

Abstract
Hippocampal area CA3 has been one of the most intensively studied brain regions for computer models of epileptiform activity. As physiological studies begin to extend outward to other hippocampal and parahippocampal areas, we must extend these models to understand more complex circuitry containing diverse elements. Study of subiculum is of particular interest in this context, as it is a structure of intermediate complexity, with an inchoate columnar and laminar organization. In addition to helping us understand seizures, modeling of these structures will also help us understand the genesis of physiological activity patterns that are below threshold for seizure generation. Such modeling can also serve as a basis for speculation regarding the non-ictal behavioral consequences of epilepsy.
Hippocampal and parahippocampal structures
The prevalence of temporal lobe epilepsy has focused attention on the cells and circuits of the hippocampal formation, the central structure of medial temporal lobe. It seems likely that major seizure generators lie among the hippocampal and parahippocampal structures. Indeed, several regions of the hippocampal formation, including entorhinal cortex, subiculum and CA3 have been shown to be potentially epileptogenic in brain slice experiments. In these in vitro experiments, hippocampal formation areas appear to interact in many of the same ways that they interact in vivo: one area may lead the seizure, or more generally the epileptiform event, in one instance and be a follower in another.
The interactions of cells within each region, the interactions of regions with one another, the alteration in roles with progression of a single seizure, and the alteration in patterns of activity with progression of epilepsy over years are complex and are only beginning to be understood. In this review, we will discuss the cells and circuits of the hippocampal formation with suggestions on how they interact to produce a seizure or to produce the behavioral anomalies seen in epilepsy. In order to reduce this enormous complexity and begin to define some basic principles of seizure generation, we rely on computer simulation to complement physiological experimentation.
The hippocampal formation, comprising hippocampus, entorhinal cortex and subicular areas, is a collection of brain regions with a variety of cell types and circuits. The dentate gyrus and Ammon’s horn (CA1–CA3), collectively referred to here as hippocampus, are characterized by a single layer of principal neurons. Each region’s cell body layer is sandwiched between apical and basal dendritic neuropil resulting in a 3-layer appearance. The neighboring “parahippocampal” structures comprise entorhinal cortex, parasubiculum, presubiculum and the subiculum. The entorhinal cortex, parasubiculum, and presubiculum are more similar to neocortex, both evolutionarily and structurally, having multiple distinct cell layers with stellate and pyramidal cells. The subiculum is morphologically intermediate between single-layered hippocampus and multilayered presubiculum with a single broad layer of pyramidal cells.
In comparison to most brain regions, the connectivity of the hippocampal formation is unusual in not featuring a reciprocal connection for every projection. Its defining characteristic, or at least the best studied one, is the trisynaptic pathway, essentially unidirectional circuit that begins in the entorhinal cortex. Stellate cells in layer II of entorhinal cortex excite dentate granule cells for what is considered the first synapse in the classic trisynaptic pathway. These entorhinal axons form the perforant pathway which crosses through the subiculum and perforates pia mater at the hippocampal fissure to enter the dentate gyrus. Other fibers from this pathway continue on to project directly to distal apical dendrites of cells in CA3, while afferents from layer III pyramidal cells of entorhinal cortex innervate the distal dendritic regions of areas CA1 and subiculum. These fibers cross the alvear white matter (angular bundle) with axons of layer II stellate cells, but the layer III projection does not perforate the pia mater, turning to run in the apical dendritic areas of subiculum and CA1.
The second synapse of the trisynaptic pathway occurs as dentate granule cells synapse on complex spines of CA3 pyramidal cells’ proximal apical dendrites. The third synapse of the pathway are the Schaffer collaterals, axons projecting from CA3 pyramidal cells to pyramidal cells of CA1. This trisynaptic circuit is embedded in a longer circuit involving subiculum. CA1 projects to subiculum and subiculum projects into entorhinal cortex with or without relays in the pre- or parasubiculum.
Although the pathway is largely unidirectional, closer analysis reveals numerous smaller subcircuits with varying degrees of reciprocity. As we will see, such reciprocity is a prerequisite for epileptiform activity. Reciprocity in hippocampus is seen in two basic types of subcircuit: intraregional and interregional. Several hippocampal formation regions possess substantial intrinsic connectivity: the pyramidal cells of CA3 are relatively heavily interconnected and the the parahippocampal regions all have some degree of intrinsic connectivity. Examples of interregional subcircuits include the mutual projections between subiculum and entorhinal cortex, a subcircuit that has been shown to generate epileptiform activity.
Strongly epileptogenic areas: CA3 and entorhinal cortex
If one applies a convulsant drug such as bicuculline (a GABAA receptor blocker) to a brain slice containing all regions of the hippocampal formation, spontaneous seizure activity will begin in area CA3 and separately in entorhinal cortex (Fig. 1). Studies of the various hippocampal regions have led to the widely held view that the combination of a critical density of recurrent excitatory connectivity and spontaneous activity in the some of the cells (pacemaker cells) is required for a brain region to generate seizure activity (Heinemann, 1987).
Fig. 1.
