

Abstract
The matrix metalloproteinases (MMPs), a family of 25 secreted and cell surface-bound neutral proteinases, process a large array of extracellular and cell surface proteins under normal and pathological conditions. MMPs play critical roles in lung organogenesis, but their expression, for the most part, is downregulated after generation of the alveoli. Our knowledge about the resurgence of the MMPs that occurs in most inflammatory diseases of the lung is rapidly expanding. Although not all members of the MMP family are found within the lung tissue, many are upregulated during the acute and chronic phases of these diseases. Furthermore, potential MMP targets in the lung include all structural proteins in the extracellular matrix (ECM), cell adhesion molecules, growth factors, cytokines, and chemokines. However, what is less known is the role of MMP proteolysis in modulating the function of these substrates in vivo. Because of their multiplicity and substantial substrate overlap, MMPs are thought to have redundant functions. However, as we explore in this review, such redundancy most likely evolved as a necessary compensatory mechanism given the critical regulatory importance of MMPs. While inhibition of MMPs has been proposed as a therapeutic option in a variety of inflammatory lung conditions, a complete understanding of the biology of these complex enzymes is needed before we can reasonably consider them as therapeutic targets.
I. INTRODUCTION
The matrix metalloproteinase (MMP) family of enzymes consists of 25 zinc-dependent endopeptidases in mice and 24 in humans, where its founding member was first discovered in metamorphosing tail skin of Xenopus in 1961 and its last member, epilysin (MMP28), was discovered some 40 years later (Fig. 1; Refs. 84, 85, 136, 152, 161). Members of the MMP family were originally identified by descriptive names that were assigned based on limited knowledge of their preferred substrate specificities, e.g., collagenases, gelatinases, stromelysins, and matrilysins (256). A sequential numbering system, loosely based on the order of discovery, was adopted when it became clear that more MMPs exist than was previously suspected and that the names based on substrate specificity type were inadequate. The missing MMPs (MMP4, MMP5, and MMP6) were removed from the list because further studies showed that either the gene products did not exist or were identical to previously described members (304). Over the past four decades, in addition to discovering additional new members, researchers have shown that members of this family undergo a multistep activation process and display distinct molecular interactions with other proteinases and substrates in vivo that make their biology decidedly complex.
FIG. 1.

Matrix metalloproteinase (MMP) discovery timeline. This diagram depicts discovery of the MMP family members chronologically with the exception of MMP1, which was actually the second MMP discovered. Over ∼40 years (1961−2001), 28 different MMP genes have been identified. The founding member of the MMP family, Xenopus collagenase, now thought to be collagenase 4 (MMP18), was discovered in 1961 by J. Gross and C. M. Lapiere (85) and the last member, epilysin (MMP28), was reported separately by two groups: W. Parks and A. Y. Strongin in 2001. With the completion of the genome project, it is clear that all members of this family have now been identified (see Refs. 13, 17, 18, 29, 50, 76, 91, 97, 101, 124, 151, 182, 209, 222, 234, 235, 239, 245, 258, 269, 294, 297, 298, 304, 309, 310).
As in all tissues, expression of MMPs in the lung is a highly regulated process, and understanding its regulation could, in part, shed light into their biological function in normal developmental processes, such as lung branching morphogenesis, and in many pathological conditions, such as asthma or chronic obstructive lung disease (58, 119, 212, 213, 246). Some of the known targets of MMPs in the lung include extracellular matrix (ECM) molecules, growth factors, chemokines, proteinases, and cell surface proteins, such as adhesion molecules. The functional significance of the interactions between MMPs and their substrates in bronchoalveolar fluid and in the lung parenchyma is a novel concept that is currently being explored, but because of the intricacy of these enzymes and their substrates, much of this type of information remains poorly understood. However, a dedicated effort is now underway to identify the many as yet undiscovered proteins that are undoubtedly critical substrates of the MMPs (83a, 278).
