samples were included for reference. A paired Wilcoxon signed rank sum test was used for statistical identification of positive signal within a sliding window centered on each probe. Transcriptionally active regions (TARs) within genomic segments of the HOXA cluster, shown in Figure 1by a P value (color scale) threshold, were identified by visual inspection for stretches of positive signals above background. The most significant intergenic transcriptional activity (red bars) mapping to the region between HOXA1 and HOXA2 was identified in mature neutrophils and in ATRA-induced NB4 cells. A more subtle induction of this intergenic transcriptional activity was detectable in ATRA-treated HL-60 cells. No significant transcription activity in this intergenic region was detected in RNA from placental, HeLa, or GM06990 cells.
Figure 1.

Identification of intergenic transcript HOTAIRM1 within the HOXA cluster. (A) Transcription mapping of the human HOXA cluster in myeloid cell lines and mature neutrophils was performed on Affymetrix ENCODE tiling arrays; each panel (labeled with cell type and conditions) presents data from at least 3 biologic repeats. Pseudo-median signals are color scaled by P value, as indicated in the right margin, to highlight the significant TARs. The median point (threshold for positive) was set to P = 10−5, based on visual inspection of well-aligned HOXA1 signals in NB4 cells. (B) Alignment of the sequence of HOTAIRM1 (mapped by 5′- and 3′-RACE), spliced ESTs, and HG_U13 probe sets with the human genome sequence for the intergenic region between HOXA1 and HOXA2 in the March 2006 human reference sequence (NCBI Build 36.1). Also shown are primer sites used for ChIP-PCR amplification of HOTAIRM1 (5′ sequence “5HOTAIRM1” and 3′ sequence “3HOTAIRM1”) in experiments presented in Figure 3. (C) Northern blot analysis shows HOTAIRM1 as a 0.5-kb RNA in NB4 and HL-60 myeloid leukemia cells and in human peripheral blood neutrophils.
In contrast to previously identified signals at HOXA1, the intergenic transcription signals were not aligned with any entries of putative genes or mRNAs in the region. The distribution of thresholded signal and P values indicates the presence of at least 2 TARs located close to the boundaries with their genic neighbors. The regions correspond roughly to 2 common exons shared by several expressed sequence tags (ESTs; Figure 1), but these could not be compiled into a common consensus form, particularly with regard to the 5′ and 3′ ends. Some additional, inconsistent intergenic signal was present, possibly because of nonspecific hybridization in a long CpG island (Figure 1B).
To determine the strand specificity of the intergenic transcripts, we used the GeneChip HG_U133 platform (Affymetrix), which includes probe sets within the region for each strand and can be hybridized with amplified single-stranded antisense RNA preparations. We examined HG_U133 profiles of NB4 cells and neutrophils, as well as 2 GEO profiles of CD34+ hematopoietic stem progenitor cells. Positive signals were only scored for the probe set that targets the plus strand of EST BC031342 (illustrated in light blue in Figure 1B). This sequence partially overlaps both intergenic TARs close to the boundaries with their genic neighbors as well as a TAR slightly further from HOXA2. Two other probe sets targeting intergenic region ESTs (AK022839, AW207863) on the minus strand were scored as absent without exception (Figure S2).
Northern blot analysis of the transcript was performed on RNA from neutrophils, HL-60 cells, NB4 cells, and ATRA-induced NB4 cells. Probes corresponding to the 2 TARs both hybridized to a band approximately 0.5 kb (Figure 1C). 5′ and 3′ RACE using RNA from differentiated NB4 cells revealed a 483 nt (Figure S3) spliced RNA consisting of 2 exons, corresponding to the 2 TARs identified by tiling array (Figure 1B). The splicing pattern of this RNA is shared by a small set of EST and mRNA deposits in GenBank27 that are aligned to the region; the group has been assembled as a putative gene “rumora” (alternative variant eApr07 in the AceView program; NCBI: http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/). We have termed this transcript HOTAIRM1 after the nomenclature for HOX region intergenic transcripts developed by Rinn et al.7 Current RNA folding algorithms, such as the minimum free-energy structure methods used by the mfold program, do not predict any distinctive secondary structure for HOTAIRM1 (Figure S3).
As shown in the conservation track of the UCSC Genome Browser (Figure 1B) and confirmed by an interspecies search in GenBank, the HOTAIRM1 sequence has not been conserved during evolution. However, similarly localized transcriptional activity in the HOXA1-A2 region occurs in some other species (eg, mouse and rat) with conservation at the level of splicing structure (apparent in the mouse), but incorporating complex gaps that break homology at the DNA sequence level (Figures S4,S5).