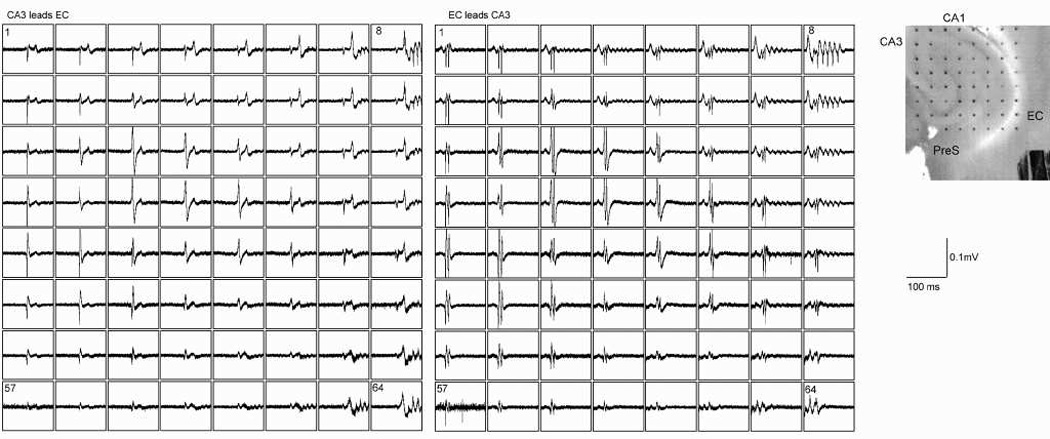
Independent seizure generation by area CA3 and entorhinal cortex after bicuculline exposure in horizontal slices from rat brain. Left panel shows a single 100 ms duration sweep on 64 electrodes that simultaneously sample electrical activity in many hippocampal formation locations. Area CA3 is sampled by electrodes in approximately the left upper quadrant. A single interictal-type epileptiform event characterizes the synchronous firing of cells in CA3. This is propagated toward entorhinal cortex (rightmost electrodes sample deep EC or underlying white matter). The EC event (seen best on electrode 8) consists of a multi-spiked event. The second panel shows another event that originates in EC (note the different appearance of both the EC and CA3 events). Photomicrograph to the far right shows the positions of the electrode contacts (300 micrometer spacing) with several structures labeled.
A combination of biological experimentation and computer simulations has been used to define the cellular and synaptic basis for epileptiform activity in CA3. [33, 34, 35, 36] These are unstructured, hence topologically zero-dimensional networks. Two major conditions have been identified as important in the formation of epileptiform activity in CA3. First, there are relatively dense recurrent excitatory connections. Second, depolarization of apical dendrites can drive repetitive dendritic calcium spiking at rates up to 10 Hz. An epileptiform burst event is terminated as the afterhyperpolarizing conductance (a slowly activating and inactivating calcium-dependent potassium conductance) grows to offset the intrinsic calcium current and the sustained NMDA receptor-mediated synaptic excitation. GABA mediated conductances (in particular GABAB, especially if the convulsant is a GABAA antagonist) can also shape the duration of the event.
In this scenario, the primary epileptiform burst and any subsequent afterdischarges are the result of sustained depolarization of the dendrites. This depolarization causes calcium spiking and subsequent afterhyperpolarization due to potassium conductances. The result is repetitive dendritic calcium spiking. Each dendritic burst triggers a somatic burst which, through axon collaterals and AMPA receptors, keeps the whole network synchronous. The string of afterdischarges stops as the afterhyperpolarization grows. Blockade of NMDA receptors shortens the primary burst and eliminates the afterdischarges, so it is thought that the NMDA receptor activation is the sustained depolarizing stimulus that drives afterdischarges.[35]
Whereas the basic elements for seizure generation seem to be in place for CA3, dentate gyrus and CA1 are normally resistant to seizure generation. Pacemaker activity does not seem to differ significantly between CA3 and CA1, so the lack of a critical density of recurrent excitatory connectivity is the preferred explanation for why CA1 does not produce epileptiform activity in response to bicuculline. Although pyramidal cells in CA1 also possess some recurrent excitatory connections, the density of these connections is apparently not sufficient to support the generation of spontaneous activity in isolated pieces of CA1 tissue. As an example, DHPG, a metabotropic glutamate receptor agonist, increases spontaneous activity in both CA3 and CA1 (Fig. 2) In CA3, this activity organizes into population events detectable as large field potentials. CA1, on the other hand, does not show synchronized population activity as connectivity is insufficient to allow a single or small set of neurons triggering a firing cascade in a substantial population. Connectivity in dentate gyrus or CA1 can, however, be enhanced by manipulations that cause axonal sprouting. Such sprouting has been shown to occur after repeated seizures in several animal models of epilepsy.[24, 31] This addition of excitatory collaterals creates sufficient internal connectivity to convert these areas into seizure generators.
Fig. 2.