Many members of the MMP family are upregulated in the lungs of humans with inflammatory diseases. To understand the biology of MMPs in a variety of relevant human pathological conditions such as allergic inflammation, tissue injury and repair, remodeling, and host defense against pathogens (9, 52, 133), the genetic approach, which uses single or complex deletion of MMP gene(s), has been particularly powerful. However, despite the use of some of these well-established models of lung diseases, no unifying theme on the advisability of inactivating these molecules in all inflammatory conditions has emerged. For instance, deletion of MMP2 is protective in allotransplant models because it significantly reduces cellular infiltration and fibrosis. But, in sharp contrast, deficiency in MMP2 increases susceptibility of mice to lethal asphyxiation in an asthma model (36, 49). Interestingly, lack of MMP2 in humans results in many abnormalities, including an osteolytic and arthritic disorder and other bone deformities (4). Thus, despite the fact that large efforts have been directed towards the development of chemical inhibitors of MMPs, it is not clear whether MMP inhibition is beneficial or harmful and in which situations it would be of clinical help. Among the critical issues that require resolution are the mechanisms of action and function of MMPs under diverse pathological conditions.
The lung has a rich vascular network and is in continuous and direct communication with the outside environment. Thus it is not surprising that besides serving as the conduit for gas exchange, one of the main functions of the lung is its role in host defense, protecting against such affronts as viral and bacterial pathogens and/or other organic/inorganic environmental insults. Although small amounts of MMP2 and MMP14 are present in the lining fluid of the lung under normal conditions, other MMPs such as MMP7, MMP8, MMP9, and MMP12 are upregulated under many pathological conditions. There is considerable evidence that MMPs in the lung play a role in host defense (9, 212, 223). In this review we present evidence from the literature that supports a direct or an indirect role for several of the MMP family members in various lung diseases. The mechanistic insight into the function of proteinases requires the use of carefully crafted models of lung injury and repair. Using examples of work done in animal models of human lung diseases, as well as in translational studies, we explore the expression of MMPs, their mechanisms of action, and their involvement in lung pathology and repair.
In section II, we compare MMPs from lower phylogenetic ranks of species, e.g., invertebrates, with those of higher orders, e.g., mammals, a comparison that may allow lessons from a simple system to provide insights into the most complex biological functions of this family of enzymes. Then, we review the molecular regulation of MMPs and their activation in the lung (see sect. III). Although not formally tested, based on their predicted cleavage sites, collectively, activated forms of MMPs can degrade most of the secreted or cell surface bound proteins in the body; thus many levels of regulation are necessary to prevent proteolytic damage. Following in section IV, we use lung development as a model system in which to explore the spatiotemporal expression of MMPs. In smoking-related human lung diseases such as chronic obstructive pulmonary disease (COPD) and emphysema, where the loss of alveolar space is the main hallmark, understanding the role of MMPs in alveolar generation/regeneration may hold the key to future treatment of these diseases. The upregulation of MMPs in the lung under various pathological conditions is explored in section V. The main issue that we address is whether there is enough evidence to support the notion that resurgence of MMPs in general is harmful in the active or chronic phases of lung diseases. We review and compare in section VI whether the actions of members of the MMP family are redundant or compensatory in nature. The role of MMPs in modulating the immune system perhaps through mediating the cross-talk between the innate and adaptive immune systems is addressed in section VII. In section VIII, we review evidence that MMP cleavage of various proteins alters their effector functions, providing a new paradigm for MMPs’ mechanism of action in inflammatory conditions in human lung. Next, we review human genetic variation in MMPs (see sect. IX). In section X, we discuss the role of MMPs in some human lung diseases. Finally, in section XI, we examine whether there is support for inhibition of MMPs in non-cancer-related lung diseases. This is a critical issue, since currently most inhibitors in the market inhibit the broad class of MMPs, which can affect multiple systems. However, there is a great interest in developing designer MMP inhibitors to target the action of individual members.