HOTAIRM1 is a ncRNA transcribed by RNA polymerase II
There is virtually no coding potential in the sequence of HOTAIRM1. The longest predicted open reading frame of HOTAIRM1 would generate a 51–amino acid product with a estimated molecular weight less than 6 kDa and no homology to any sequences in current protein databases. To test experimentally for translation of the transcript, we performed sucrose gradient fractionation of ATRA-induced NB4 cells. As shown in Figure 2, we used multiplex RT-PCR to screen sucrose density gradient fractions for transcripts representing HOXA1 (423 bp amplicon), intergenic HOTAIRM1 (246 bp), and α-tubulin (TUBA1B, 101 bp). Fractions containing free RNA, ribosomal subunits, monoribosomes, and polyribosomes (indicated in Figure 3A) were identified (respectively, left to right in the figure) by optical absorbance at 254 nm and 280 nm and by the relative intensities of 18s and 28s rRNA bands on agarose gel electrophoresis. Intergenic HOTAIRM1 transcripts appeared largely in nonribosomal or monoribosomal fractions, in contrast to HOXA1 and α-tubulin transcripts, which are represented in the higher density polysomal fractions to the right. Both HOXA1 and α-tubulin proteins can be detected in NB4 cells (data not shown). This polysome profile, along with sequence data, indicates that HOTAIRM1 is most probably an ncRNA.
Figure 2.

Polyribosome distribution of HOTAIRM1 and HOXA1 transcripts in ATRA-induced NB4 cells. (A) Polyribosome distribution among fractions from top to bottom (1-22) along the gradient of increasing sucrose density. (B) Multiplex RT-PCR analysis of HOTAIRM1 transcripts in gradient fractions, compared with the distributions of HOXA1 and α-tubulin (TUBA1B) mRNAs, as indicated.
Figure 3.

ChIP assay of RNAP II binding to the HOTAIRM1 gene. The 5′ and 3′ ends of the HOTAIRM1 gene (HOTAIRM1-5 [557 bp] and HOTAIRM1-3 [267 bp], respectively), along with 3′ end of the α-tubulin (TUBA1B) gene (484 bp) and the distal U6 snRNA gene promoter (118 bp), were amplified from anti-RNAP II pull-downs and from input DNA of NB4, ATRA induced NB4, and HeLa cells. (Top panel) Agarose gel electrophoresis of multiplex PCR products of the indicated DNA species and cell types. (Bottom panels) Positions of the U6 snRNA and α-tubulin amplicons are shown schematically as shaded bars. (The HOTAIRM1 amplicons are illustrated in Figure 2.)
RNA ligase–mediated RACE demonstrated 3′-polyadenlyation of the HOTAIRM1 transcript and revealed a canonical polyadenylation signal 18 nt upstream of the 3′ end (Figure S3). To test whether RNA polymerase II (RNAP II) is responsible for its transcription, we performed chromatin immunoprecipitation of HOTAIRM1 DNA from NB4 cells, using a mouse monoclonal antibody to RNAP II. PCR detection of the transcript from both total DNA (input) and DNA pulled down with anti–RNAP II antibody was performed using primers directed to junction of first exon and intron (primer 5′HOTAIRM1) and the 3′ end of HOTAIRM1 gene (primer 3′HOTAIRM1), as illustrated in Figure 1B. Sequences from the distal promoter of the U6A snRNA gene (RNU6A)28 and the 3′ end of the α-tubulin gene (TUBA1B), as illustrated in Figure 3, were concurrently amplified as positive controls in multiplex PCR. The 3′ end of HOTAIRM1 was amplified from DNA immunoprecipitated by anti–RNAP II antibody from ATRA-induced NB4 cells (Figure 3), but much less signal was detected in immunoprecipitated DNA from native NB4 cells and none from HeLa cells, which do not express HOTAIRM1. The 5′ end of HOTAIRM1 was amplified in DNA immunoprecipitated from both untreated and ATRA-induced NB4 cells, but not detected in HeLa cells. This apparent RNAP II binding does not exclusively indicate active transcription, as the signal could also reflect the frequency of RNAP II stalling within proximal promoter regions throughout the genome29 or the expression of HOXA1 from the opposite strand in NB4 cells.
Retinoid-responsive expression
Exogenous RA, via RARs, drives terminal granulocytic differentiation of committed myeloid progenitors by targeting myeloid development regulators and interacting with relevant cytokine pathways.30,31 To further investigate ATRA-induced expression of HOTAIRM1 during myeloid differentiation, we examined the time course of induction by ATRA (1 μM) in NB4 cells and the ATRA-resistant subclone NB4r2 by real-time RT-PCR. NB4 cells showed a latency of at least 6 hours in the rise of HOTAIRM1 expression after ATRA treatment (Figure 4A). HOTAIRM1 induction was abrogated in the ATRA-resistant NB4r2 subline that bears a mutation in the ligand-binding domain of the promyelocytic leukemia–retinoic acid receptor-α (PML-RARα) fusion gene (Figure 4B).32 To examine the RAR dependence of this induction, we examined gene expression in response to 9-cis-RA, a ligand that activates both the retinoid X receptor (which is not stimulated by ATRA) as well as all RAR isotypes.33 9-cis-RA proved to be as potent as ATRA for induction of HOTAIRM1 expression in NB4 cells (Figure 4A). In ATRA-resistant NB4r2 cells, 9-cis-RA was similarly unable to induce expression of HOTAIRM1 (Figure 4B), indicating dependence on RAR for expression of these transcripts.
Figure 4.