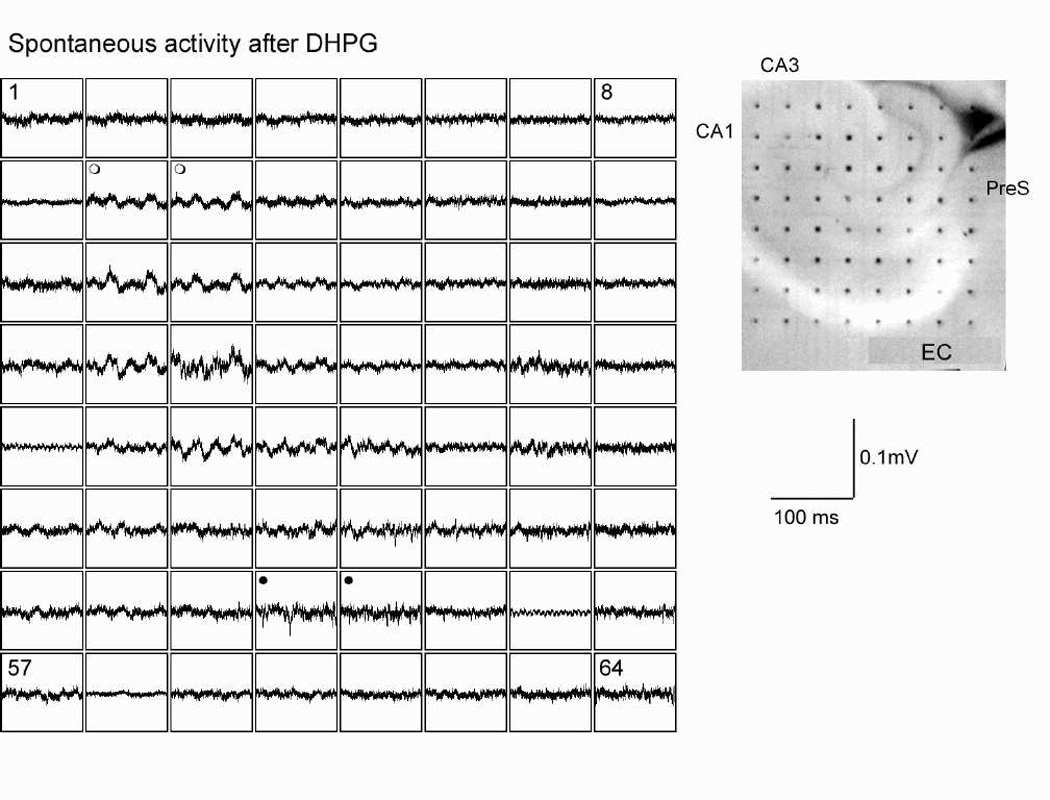
Metabotropic glutamate receptor activation increases spontaneous activity in hippocampal formation, but synchrony is clearest in area CA3. Bath application of DHPG increases spontaneous firing in many parts of the hippocampal formation. In area CA1, firing is desynchronized (e.g. sweeps labeled with filled circles). In CA3, activity organizes into gamma frequency oscillations. Far right shows locations of recording electrodes with several regions labeled.
At the other end of the complexity spectrum is entorhinal cortex. The isolated entorhinal cortex has also been shown to be an initiator of seizure activity, but the epileptiform events that originate in the entorhinal cortex are distinctly more complex than events originating in CA3.[8] The combination of intralaminar and interlaminar circuitry in entorhinal cortex leads to epileptiform events with multiple afterdischarges in the entorhinal cortex after disinhibition (see Fig. 1). [8, 28, 29] By contrast, a similar intralaminar circuitry with limited interlaminar circuitry in presubiculum and parasubiculum is the reason for simpler epileptiform events in isolated pre- and parasubiculum preparations.[9]
Secondary epileptiform areas: the role of the subicular complex
The production of epileptiform activity by subiculum is of particular interest since its circuitry possesses a crude laminar and columnar connectivity, making it more complex compared to CA3, by requiring a distributed network topology in simulations. Like CA3, subiculum has sufficient intrinsic connectivity and burst firing neurons to generate seizure activity. Harris and Stewart described morphological and electrophysiological evidence for excitatory interactions among rat subicular neurons in its much broader, but still single, layer of principal cells.[13, 14, 15] Unlike CA3, where reciprocal connections connect cells with similar firing patterns, there appear to be two distinct classes of principal cells in the subiculum. Burst firing neurons fire at threshold a brief series of action potentials which includes a calcium-dependent component.[17, 30, 32] Small depolarizations of the soma can convert the threshold response to regular spiking.[30] In spite of these differences, epileptiform events generated by the isolated subiculum resemble the events generated by the isolated CA3.[2, 14]
In laminar slice preparations, whether of isolated subiculum or whole hippocampus, both burst firing and regular spiking neurons of subiculum discharge during spontaneous epileptiform events. Burst firing neurons are active in the absence of synchronous activity in field recordings.[14] This suggests that the burst firing neurons may serve as pacemaker neurons for the generation of epileptiform activity in subiculum. Interestingly, multiple locations within subiculum could serve as the seizure focus in isolated pieces of subiculum tissue. Greene and Totterdell (1997) found relatively more burst firing neurons closer to the borders of subiculum with CA1 and presubiculum. A concentration of intrinsic bursting neurons at the borders would suggest that these regions might be more likely to serve as seizure foci which was confirmed by the finding that spontaneous epileptiform events tended to originate nearer to one or both borders than toward the midpoint of the CA1-presubiculum axis. Remarkably, some slices had independent seizure foci at both sides and events that propagated back and forth across the slice.
Recordings from apical dendrites of subicular cells and recordings taken during epileptiform activity in different slice preparations that include subiculum have also shed light on the cellular and circuit properties of subicular cells. In contrast to recordings from the soma, sustained depolarization through a dendritic impalement caused repetitive burst firing in some cells.[7] Repetitive burst firing was also seen during spontaneous or triggered events in combined subiculum/presubiculum circuits where pairwise intracellular recordings showed sequential and repetitive activation of subiculum, deep presubiculum and superficial pre/parasubiculum.[7]
What makes this especially interesting is the possibility that activity may be relayed back and forth between different parts of the subiculum. Just as action potential generation in dendrites that propagate to the soma or spike generation at the cell soma with backpropagation into the dendrites has led to many new possibilities for cell function, the ability of subiculum to generate epileptiform events on one locus or another may determine which subcircuits get activated. Parts of subiculum located closer to the presubiculum are more heavily interconnected with presubiculum and medial entorhinal cortex and get input from different parts of CA1 compared with subiculum that is located closer to CA1, which is more heavily influenced by lateral entorhinal cortex.