II. EVOLUTION OF MATRIX METALLOPROTEINASES
The MMPs belong to the metzincin superfamily that includes enzymes with similar metalloproteinase domains such as astacins, ADAMs, and ADAM-TS proteinases (256). There is little consensus as to how the MMPs should be grouped, and different experts in the field classify them based on their structural similarities, substrate specificity, or tissue expression (Fig. 1; Refs. 213, 256). The MMPs have a catalytic domain and a hemopexin-like domain. Among MMPs, the zinc-binding motif (HEXXHXXGXXH, where X is a variable amino acid) and the propeptide cysteine switch (PRCGXPD) are highly conserved (164). Another highly conserved region is the “methionine turn” (XXMXP) thought to maintain the structural integrity of the zinc-binding site (255, 256). MMPs are formed as zymogens and activated following secretion either by cleavages in the hydrophobic propeptide domain that is located in the NH2 terminus or in the trans-Golgi network by furin-like protein convertases (as detailed in sect. III).
Multiple sequence analyses of the different domains indicate that modern, multidomain MMPs evolved early on, before the divergence of vertebrates from invertebrates, from an enzyme with a simpler structure (164). One putative candidate, enterotoxin from Bacteroides fragilis (BFT; GenBank accession no. U90931) has a zinc-binding consensus motif, characteristic of the metalloproteinase family (75), and shares 59% sequence identity with the 27 amino acid stretch in human MMP-1, which includes the catalytic zinc-binding domain and the “Met turn” (75, 307). In addition, purified BFT can cleave monomeric G-actin, gelatin, and azocoll in vitro. Because bacteria first appeared more than 3.5 billion years ago, and eukaryotes around 1.8 billion years ago, it is possible that the metalloproteinases evolved at that time and apparent that MMPs are ancient (164).
Because most of the literature on MMPs is focused on mammals, many comparative studies focus first on identifying the presence of a proteinase and then classifying the proteinase as a matrix-degrading enzyme. This classification is done in several ways: analysis of protein sequence and putative tertiary structures, determination of the substrate specificity, and inhibition of degradatory capacity by known MMP inhibitors. These necessary studies have led to hypotheses that MMPs are critical for regeneration of tissues and development. Now that the presence of MMPs has been unequivocally established in lower phyla, it is time to focus on the structure-function relationship of these MMPs and utilize new model systems for investigating mechanisms for MMP action.
To better understand the roles that MMPs play today in development and disease, it is helpful to understand their historical functions and their function in other organisms. MMPs are widely distributed across phylogenies, but their structures are fairly conserved. Members of the MMP family have been found in soybean (167) and mustard (155) plants and algae (125). In invertebrates, MMP-like activity has been noted in Balanus amphitrite barnacle larvae (158), Strongylocentrotus purpuratus sea urchin embryos (228), Scylla serrata crabs (250), Crassostrea virginica (321) and C. gigas (268) oysters, and Mytilus galloprovincialis mussels (159). The nematodes, Caenorhabditis elegans (288) and Gnathostoma spinigerum (280), have MMPs. Drosophila has two MMPs (149, 150, 206). What remains to be fully answered is whether MMP functions in complex biological processes such as development, tissue repair, and immune response are also conserved.
A. Development
In vitro, as a group MMPs are able to degrade every type of ECM protein yet tested (256). Conceivably based on this observation, MMPs could be viewed as critical players in development and repair processes. It is clear that Drosophila melanogaster1-MMP (DM1-MMP) is required for proper development of the larval tracheal respiratory system and for pupal head eversion, while DM2-MMP is important for metamorphosis and neural development. Mutant Drosophila, lacking functional DM1-MMP, are smaller in size, grow tracheae that appear to be overstretched and broken in spots, and wander earlier than their wild-type counterparts (206). These researchers also note that at ecdysis from larval/larval molting, flies appear to have difficulty shedding their exoskeleton, which appears to be stuck at the ends of the tracheae. Because new tracheae are formed inside the old tracheae, separated by tracheal fluid, this could imply that there are cuticular defects or possibly a defect in the tracheal lining fluid.