Retinoid induction of HOTAIRM1 transcription. (A) Induction of HOTAIRM1 transcripts in NB4 cells incubated for up to 96 hours with ATRA (1 μM) or 9-cis-RA (1 μM), as indicated above each graph. Induction of HOTAIRM1 by ATRA was also measured in the presence of cycloheximide (100 μg/mL) in the culture media, as indicated. (B) Induction of the ATRA-resistant subline NB4r2, under the same conditions. (C) Comparison of CEBPE, HOXA1, and HOTAIRM1 induction in NB4 cells during 12-hour incubation with ATRA (1 μM). Relative expression levels of targets were measured by quantitative real-time RT-PCR, with results shown as ratios to the levels of uninduced reference cells. Bars and error lines represent means and SDs of results, normalized to an endogenous reference gene (TUBA1B or RPS6), of at least triplicate experiments for each group.
To test whether regulation of HOTAIRM1 expression represents a direct effect of retinoic acid receptor ligation, we pretreated NB4 cells with the translation inhibitor cycloheximide for 30 minutes before addition of ATRA. Induction of HOTAIRM1 was largely abolished by cycloheximide treatment under these conditions (Figure 5A), suggesting that its up-regulation depends at least in part on newly synthesized intermediary protein(s), which may explain its relatively late response to ATRA. Indeed, during differentiation of NB4 cells by ATRA, induction of CEBPE occurred before HOTAIRM1 expression, which paralleled that of HOXA1 (Figure 4C). CEBPE encodes an ATRA-responsive transcription factor critical for terminal differentiation of granulocytes, and its expression is not inhibited by cycloheximide.34
Figure 5.

Tissue distribution of HOTAIRM1 expression. (A) Multiplex RT-PCR analysis of HOTAIRM1 and internal reference GAPDH transcripts on a multiple tissue human cDNA panel, with each tissue type indicated above the image of a representative agarose gel. (B) Multiplex RT-PCR analysis of HOTAIRM1 and internal reference TUBA1B (101 bp) transcripts on a multiple cell line panel, with each cell type and its treatment with ATRA (− indicates untreated; + indicates treated with 1 μM ATRA for 1 day) shown above the image of a representative agarose gel. Vertical line indicates the location of redundant lanes cropped from the gel image.
Expression in the myeloid lineage
In the initial tiling array data, HOTAIRM1 expression appeared to be related or restricted to the myeloid lineage. To better define its lineage specificity, we examined cell line and tissue cDNA panels for HOTAIRM1 transcripts. Multiplex RT-PCR assays measured HOTAIRM1 and HOXA1 expression relative to the ubiquitously expressed reference genes TUBA1B or glyceraldehyde 3-phosphate dehydrogenase (GAPDH). HOTAIRM1 expression demonstrated myeloid lineage specificity (Figure 5A,B). Strong positive signals were observed only in leukocytes on the tissue panel and in myeloid NB4 and K562 cells in the cell line panel. A significant increase of expression was induced by ATRA in both NB4 and K562 cells. Low-level HOTAIRM1 expression and very subtle induction by ATRA could be detected in HL-60 cells, which does not respond to ATRA efficiently and lacks a PML-RARα fusion gene. Embryonic kidney HEK293 cells showed strong expression of HOXA1 but only very low signal for HOTAIRM1, indicating discordant expression of the 2 neighboring genes in another context.
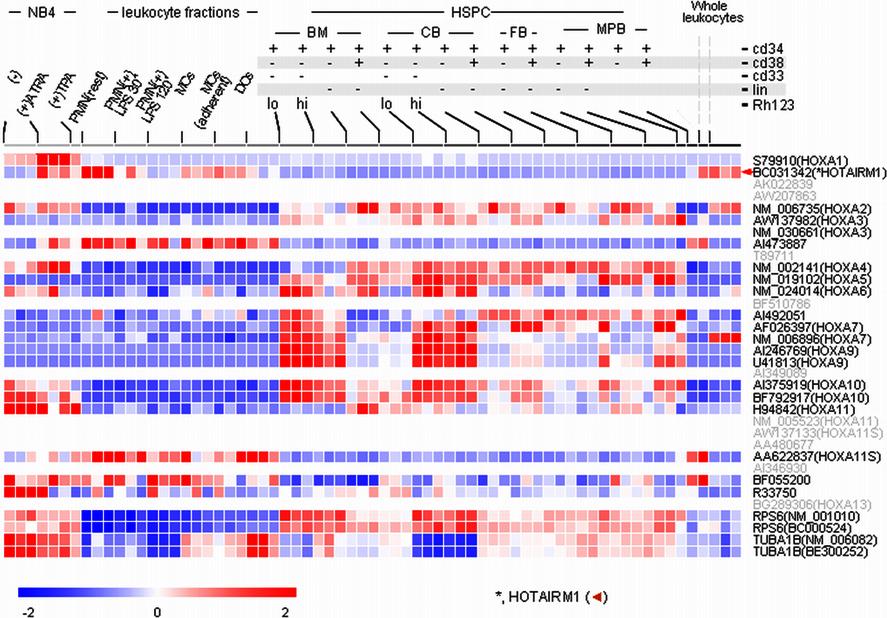
We further assembled a comprehensive gene expression dataset from human genome array studies of hematopoietic cells at different stages of differentiation. Public GEO data series for hematopoietic progenitor cell subpopulations (GSE2666, GSE3823) and human whole blood leukocytes (GSE3284) were compiled with our transcription profiling datasets from peripheral blood progenitor subpopulations, ATRA-induced NB4 cells, and circulating human neutrophils, monocytes, and dendritic cells.35–37 Only GEO datasets available as probe level intensity “raw” data were included. To ensure the cross-laboratory comparability, we applied the same quantile approach for normalization at the probe level and other statistics described in “Methods” for the cross-laboratory analysis of tiling array data. As shown in Figure 6, HOTAIRM1 expression showed a striking association with myeloid differentiation. Relative levels of HOTAIRM1 in cell populations enriched in hematopoietic stem and progenitor cells were strikingly lower than in myeloid leukemia cells and mature leukocytes. Quantitatively, HOTAIRM1 expression among the progenitor populations was marginal or absent, suggesting that the transcript is not involved in earlier stages of stem or progenitor cell development and lineage commitment.