In addition to its intrinsic connections, subiculum also has reciprocal interconnections with other parahippocampal regions, permitting it to participate in more complex patterns of seizure activity. This gives it an unusual richness of epileptiform activity patterns. For example, subiculum is a target of projections from CA1, which can relay epileptiform activity that originates in area CA3.[30] Subiculum is also the target of inputs from entorhinal cortex and presubiculum allowing subicular neurons to follow activity originating in CA3 or entorhinal cortex.[42] Reciprocal connections between subiculum and presubiculum can also lead to re-entrant seizure activity as noted above.[7] Taking these regional interconnections as a whole, we can consider subiculum to be the way station that closes the loop that begins with entorhinal cortex and the trisynaptic pathway.
The entorhinal cortex is the primary input structure of the hippocampal formation, relaying virtually all neocortical activity that will eventually reach the hippocampus. Subiculum projects back to entorhinal cortex returning this cortical information flow through a loop. Subiculum also serves as the primary output area of the hippocampal formationto subcortical brain areas. Through these cortical and subcortical output paths, epileptic activity can spread from subiculum to other areas of the brain. Similarly, any baleful influences of abberent interictal hippocampal activity will spread from subiculum to the rest of the brain.
Role of computer modeling for understanding networks
As we consider the interaction of physiology and computer simulation in understanding how cells interact in networks, and how networks interact with each other, it is helpful to refer back to the history of physiological understanding of the single neuron. Simulation permitted Hodgkin and Huxley to fit together their physiological observations, providing not only understanding of the squid axon action potential but also a critical prediction of the existence of voltage-sensitive ion channels. In the years that followed, a variety of experiments confirmed the existence of these channels and provided data on channel types, behavior and distribution. At the same time, single neuron simulation has grown in complexity from simple spherical cells (point neurons), to distributed cells having passive dendrites and axons, to cells having active dendrites.[21]
This history provides metaphor and outline for the continuing development of network simulation. At each stage, computer models served first to organize data from different experiments, second to teach about structure-function relations, third to make predictions that could be confirmed experimentally. In producing single neuron simulations with greater and greater firing complexity and verisimilitude, one adjusts both the geometry of the cell (the dendrites) and the variety of ion channels. Similarly, in developing a network, we alter both the complexity of the network circuitry and the variety of the single cell types used. A simple, randomly connected network with simple single cell models can produce an interictal spike but nothing more. By making the cell model more complex, still using a single neuron type and undifferentiated connectivity, one can produce more prolonged epileptiform activity. This is the type of model that has been typically used to model CA3.
Further development of network simulation will involve producing distributed networks within a single region with more detailed connectivity based on the neuroanatomy and diverse cell types based on the neurophysiology of individual cells. This may be viewed as analogous to the development of single neuron models with active dendrites. From there we can build networks of networks in order to better understand how the different regions interact. Simulating observed epileptiform events for individual regions will constrain parameter sets and permit questions to be asked about behaviors that are below threshold for generation of epileptiform activity. Just as cells can have simple action potentials or action potential bursts, epileptiform events from individual regions vary in their complexity, depending on cellular and circuit properties. Once configured to simulate epileptiform events of a region, computer models can teach us about non-epileptiform activity as well, just as understanding of the action potential led to a broader understanding of cellular electrophysiology.
Modeling seizure generation and filtering in cortical structures
Computer modeling of epileptiform activity has generally followed the example provided by the in vitro slice model: reducing the in vivo complexity of a network of networks by focusing on an isolated area with only internal connectivity. A key finding of early studies was the notion that epileptic phenomenology is likely due to network properties rather than to abnormalities that could be traced to individual “epileptic neurons.” The concept of the epileptic neuron had the appeal of reductionism, holding out the promise of tracking down a particular malefactor. (This notion has come back into vogue lately but with the epileptic gene replacing the epileptic neuron.) By contrast, simulation studies have tended to follow an opposing fashion – stressing the importance of emergent properties of complex systems. The strong notion of an emergent property is that it cannot be explained by reference to constituent elements.
We must continually face this forest/trees dichotomy as we try to understand epilepsy or other brain functions and dysfunctions. For the purposes of developing therapies it is necessary to make the connections with specific genetic and cellular anomalies at the reductionist level. However, to understand the phenomenology of epilepsy, we must work with large scale interactions. The advantage of computer simulation is that we can flip back and forth between the forest and the trees at will to understand how the details of the latter effect the overall appearance of the former.
In performing simulations, we use the epileptiform event as a tool to constrain our simulations so that we can query these models about behavior below threshold for an epileptiform event. In terms of therapeutic strategies for epilepsy, an understanding of the threshold for seizure activity can lead to rational drug strategies to alter this threshold. In terms of normal activity, one can view normal function by any of these structures as below this threshold.
As noted above, Traub’s early findings demonstrated factors that appeared critical for the production of epileptiform activity in the CA3 model.[33, 34, 35, 36] The single critical factor was a relatively dense interconnectivity that permits activation to spread readily in the absence of restraining or the presence of enhancing factors. Note that dense connectivity in a biological context implies connections from about 1% to 10% of the network. (In the context of non-biological artificial neural networks such figures would be considered sparse connectivity, a discrepancy in approach that severely limits the applicability of most artificial neural networks to biological problems.) This factor by itself is sufficient to explain the interictal burst. A second major factor is the presence of some type of pacemaker, required to produce ongoing activity.