In contrast, MMP2 has been found in developing zebrafish throughout embryonic development and is required for normal embryogenesis (316). In addition, using a novel technique that would best be described as in vivo zymography in the zebrafish model, MMP activity has been visualized throughout embryogenesis in these fish; however, its role in the developing respiratory system remains unclear (54).
XMMP7, the Xenopus homolog to human MMP7, is expressed in macrophages during embryonic development (98) along with XMMP9 (38) and is hypothesized to play a role in macrophage migration.
B. Tissue Repair
In organisms from hydra to fish, MMPs are involved with tissue repair. For instance, if MMPs are blocked using the MMP-specific, zinc-chelating inhibitor GM6001, limb regeneration stops in newts (285). In hydra, foot regeneration inhibited by GM6001 is reversible, and upon removal of the inhibitor, foot regeneration resumes (143). Zebrafish also cannot regenerate fins if MMPs are blocked with GM6001 (10). Whether MMPs are involved in gill tissue repair in the fish is unclear. However, in oysters, one study demonstrated increased MMP mRNA expression in gill tissue 18−24 h after exposure to hypoxia. This could represent tissue degradation after oxidative destruction; however, MMPs are often upregulated during angiogenesis. Given their role in tissue regeneration, it seems more likely that they are working to increase gill area or to replace damaged tissue (268). This response is similar to that seen in rats, where systemic treatment with GM6001 delayed epithelial wound healing (175).
C. Immune Response
The function of MMPs in the innate immune response is a role that evolved before the divergence of vertebrates and invertebrates. In the oyster Perkinsus marinus, MMP-like activity increases after infection with a protozoan parasite (183). In Crassostrea gigas, bacterial challenge results in increased mRNA expression of cg-MMP (89). In response to shell damage and bacterial challenge, cg-TIMP (tissue inhibitor of metalloproteinases) is upregulated in hemocytes in gill (178), suggesting not only that the complexity of MMP regulation seen in mammals evolved early on, but also that the role of MMPs in respiratory system function has long been important. In support of this, adult hydra injected with the MMP inhibitor GM6001 exhibit decreased mucus production, as evidenced by a decrease in cell adhesion (hydra did not stick to a glass rod). The researchers also found decreased peroxidase activity (143). This is suggestive of immune system activity; however, more studies of MMP function in plants and invertebrates will be necessary to answer this question.
III. REGULATION OF MATRIX METALLOPROTEINASES IN LUNG: TRANSCRIPTION, TRANSLATION, AND POSTTRANSLATIONAL REGULATION
MMPs are tightly regulated in the lung at the transcriptional and translational levels of gene expression via multiple transcription factors (activators and repressors). These factors include a large list of growth factors, hormones, cytokines, cell adhesion molecules, ECM proteins and their bioactive fragments, intracellular signaling factors (GTPases), and agents that cause actin cytoskeletal reorganization, all of which can directly or indirectly upregulate the expression of MMPs (121). Additionally, mRNA stability has been suggested in some studies to play a role in regulation of MMP1, MMP2, and MMP9 (200, 231). Finally, MMPs are regulated at the protein level through their activation state and cellular location.
A. Transcriptional and Translational Regulation
As emphasized in this review, MMPs are made in different cell types under unique conditions, and thus distinctive inductive and suppressive factors regulate the expression of each of the members of the MMP family. These are detailed comprehensively in several recent reviews; therefore, we will briefly review the main types of regulating factors and provide recent examples (61, 256). Interestingly, most of the same factors that regulate the expression of MMPs in the lung can also act as their substrates, providing a built-in and, most likely, essential component of the regulatory cascades. This coordination between MMP and substrate ensures efficient activation and control of MMPs in vivo (173, 283).