Figure 6.

Expression pattern of HOTAIRM1 during myelopoiesis. HG_U133 transcription profiles of the human HOXA cluster region were assembled for human hematopoietic stem/progenitor cells (HSPC), primary leukocytes, and myeloid cell line NB4. The stem/progenitor cell subpopulations are labeled according to the presence (+) or absence (−) of lineage development markers (CD33, CD34, CD38, or Lin) and low or high rhodamine 123 retention (Rhlow, Rhhigh). Also indicated are the sources of the stem/progenitor cells: bone marrow (BM), cord blood (CB), fetal blood (FB), and G-CSF–mobilized peripheral blood (MPB). NB4 cells were examined either without treatment or after incubation with ATRA and TPA, as indicated. Primary leukocyte populations included total monocytes (MCs), adherent monocytes, monocyte-derived dendritic cells (DCs), resting neutrophils (PMNs), and neutrophils incubated with lipopolysaccharide (10 ng/mL) for 30 or 120 minutes, as indicated. The bar plot presents means and SDs for HOTAIRM1 and HOXA1 transcript levels in each sample group.
A significant increase in HOTAIRM1 expression also occurred during differentiation of the NB4 cell line, as observed in our tiling array and reverse transcription–PCR experiments. Uninduced promyelocytic NB4 cells expressed a low but detectable baseline level of HOTAIRM1 transcripts and showed dramatically increasing expression during neutrophilic differentiation induced by ATRA. A much lesser degree of induction was observed during monocytic differentiation induced by TPA, confirming the results obtained with tiling arrays (Figure 1). Among primary leukocytes, HOTAIRM1 expression was 4- to 5-fold higher in neutrophils than in monocytes or dendritic cells. Escherichia coli lipopolysaccharide, a strong stimulus of the neutrophil inflammatory response,22,38 dramatically down-regulated HOTAIRM1 expression (Figures 1, S2). Although similar expression patterns for HOTAIRM1 and HOXA1 were observed in NB4 cells, their expression was not coordinated in either progenitor cells or mature neutrophils.
shRNA-mediated silencing of HOTAIRM1 RNA attenuates expressions of 3′ HOXA genes
The intergenic ncRNAs within HoxA cluster have been proposed to function as epigenetic regulators of neighboring HOX genes or remote paralogs. To assess the potential impact of HOTAIRM1on the transcriptional regulation of local HOXA cluster members and test for functional effects on myeloid differentiation, we transduced promyelocytic NB4 cells with lentiviral vectors that express shRNAs targeting the HOTAIRM1 transcript. Figure 7A shows more than 70% reduction of HOTAIRM1 RNA, in 2 representative ATRA-treated stably shRNA-transduced clones, compared with nontransduced cells or cells transduced by the viral vector expressing a “scramble” shRNA.
Figure 7.

shRNA-mediated silencing of HOTAIRM1 RNA. (A) Stable knockdown of HOTAIRM1 transcripts in 2 NB4 cell clones transduced with pLKO.1 lentiviral vector expressing shRNA targeting HOTAIRM1 transcripts and incubated with ATRA (1 μM). The effectiveness of silencing is compared with pLKO.1 expressing negative control “scramble” shRNA. Quantitative RT-PCR results are shown as the percentage of HOTAIRM1 expression in each transduced cell type relative to the level in nontransduced NB4 cells. (B) Expression of myelopoiesis-relevant HOXA genes by HOTAIRM1 knockdown cells during 3 days of ATRA (1 μM)-induced granulocytic differentiation of NB4 cells. Relative expression levels of the indicated targets were measured by quantitative real-time RT-PCR in 2 stable knockdown NB4 clones. The bars and error lines represented means and SDs of at least triplicate experiments for each group, normalized to the geometric mean of a panel of endogenous reference genes.
To test the effects of HOTAIRM1 knockdown on Hox gene expression, we measured transcript levels for other HOXA cluster genes by quantitative real-time RT-PCR during ATRA-induced myeloid differentiation in the 2 clones of transduced NB4 cells (Figure 7A). HOXA1, A4, A5, A9, A10, and A11 were selected as the most relevant to myelopoiesis in NB4 cells, based on our genomics data (Figure S2) and the published literature.39–42 As shown in Figure 7B, HOXA1 and A4 demonstrated striking, significant induction of expression in native and scramble shRNA-transduced NB4 cells, but not in HOTAIRM1 knockdown cells. HOXA5 showed a subtle increase in expression during 3 days of ATRA induction in native and negative control cells, but transcript levels fell approximately 3-fold in HOTAIRM1 knockdown cells (Figure 7B). In contrast to the suppression of these genes from the 3′ end of the HOXA cluster, knockdown of HOTAIRM1 had no significant effect on the induced responses of HOXA9, A10, or A11 on the 5′ end of the HOXA cluster (Figure 7B). Accordingly, no significant impact of its knockdown was observed on the induction of the phagocyte oxidase gene CYBB (Figure S6), a known target of HOXA9 and HOXA10 modulation.43,44 There was no apparent effect of HOTAIRM1 knockdown on morphologic maturation detectable by Wright-Giemsa staining of ATRA-induced NB4 cells (not shown).