From this basis, one can begin to consider restraining and enhancing factors for epileptiform activity. The enhancing factors are precisely what one would expect from a knowledge of the corresponding slice models of epilepsy: reduction in inhibition (bicuculline and picrotoxin) increase in bursting or burst tendency (metabotropics), general increase in excitability (high K+, 4−AP), increase in long-acting NMDA synaptic effects (low Mg++). In general the corresponding modeling results are as expected. Something that is more surprising is how hard it is to get sustained epileptiform activity: to balance the strength and excitability of the pacemaking mechanisms with the excitability of the network as a whole. On the one hand, it is of course easy to get the model slice to be quiet. On the other, it is easy to get the model slice to fire off a brief interictal spike and then be quiet. However, it is not easy to get powerful activity that neither quickly resolves itself by activating all elements nor maintains itself by continuous maximal activation of all the neurons in the circuit. Note that simulated networks, and the slices on which they are based, are both too sparsely and too rapidly interconnected to sustain the type of circus rhythms that can produce arrhythmias in the heart.[25]
The difficulty of avoiding an interictal spike is in itself an interesting observation that has been commented upon by modelers.[5, 22] In model networks, the interictal spike precludes further activity through two mechanisms. First it exhausts the spiking potential of the network: all cells are in their refractory periods over a short period, leaving no cells to respond to additional activation. Second, in the presence of inhibition, such a massive volley will produce substantial post-population-spike inhibition, further reducing responsiveness. In this way the inter-ictal spike can be regarded as a protective mechanism preventing prolonged aberrant activity.
The major modeling approach to circumventing the premature termination of an interictal spike is to activate long-lasting postsynaptic excitatory mechanisms that can carry the activity over gaps in spiking. Such excitatory activity is often imposed by assuming that random miniature potentials or aberrant back-firing action potentials are producing a background of random noise that serves to drive the overexcitable network. This is likely to correspond to situations where cells are uniformly depolarized through high K+ or K+-channel blockade. The other mechanism that has been explored for maintaining activity is activation of NMDA with its prolonged postsynaptic effects. Such a model has been proposed to explain post-traumatic epilepsy, a condition in which there does not appear to be any decrease in inhibition.[5] A third mechanism which has not been explored extensively but appears relavant to subiculum, is the distributed network, which introduces delays between active cells and me be a fundamental requirement for the generation of sustained activity.
Implications for behavior – when good circuits go bad
Mesiotemporal lobe epilepsy (MTLE), with or without hippocampal sclerosis, appears to be associated with memory, emotional, behavioral and likely cognitive anomalies that cannot be fully explained by drug side-effects or the general stress of coping with chronic illness.[11] Memory difficulties have been clearly documented. Other reported difficulties include depression, schizophreniform disorders and personality anomalies.[18, 1] These additional psychological and psychiatric manifestations are somewhat controversial and hard to pin down. Of course, computer models of brain function and dysfunction are still more uncertain and controversial.
One conclusion seems likely: in the presence of hippocampal sclerosis the loss of neurons will itself cause dysfunction in processes involving the hippocampus, notably memory.[26] Such specific localized anomalies would be expected in any ablative disease, notably those associated with stroke.[23]
Beyond the detrimental effects associated with neuronal loss, it is likely that further impairment of brain function will occur due to abnormal activity in the remaining neurons. The neurons that are directly or proximally involved in epileptiform activity and seizures maintain their connections with the rest of the brain. Although some interictal pathological firing is apparent (interictal spikes), it is generally uncertain how these neurons’ firing patterns relate to firing in the normal brain. We propose three alternative hypotheses for the interictal activity of these neurons. 1. They produce normal patterns of activity and do not contribute to the interictal abnormalities which are purely due to missing neurons. 2. They are walled-off from the brain, whether through inhibition or though involvement in some non-epileptiform abnormal oscillation which prevents their normal functional interactions with other brain areas. The functional effect would be comparable to that of an ablation. 3. They are active and involved in brain function but are unable to fully produce the signals or oscillations that are required for normal brain function.
Of course, all 3 situations may apply to a greater or lesser extent in particular patients or particular interictal symptoms. With regard to the origins of behavioral disturbances, the third hypothesis from our list, that brain areas are “active-but-abnormal” would appear to best explain positive symptoms such as those seen in the personality disorders or psychoses associated with MTLE.[11] Additional support for this hypothesis comes from non-linear analyses of the pre-ictal electroencephalogram (EEG).[27] These analyses suggest that the area surrounding the epileptic focus produces normal synchronization and phase-locking with the rest of the brain up until a period several hours prior to the seizure. At this time the epileptiform area appears to “unlink” from the rest of the brain (phase scattering) prior to relinking at the pathological frequencies just prior to the onset of the clinical seizure. One can extrapolate from this to suggest that the unlinking is the initial pathophysiological phase. This unlinking would presumably produce mild brain dysfunctions. Such a propensity to unlinking would be a candidate cause for interictal disturbances of behavior and cognitive and emotional function even under circumstances where progression to seizure did not occur.
Complicating things even further is a seizure generating network that is not stable. Repeated seizures may damage a brain region that was part of the seizure generating mechanism, while at the same time, other brain regions acquire seizure generating capabilities (as described for areas CA1 and dentate gyrus earlier) and thereby show reduced or aberrent participation in normal activity patterns.