In addition, knockdown of HOTAIRM1 during NB4 granulocytic differentiation significantly attenuates the induction of beta2 integrin CD11b and CD18 transcripts (Figure S6), 2 hallmark granulocyte maturation genes.45–47 Adhesion molecule genes, such as beta3 integrin,48 are well-documented HOX targets, but CD11b and CD18 genes have not yet been linked to HOX modulation. However, these results suggest that HOTAIRM1 also has a potential role in myeloid transcriptional regulation involving, directly or indirectly, targets outside the HOXA cluster.
Discussion
In this study, we present evidence that HOTAIRM1, a small intergenic transcript from the plus (opposite) strand between the HOXA1 and HOXA2 genes, is a ncRNA expressed specifically in the myeloid lineage. We have demonstrated its expression pattern in mature myeloid cells and during granulocytic differentiation using a comprehensive collection of transcription mapping data from tiling array analyses of hematopoietic cells and circulating leukocytes. In myeloid cell lines, expression of HOTAIRM1 was induced by ATRA and this response was abrogated in an ATRA-resistant NB4 subclone. We further showed that high-level expression of HOTAIRM1 in circulating neutrophils could be decreased by lipopolysaccharide stimulation. These data indicate a lineage-specific pattern of regulated expression for HOTAIRM1 that parallels functional genes associated with late granulocytic maturation and with inflammatory or apoptotic functions. The lack of an open reading frame or association with polyribosomes indicates that the transcript probably represents a ncRNA. shRNA-mediated knockdown experiments indicate a functional role for HOTAIRM1 in regulating the expression of neighboring genes at the 3′ end of the HOXA cluster and impact the expression of genes encoding Beta2 integrins CD11b and CD18.
HOX genes of the A and B paralog groups have emerged as a class of key transcriptional regulators in definitive hematopoiesis.48 Functions of homeobox pathways have been well documented in normal hematopoiesis and acute myeloid leukemia.40,50,51 However, the general collinearity rule of Hox gene expression seen in body axis patterning during embryogenesis does not hold for hematopoiesis. Although there is a 3′ to 5′ orderly activation sequence, with 3′ genes expressed during hematopoietic stem cell renewal and expansion and 5′ genes (HoxA7-10) expressed mainly in committed progenitors and mature cells, the “HOX code” associated with adult hematopoietic lineage development remains unclear. For example, both the 3′-located HOXB4 and 5′ gene HOXA9 are required for normal hematopoietic stem cell function both in vitro and in vivo.52,53 Moreover, the HOXA9 and HOXA10 genes remain detectable in mature myeloid cells and regulate the transcription of genes encoding the phagocyte proteins gp91-phox, p67-phox, and β3 integrin.43,48,49
The ncRNAs have emerged as an important new feature of the human transcriptome, representing a surprisingly high proportion of all transcripts identified from the 1% of the human genome analyzed in the ENCODE project.12 The particularly extensive intergenic transcriptional activity within the HOX gene clusters has been proposed to generate ncRNAs with transcriptional regulatory functions.7,9,10 In the HOXA region, Rinn et al7 have identified a class of transcripts, termed HOTAIR, which act in trans on remote targets as recruiters of histone methyltransferases.7 Other HOXA intergenic transcripts have been speculated to serve in cis control of coordinately expressed neighboring HOXA genes.9,10 Although miRNAs appear to exert their regulatory influence through processes involving sequence homology, both the molecular mechanisms and functions of other ncRNAs in gene regulation remain largely unknown.
In addition to the burgeoning roles of miRNAs in hematopoiesis and leukemogenesis,54 recent evidence has associated other forms of ncRNA with leukocyte development. A long ncRNA, termed EGO, which is transcribed from an intron of the inositol triphosphate receptor type 1 gene (ITPR1), transcriptionally regulates secondary granule protein expression during eosinophil development.55 In T-cell leukemia cell lines, expression of a transcript paired divergently with a homeobox gene TLX1/HOX11 (T-cell leukemia homeobox 1) and hence termed TDI (TLX1 divergent), correlates with the expression of TLX1, a proto-oncogene inappropriately activated in human T-cell acute lymphoblastic leukemia and reported to inhibit hematopoietic differentiation in several murine models.56,57 No direct targets have yet been established for those regulatory hematopoietic ncRNAs.