There is certainly no concensus on how neurons and neural circuits produce memory, cognition, perception or behavior. However, there is at least one widely accepted principal, that of Hebb. There are also a number of general notions and a few recent hypotheses that enjoy some experimental support. This is not the place to review proposed mechanisms of brain function in detail. We can however suggest how epileptiform discharges and the consequences of seizures are likely to alter information processing by disrupting the patterns of brain organization and activity that these mechanisms require.
The Hebbian synapse is currently believed to underly memory storage in the brain. It is though to be implemented in mechanisms involving glutamate receptor interactions that produce long-term potentiation (LTP).[4] LTP can be produced in vitro using strong repetitive stimulation or by precise timing of pre- and postsynaptic activation, a process referred to as spike-timing dependent plasticity (STDP).[3, 19] In vivo, it may be that strong simultaneous activation of multiple synapses during sharp waves is responsible for “imprinting” memories. If this is the case, the interictal spike would be a pathological version of a sharp wave that could imprint false memories. If, on the other hand, the precise timing of incoming activity is the critical factor in altering synaptic strengths in vivo, anomalous activity from a seizure focus would not need to be powerful to disrupt memory formation. Whether due to powerful excitation or to precise timing, these mechanisms would be expected to disrupt existing memories and perhaps produce anomalous memories resulting in the delusions of the schizophrenia-like psychosis of epilepsy.[11]
The plasticity of the Hebb synapse results in memory stored in static connectivity strengths. The memories are not however directly read-out from these strengths. Instead, the synaptic strength pattern (the connectivity matrix) alters the dynamics of neuronal firing, producing coincident activity of sets of neurons, the Hebb assembly. The Hebb cell assembly is a distinct hypothesis which gives a theory of representation based on the activity of neurons. Unlike the Hebb synapse hypothesis, the cell assembly hypothesis is not clearly supported by data, due to the current impossibility of recording from vast numbers of neurons simultaneously in an awake behaving animal. In the simplest version of the cell assembly model, simultaneous high-frequency firing of a set of cells represents something in the world. Such a representation would be prone to disruption by a powerful incoming signal that simultaneously activates multiple interconnected cells. In Fig. 3, we show some sample cells from a simple model of a sparsely interconnected 1250-neuron network which is subject to an interictal spike that activates 60% of the neurons. This powerful activation starts an activation cascade which, by recruiting via long-acting NMDA receptors, produces prolonged simultaneous firing of all the cells in the circuit, six of which are shown here. Representations based on simultaneous activation of neurons would be overwhelmed by confused coincidence through such a barrage of activity.
Fig. 3.

Six representative excitatory cells from a 3 s simulation of a 1250-neuron network. A single spike given at t=1 s directly activates 60% of the excitatory cells with subsequent activation of the remaining population.
The extreme case shown in Fig. 3 depends on a powerful stimulation and a network with random interconnections. The potential for disruption of memories is likely to be far more subtle – requiring less powerful aberrant stimuli and resulting in only moderate alterations in normal activity pattern. The governing hypothesis for more realistic models is that of the attractor network, first proposed by Hopfield,[16] and recently elaborated in a more biological context by Wang and colleagues.[20, 40, 41] The key concept here is that patterns of elevated firing across sets of interconnected neurons can remain stable for prolonged periods. However, this notion of stability is a tricky one, shown for Hopfield in the case of fully interconnected networks (every neuron is connected to every other one) used to store relatively few memories. In a biological context, stability can be harder to achieve and may be quite prone to disruption. It is possible that even relatively mild anomalies in incoming signal could disrupt these stable attractors, forcing the system into other attractors. Alternatively, these input anomalies might actually alter the attractor surface, creating new memories or perceptions that could form the basis of delusions or hallucinations.
A widely discussed problem with attractor theory is relevant to our discussion here. The activated neurons in an attractor model are considered to represent particular objects or attributes of objects. If you simultaneously activate cell assemblies that represent e.g., a square and a circle, along with cell assemblies that represent the color blue and the color red, you have an ambiguity. Is the system perceiving a red circle and a blue square, or a blue circle and a red square? This is the binding problem. Von der Malsburg suggested a solution that would allow networks to disambiguate the associations between simultaneously active neurons by utilizing the temporal structure of the spike trains: neurons which spike at approximately the same time are bound together.[38, 37, 39] This hypothesis has gained further currency since long-distance gamma-frequency phase locking was demonstrated by Gray and Singer[12, 6] Similar long distance phase locking has been seen in parahippocampal areas in slices.[10] If attribute binding requires phase locking and if the pre- or interictal state involves phase scattering then the process of “unlinking” preceding the seizure would extend from the physiological to the cognitive domain, again with relevance for schizophreniform symptomatology.
The hippocampus and parahippocampus together form a complex system whose critical roles in memory formation and consolidation can relatively readily be perverted to produce seizure activity. Although a unified understanding of the interactions of the various sub-areas involved is still a long ways off, we are beginning to classify the various areas in terms of their propensity to produce epileptiform activity. However, it is important to emphasize that these classifications are not static but that they change during a seizure and will change over longer periods of time with evolution of the epileptic disorder. Similarly, the influence of the epileptic focus on behavior will shift over time, likely involving loss neurons, dynamical walling off of living neurons, and abberent activity projecting to other brain areas. Further understanding will come as we map activity spread in slice and in vivo.