DNA sequence analysis of HOTAIRM1 shows poor evolutional conservation at the sequence level even among mammals, with no homologs found in current genome assemblies. However, the similarity of localized ESTs in other mammals, including murine ESTs with similar splicing patterns suggest the more general conservation of intergenic transcription activity. In addition, a long CpG island is associated with the transcription start site, as seen in almost all mammalian intergenic RNAs. This structure, suggesting the presence of a bi-directional promoter shared by the divergent coding and noncoding RNAs, has been proposed to facilitate the cis action of intergenic ncRNAs on their genic partners.9,10 However, the expression of HOTAIRM1 and HOXA1 is not always synchronized, suggesting that HOTAIRM1 may also function independently from the immediately adjacent gene.
Expression of early HOX paralogs in anterior-posterior body patterning during embryogenesis is mediated by flanking retinoic acid response elements, including a site downstream of HOXA1 that has been verified experimentally.58 Additional putative retinoid response elements have been predicted within the 3′ side of the HOXA cluster, including one within the CpG island embedded the shared promoter region of HOXA1 and HOTAIRM1.10 Our current finding that cycloheximide did not entirely abrogate the induction of HOTAIRM1 suggests that retinoid receptors or other transcription factors may also mediate induction of HOTAIRM1.
In conclusion, the current studies indicate a close association of the HOXA intergenic transcript HOTAIRM1 with differentiation and function in the myeloid lineage and lay the foundation for further analysis of its potential regulatory function in hematopoiesis and leukemogenesis.
Acknowledgments
This work was supported by the National Institutes of Health (grant DK54369; P.E.N., S.M.W.), National Cancer Institute (contract N01-CO-12400; T.R.G.), National Human Genome Research Institute (grant U01-HG003147; T.R.G.), and Affymetrix.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: X.Z. designed research, collected, analyzed, and interpreted data, performed statistical analysis, and wrote the manuscript; Z.L. and C.P. collected data; M.B.G., J.R., M.S., T.R.G., and P.K. contributed vital analytical tools, analyzed data, and performed statistical analysis; and S.M.W. and P.E.N. designed research, analyzed and interpreted data, and reviewed the manuscript.
Conflict-of-interest disclosure: T.R.G. and P.K. were employees of Affymetrix and own stock in the company. The remaining authors declare no competing financial interests.
Correspondence: Peter E. Newburger, Department of Pediatrics, University of Massachusetts Medical School, Worcester, MA 01655; e-mail: peter.newburger@umassmed.edu.
References
- 1.Mainguy G, Koster J, Woltering J, Jansen H, Durston A. Extensive polycistronism and antisense transcription in the Mammalian hox clusters. PLoS ONE. 2007;2:e356. doi: 10.1371/journal.pone.0000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miles J, Mitchell JA, Chakalova L, et al. Intergenic transcription, cell-cycle and the developmentally regulated epigenetic profile of the human beta-globin locus. PLoS ONE. 2007;2:e630. doi: 10.1371/journal.pone.0000630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haddad F, Qin AX, Bodell PW, Jiang W, Giger JM, Baldwin KM. Intergenic transcription and developmental regulation of cardiac myosin heavy chain genes. Am J Physiol Heart Circ Physiol. 2008;294:H29–H40. doi: 10.1152/ajpheart.01125.2007. [DOI] [PubMed] [Google Scholar]
- 4.Rogan DF, Cousins DJ, Santangelo S, et al. Analysis of intergenic transcription in the human IL-4/IL-13 gene cluster. Proc Natl Acad Sci U S A. 2004;101:2446–2451. doi: 10.1073/pnas.0308327100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemons D, McGinnis W. Genomic evolution of Hox gene clusters. Science. 2006;313:1918–1922. doi: 10.1126/science.1132040. [DOI] [PubMed] [Google Scholar]
- 6.Tanzer A, Amemiya CT, Kim CB, Stadler PF. Evolution of microRNAs located within Hox gene clusters. J Exp Zool B Mol Dev Evol. 2005;304:75–85. doi: 10.1002/jez.b.21021. [DOI] [PubMed] [Google Scholar]
- 7.Rinn JL, Kertesz M, Wang JK, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner GP, Amemiya C, Ruddle F. Hox cluster duplications and the opportunity for evolutionary novelties. Proc Natl Acad Sci U S A. 2003;100:14603–14606. doi: 10.1073/pnas.2536656100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sasaki YT, Sano M, Kin T, Asai K, Hirose T. Coordinated expression of ncRNAs and HOX mRNAs in the human HOXA locus. Biochem Biophys Res Commun. 2007;357:724–730. doi: 10.1016/j.bbrc.2007.03.200. [DOI] [PubMed] [Google Scholar]
- 10.Sessa L, Breiling A, Lavorgna G, Silvestri L, Casari G, Orlando V. Noncoding RNA synthesis and loss of Polycomb group repression accompanies the colinear activation of the human HOXA cluster. RNA. 2007;13:223–239. doi: 10.1261/rna.266707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rauch T, Wang Z, Zhang X, et al. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci U S A. 2007;104:5527–5532. doi: 10.1073/pnas.0701059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertone P, Stolc V, Royce TE, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 14.Babak T, Blencowe B, Hughes T. A systematic search for new mammalian noncoding RNAs indicates little conserved intergenic transcription. BMC Genom. 2005;6:104. doi: 10.1186/1471-2164-6-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khaitovich P, Kelso J, Franz H, et al. Functionality of intergenic transcription: an evolutionary comparison. PLoS Genet. 2006;2:e171. doi: 10.1371/journal.pgen.0020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodrich JA, Kugel JF. Non-coding-RNA regulators of RNA polymerase II transcription. Nat Rev Mol Cell Biol. 2006;7:612–616. doi: 10.1038/nrm1946. [DOI] [PubMed] [Google Scholar]
- 17.Subrahmanyam YV, Baskaran N, Newburger PE, Weissman SM. A modified method for the display of 3′-end restriction fragments of cDNAs: molecular profiling of gene expression in neutrophils. Methods Enzymol. 1999;303:272–297. doi: 10.1016/s0076-6879(99)03018-9. [DOI] [PubMed] [Google Scholar]
- 18.Kluger Y, Tuck DP, Chang JT, et al. Lineage specificity of gene expression patterns. Proc Natl Acad Sci U S A. 2004;101:6508–6513. doi: 10.1073/pnas.0401136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cathcart MK, Morel DW, Chisolm GM., 3rd Monocytes and neutrophils oxidize low density lipoprotein making it cytotoxic. J Leukoc Biol. 1985;38:341–350. doi: 10.1002/jlb.38.2.341. [DOI] [PubMed] [Google Scholar]
- 20.Debelak J, Shlomchik MJ, Snyder EL, et al. Isolation and flow cytometric analysis of T-cell-depleted CD34+ PBPCs. Transfusion. 2000;40:1475–1481. doi: 10.1046/j.1537-2995.2000.40121475.x. [DOI] [PubMed] [Google Scholar]
- 21.Emanuelsson O, Nagalakshmi U, Zheng D, et al. Assessing the performance of different high-density tiling microarray strategies for mapping transcribed regions of the human genome. Genome Res. 2007;17:886–897. doi: 10.1101/gr.5014606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang XQ, Kluger Y, Nakayama Y, et al. Gene expression in mature neutrophils: early responses to inflammatory stimuli. J Leukoc Biol. 2004;75:358–372. doi: 10.1189/jlb.0903412. [DOI] [PubMed] [Google Scholar]
- 23.Thomas PS. Hybridization of denatured RNA and small DNA fragments transferred to nitrocellulose. Proc Natl Acad Sci U S A. 1980;77:5201–5205. doi: 10.1073/pnas.77.9.5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang G, Balamotis MA, Stevens JL, Yamaguchi Y, Handa H, Berk AJ. Mediator requirement for both recruitment and postrecruitment steps in transcription initiation. Mol Cell. 2005;17:683–694. doi: 10.1016/j.molcel.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 25.Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. siRNA selection server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004;32:W130–W134. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hattori H, Zhang X, Jia Y, et al. RNAi screen identifies UBE2D3 as a mediator of all-trans retinoic acid-induced cell growth arrest in human acute promyelocytic NB4 cells. Blood. 2007;110:640–650. doi: 10.1182/blood-2006-11-059048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.National Center for Biotechnology Information. Genbank database. [Accessed December 23, 2008]. http://www.ncbi.nlm.nih.gov/Genbank/
- 28.Listerman I, Bledau AS, Grishina I, Neugebauer KM. Extragenic accumulation of RNA polymerase II enhances transcription by RNA polymerase III. PLoS Genet. 2007;3:e212. doi: 10.1371/journal.pgen.0030212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawson ND, Berliner N. Neutrophil maturation and the role of retinoic acid. Exp Hematol. 1999;27:1355–1367. doi: 10.1016/s0301-472x(99)00085-5. [DOI] [PubMed] [Google Scholar]
- 31.Evans T. Regulation of hematopoiesis by retinoid signaling. Exp Hematol. 2005;33:1055–1061. doi: 10.1016/j.exphem.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Duprez E, Benoit G, Flexor M, Lillehaug JR, Lanotte M. A mutated PML/RARA found in the retinoid maturation resistant NB4 subclone, NB4-R2, blocks RARA and wild-type PML/RARA transcriptional activities. Leukemia. 2000;14:255–261. doi: 10.1038/sj.leu.2401683. [DOI] [PubMed] [Google Scholar]
- 33.Kizaki M, Ikeda Y, Tanosaki R, et al. Effects of novel retinoic acid compound, 9-cis-retinoic acid, on proliferation, differentiation, and expression of retinoic acid receptor-alpha and retinoid X receptor-alpha RNA by HL-60 cells. Blood. 1993;82:3592–3599. [PubMed] [Google Scholar]