Acknowledgements
Research supported by NIH (NS045612)
References
- 1.Bear DM, Fedio P. Quantitative analysis of interictal behavior in temporal lobe epilepsy. Archives of Neurology. 1977;34:454–467. doi: 10.1001/archneur.1977.00500200014003. [DOI] [PubMed] [Google Scholar]
- 2.Behr J, Empson RM, Schmitz D, Gloveli T, Heinemann U. Electrophysiological properties of rat subicular neurons in vitro. Neuroscience Letters. 1996;220:41–44. doi: 10.1016/s0304-3940(96)13242-0. [DOI] [PubMed] [Google Scholar]
- 3.Bi GQ, Wang HX. Temporal asymmetry in spike timing-dependent synaptic plasticity. Physiology & Behavior. 2002;77:551–555. doi: 10.1016/s0031-9384(02)00933-2. [DOI] [PubMed] [Google Scholar]
- 4.Brown TH, Kairiss EW, Keenan CL. Hebbian synapses: biophysical mechanisms and algorithms. Ann Rev Neurosci. 1990;13:475–511. doi: 10.1146/annurev.ne.13.030190.002355. [DOI] [PubMed] [Google Scholar]
- 5.Bush PC, Prince DA, Miller KD. Increased pyramidal excitability and nmda conductance can explain posttraumatic epileptogenesis without disinhibition: a model. Journal of Neurophysiology. 1999;82:1748–1758. doi: 10.1152/jn.1999.82.4.1748. [DOI] [PubMed] [Google Scholar]
- 6.Engel A, Konig P, Kreiter A, Gray C, Singer W. Temporal coding by coherent oscillations as a potential solution to the binding problem: physiological evidence. In: Schuster HG, editor. Nonlinear dynamics and neural networks. Weinheim: VCH Verlagsgesellschaft; 1991. [Google Scholar]
- 7.Funahashi M, Harris E, Stewart M. Re-entrant activity in a presubiculum-subiculum circuit generates epileptiform activity in vitro. Brain Res. 1999;849:139–146. doi: 10.1016/s0006-8993(99)02045-4. [DOI] [PubMed] [Google Scholar]
- 8.Funahashi M, Matsuo R, Stewart M. Propagation of synchronous burst discharges from entorhinal cortex to morphologically and electrophysiologically identified neurons of rat lateral amygdala. Brain Res. 2000;884:104–115. doi: 10.1016/s0006-8993(00)02854-7. [DOI] [PubMed] [Google Scholar]
- 9.Funahashi M, Stewart M. Presubicular and parasubicular cortical neurons of the rat: functional separation of deep and superficial neurons in vitro. J Physiol (Lond) 1997;501:387–403. doi: 10.1111/j.1469-7793.1997.387bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funahashi M, Stewart M. Properties of gamma-frequency oscillations initiated by propagating population bursts in retrohippocampal regions of rat brain slices. J Physiol (Lond) 1998;510:191–208. doi: 10.1111/j.1469-7793.1998.191bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaitatzis A, Trimble MR, Sander JW. The psychiatric comorbidity of epilepsy. Acta Neurologica Scandinavica. 2004;110:207–220. doi: 10.1111/j.1600-0404.2004.00324.x. [DOI] [PubMed] [Google Scholar]
- 12.Gray C, Singer W. Stimulus-specific neuronal oscillations in orientation columns of cat visual cortex. Proc Nat Acad Sci USA. 1989;86:1698–1702. doi: 10.1073/pnas.86.5.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris E, Stewart M. Propagation of synchronous epileptiform events from subiculum backward into area CA1 of rat brain slices. Brain Res. 2001;895:41–49. doi: 10.1016/s0006-8993(01)02023-6. [DOI] [PubMed] [Google Scholar]
- 14.Harris E, Stewart M. Intrinsic connectivity of the rat subiculum: II. properties of synchronous spontaneous activity and a demonstration of multiple generator regions. J Comp Neurol. 2001b;436:506–518. doi: 10.1002/cne.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris E, Witter MP, Weinstein G, Stewart M. Intrinsic connectivity of the rat subiculum: I. dendritic morphology and patterns of axonal arborization by pyramidal neurons. J Comp Neurol. 2001a;436:490–505. doi: 10.1002/cne.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hopfield JJ. Neural neworks and physical systems with emergent collective computational abilities. Proc Nat Acad Sci USA. 1982;79:2554–2558. doi: 10.1073/pnas.79.8.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung HY, Staff NP, Spruston N. Action potential bursting in subicular pyramidal neurons is driven by a calcium tail current. J Neurosci. 2001;21:3312–3321. doi: 10.1523/JNEUROSCI.21-10-03312.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalynchuk LE. Long-term amygdala kindling in rats as a model for the study of interictal emotionality in temporal lobe epilepsy. Neuroscience & Biobehavioral Reviews. 2000;24:691–704. doi: 10.1016/s0149-7634(00)00031-2. [DOI] [PubMed] [Google Scholar]
- 19.Kistler WM, Hemmen JL. Modeling synaptic plasticity in conjuction with the timing of pre- and postsynaptic action potentials. Neural Computation. 2000;12:385–405. doi: 10.1162/089976600300015844. [DOI] [PubMed] [Google Scholar]
- 20.Lisman JE, Fellous JM, Wang XJ. A role for NMDA-receptor channels in working memory. Nature Neuroscience. 1998;1:273–275. doi: 10.1038/1086. [DOI] [PubMed] [Google Scholar]