- 34.Park DJ, Chumakov AM, Vuong PT, et al. CCAAT/enhancer binding protein ϵ is a potential retinoid target gene in acute promyelocytic leukemia treatment. J Clin Invest. 1999;103:1399–1408. doi: 10.1172/JCI2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eckfeldt CE, Mendenhall EM, Flynn CM, et al. Functional analysis of human hematopoietic stem cell gene expression using zebrafish. PLoS Biol. 2005;3:e254. doi: 10.1371/journal.pbio.0030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez-Iratxeta C, Palidwor G, Porter CJ, et al. Study of stem cell function using microarray experiments. FEBS Lett. 2005;579:1795–1801. doi: 10.1016/j.febslet.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 37.Calvano SE, Xiao W, Richards DR, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 38.Tsukahara Y, Lian Z, Zhang XQ, et al. Gene expression in human neutrophils during activation and priming by bacterial lipopolysaccharide. J Cell Biochem. 2003;89:848–861. doi: 10.1002/jcb.10526. [DOI] [PubMed] [Google Scholar]
- 39.Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26:6766–6776. doi: 10.1038/sj.onc.1210760. [DOI] [PubMed] [Google Scholar]
- 40.Rice KL, Licht JD. HOX deregulation in acute myeloid leukemia. J Clin Invest. 2007;117:865–868. doi: 10.1172/JCI31861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson A, Quinn MF, Grimwade D, et al. Global down-regulation of HOX gene expression in PML-RARalpha + acute promyelocytic leukemia identified by small-array real-time PCR. Blood. 2003;101:1558–1565. doi: 10.1182/blood.V101.4.1558. [DOI] [PubMed] [Google Scholar]
- 42.Strathdee G, Holyoake TL, Sim A, et al. Inactivation of HOXA genes by hypermethylation in myeloid and lymphoid malignancy is frequent and associated with poor prognosis. Clin Cancer Res. 2007;13:5048–5055. doi: 10.1158/1078-0432.CCR-07-0919. [DOI] [PubMed] [Google Scholar]
- 43.Bei L, Lu Y, Eklund EA. HOXA9 activates transcription of the gene encoding gp91Phox during myeloid differentiation. J Biol Chem. 2005;280:12359–12370. doi: 10.1074/jbc.M408138200. [DOI] [PubMed] [Google Scholar]
- 44.Lu Y, Goldenberg I, Bei L, Andrejic J, Eklund EA. HoxA10 represses gene transcription in undifferentiated myeloid cells by interaction with histone deacetylase 2. J Biol Chem. 2003;278:47792–47802. doi: 10.1074/jbc.M305885200. [DOI] [PubMed] [Google Scholar]
- 45.Wu JJ, Cantor A, Moscinski LC. [beta]2 integrins are characteristically absent in acute promyelocytic leukemia and rapidly upregulated in vivo upon differentiation with all-trans retinoic acid. Leuk Res. 2007;31:49–57. doi: 10.1016/j.leukres.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 46.Mayadas TN, Cullere X. Neutrophil [beta]2 integrins: moderators of life or death decisions. Trends Immunol. 2005;26:388–395. doi: 10.1016/j.it.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 47.Zang C, Liu H, Ries C, Ismair MG, Petrides PE. Enhanced migration of the acute promyelocytic leukemia cell line NB4 under in vitro conditions during short-term all-trans-retinoic acid treatment. J Cancer Res Clin Oncol. 2000;126:33–40. doi: 10.1007/PL00008462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bei L, Lu Y, Bellis SL, Zhou W, Horvath E, Eklund EA. Identification of a HoxA10 activation domain necessary for transcription of the gene encoding beta3 integrin during myeloid differentiation. J Biol Chem. 2007;282:16846–16859. doi: 10.1074/jbc.M609744200. [DOI] [PubMed] [Google Scholar]
- 49.Eklund EA. The role of HOX genes in myeloid leukemogenesis. Curr Opin Hematol. 2006;13:67–73. doi: 10.1097/01.moh.0000208467.63861.d6. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Yates F, Naveiras O, Ernst P, Daley GQ. Embryonic stem cell-derived hematopoietic stem cells. Proc Natl Acad Sci U S A. 2005;102:19081–19086. doi: 10.1073/pnas.0506127102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Owens BM, Hawley RG. HOX and non-HOX homeobox genes in leukemic hematopoiesis. Stem Cells. 2002;20:364–379. doi: 10.1634/stemcells.20-5-364. [DOI] [PubMed] [Google Scholar]
- 52.Lawrence HJ, Christensen J, Fong S, et al. Loss of expression of the Hoxa-9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood. 2005;106:3988–3994. doi: 10.1182/blood-2005-05-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horton SJ, Grier DG, McGonigle GJ, et al. Continuous MLL-ENL expression is necessary to establish a “Hox Code” and maintain immortalization of hematopoietic progenitor cells. Cancer Res. 2005;65:9245–9252. doi: 10.1158/0008-5472.CAN-05-1691. [DOI] [PubMed] [Google Scholar]
- 54.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 55.Wagner LA, Christensen CJ, Dunn DM, et al. EGO, a novel, noncoding RNA gene, regulates eosinophil granule protein transcript expression. Blood. 2007;109:5191–5198. doi: 10.1182/blood-2006-06-027987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greene WK, Sontani Y, Sharp MA, Dunn DS, Kees UR, Bellgard MI. A promoter with bidirectional activity is located between TLX1/HOX11 and a divergently transcribed novel human gene. Gene. 2007;391:223–232. doi: 10.1016/j.gene.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 57.Riz I, Akimov SS, Eaker SS, et al. TLX1//HOX11-induced hematopoietic differentiation blockade. Oncogene. 2007;26:4115–4123. doi: 10.1038/sj.onc.1210185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langston AW, Gudas LJ. Identification of a retinoic acid responsive enhancer 3′ of the murine homeobox gene Hox-1. 6. Mech Dev. 1992;38:217–227. doi: 10.1016/0925-4773(92)90055-o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}