- 21.Lytton WW. From Computer to Brain. New york: Springer Verlag; 2000. [Google Scholar]
- 22.Lytton WW, Hellman KM, Sutula TP. Computer models of hippocampal circuit changes of the kindling model of epilepsy. Artificial Intelligence in Medicine. 1998;13:81–98. doi: 10.1016/s0933-3657(98)00005-0. [DOI] [PubMed] [Google Scholar]
- 23.Lytton WW, Sejnowski TJ. Computational neuroscience, chapter 120. In: Asbury AK, McKhann GM, McDonald WI, editors. Diseases of the Nervous System: Clinical Neurobiology. 2nd edition. Orlando Fl: W.B. Saunders Co.; 1992. [Google Scholar]
- 24.Meier CL, Dudek FE. Spontaneous and stimulation-induced synchronized burst afterdischarges in the isolated CA1 of kainate-treated rats. J Neurophysiol. 1996;76:2231–2239. doi: 10.1152/jn.1996.76.4.2231. [DOI] [PubMed] [Google Scholar]
- 25.Noble D, Rudy Y. Models of cardiac ventricular action potentials: iterative interaction between experiment and simulation. Phil. Trans. R. Soc. Lond. A. 2001;359:1127–1142. [Google Scholar]
- 26.Ozkara C, Hanoglu L, Keskinkilic C, Yeni N, Aysal F, Ozyurt E, Karaagac N. Memory in patients with drug-responsive mesial temporal lobe epilepsy and hippocampal sclerosis. Epilepsia. 2004;45:1392–1396. doi: 10.1111/j.0013-9580.2004.23304.x. [DOI] [PubMed] [Google Scholar]
- 27.Quyen M, Navarro V, Martinerie J, Baulac M, Varela FJ. Toward a neurodynamical understanding of ictogenesis. Epilepsia. 2003;44:30–43. doi: 10.1111/j.0013-9580.2003.12007.x. [DOI] [PubMed] [Google Scholar]
- 28.Scharfman HE. Epileptogenesis in the parahippocampal region. parallels with the dentate gyrus. In: Scharfman HE, Witter MP, Schwarcz R, editors. The parahippocampal region. Implications for neurological and psychiatric diseases. volume 911. New York: NYAS; 2000. pp. 305–327. [Google Scholar]
- 29.Stewart M. Columnar activity supports propagation of population bursts in slices of rat entorhinal cortex. Brain Res. 1999;830:274–284. doi: 10.1016/s0006-8993(99)01404-3. [DOI] [PubMed] [Google Scholar]
- 30.Stewart M, Wong RK. Intrinsic properties and evoked responses of guinea pig subicular neurons in vitro. J Neurophysiol. 1993;70:232–245. doi: 10.1152/jn.1993.70.1.232. [DOI] [PubMed] [Google Scholar]
- 31.Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- 32.Taube JS. Electrophysiological properties of neurons in the rat subiculum in vitro. Expt Brain Res. 1993;96:304–318. doi: 10.1007/BF00227110. [DOI] [PubMed] [Google Scholar]
- 33.Traub v, Jefferys JGR, Miles R. Analysis of the propagation of disinhibition-induced after-discharges along the guinea-pig hippocampal slice invitro. J Physiol (Lond) 1993;472:267–287. doi: 10.1113/jphysiol.1993.sp019946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Traub RD, Jefferys JGR, Whittington MA. Enhanced NMDA conductance can account for epileptiform activity induced by low Mg++ in the rat hippocampal slice. J Physiol (Lond) 1994;478:379–393. doi: 10.1113/jphysiol.1994.sp020259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Traub RD, Miles R, Jefferys JGR. Synaptic and intrinsic conductances shape picrotoxin-induced synchronized after-discharges in the guinea-pig hippocampal slice. J Physiol (Lond) 1993;461:525–547. doi: 10.1113/jphysiol.1993.sp019527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Traub RD, Whittington MA, Stanford IM, Jefferys JG. A mechanism for generation of long-range synchronous fast oscillations in the cortex. Nature. 1996;383:621–624. doi: 10.1038/383621a0. [DOI] [PubMed] [Google Scholar]
- 37.von der Malsburg C. Binding in models of perception and brain function. Neuron. 1995;5:95–104. doi: 10.1016/0959-4388(95)80014-x. [DOI] [PubMed] [Google Scholar]
- 38.von der Malsburg C. The what and why of binding: the modeler’s perspective. Neuron. 1999;24:95–104. doi: 10.1016/s0896-6273(00)80825-9. [DOI] [PubMed] [Google Scholar]
- 39.Von der Malsburg C, Schneider W. A neural cocktail-party processor. Biological Cybernetics. 1986;54:29–40. doi: 10.1007/BF00337113. [DOI] [PubMed] [Google Scholar]
- 40.Wang XJ. Synaptic basis of cortical persistent activity: the importance of NMDA receptors to working memory. Journal of Neuroscience. 1999a;19:9587–9603. doi: 10.1523/JNEUROSCI.19-21-09587.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang XJ. Synaptic reverberation underlying mnemonic persistent activity. Trends in Neurosciences. 2001;24:455–463. doi: 10.1016/s0166-2236(00)01868-3. [DOI] [PubMed] [Google Scholar]
- 42.Wong RK, Stewart M. Different firing patterns generated in dendrites and somata of CA1 pyramidal neurones in guinea-pig hippocampus. J Physiol (Lond) 1992;457:675–687. doi: 10.1113/jphysiol.1992.sp019401. [DOI] [PMC free article] [PubMed] [Google Scholar]