1. Transcription factors
The first step in regulating MMP production occurs at the transcriptional level. Several proteins, including early-growth response gene product 1 (EGR1), nuclear factor kappa B (NFκB), globin transcription factor 1 (GATA1), activator protein 1 family members (AP-1), and signal transducer and activator of transcription 3 (STAT3C), have been shown to affect several members of the MMP gene family. For example, lung fibroblasts from EGR1 null mice fail to upregulate expression of MMP2 in response to stimulation with a mixture of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and interferon (IFN)-γ (189). Interestingly, in endothelial cells EGR1 binds to the MMP14 promoter, also regulating its expression (94). Because MMP14 activates MMP2, this provides a positive-feedback loop for upregulation of MMP2. With the use of a luciferase promoter/reporter assay in cultured fibroblasts, MMP1 transcription increases during cell shape changes and depends on NFκB-mediated IL-1 secretion (121, 295). Similarly, GATA-1 in endothelial cells and members of the AP-1 family, c-Jun, JunD, c-Fos, FosB, and Fra-1 proteins in lung cancer cells activate the MMP2 promoter (95. 41). In a mouse model of hyperoxia-induced lung injury, the development of lung damage, including capillary leakage and neutrophil infiltration into alveoli, is mediated by the synthesis and release of MMP9 and MMP12 from neutrophils and alveolar resident cells. This deleterious effect is prevented by overexpression of STAT3C in alveolar epithelial cells, which decreases both transcription and activity of MMP9 and MMP12 (146).
2. Growth factors
Epidermal growth factor receptor (EGFR) activation upregulates MMP14 in lung fibroblasts (122). MMP2 and MMP9 are upregulated in response to treatment of mice with hepatocyte growth factor (HGF), a treatment that also improves lung fibrosis through a mechanism involving increased myofibroblast apoptosis (176). Deletion of the α1,6-fucosyltransferase (Fut8) gene in mice is associated with abnormal lung parenchyma development consistent with emphysema-like lesions and overexpression of MMP12 and MMP13 (290). Although most likely abnormal lung development plays a significant role in lung lesions seen in mice deficient in Fut8, the unexpected dysregulation of MMPs in the absence of Fut8 is mediated through aberrant transforming growth factor (TGF)-β1 receptor signaling, which is known to downregulate MMP1, MMP12, and MMP13 through negative transcriptional regulatory mechanisms (290).
3. Cytokines/chemokines
Chemokines have been shown to regulate several members of the MMP family. Indirect evidence shows that activation of CCR1, the receptor for RANTES/CCL5, MCP-1/CCL2, and SDF-1/CXCL12, induces secretion of MMP9 in primary human monocytes (126). In fibroblasts, CCL2 increases expression of MMP1 and MMP2. These increases are mediated by the proinflammatory cytokine IL-1α (308).
In cultured cells, the proinflammatory cytokines IL-1, IL-6, and TNF-α upregulate MMPs (132, 147, 308, 311). More importantly, this is also observed in vivo. IL-1β is produced in a biologically inactive form and can be activated by cleavage with MMP9 (237). When IL-1β production is ectopically expressed in the lung epithelium using a Clara cell secretory protein promoter (CCSP) as driver, it causes pulmonary inflammation, spontaneous overexpression of MMP9 and MMP12, disruption of elastin fibers in alveolar septa, and fibrosis in airway walls, a pathology resembling that of emphysema (137).
When IL-13, another proinflammatory cytokine, is administered intranasally, it induces airway hyperresponsiveness (AHR) and increases MMP2 mRNA in lung and both pro- and active MMP2 in bronchoalveolor lavage (49). IL-13 works through STAT6 to induce expression of MMP2 and MMP9 in the lung, and it requires the IL-4/IL-13 receptor-specific signaling chain IL-4Rα (46, 284; F. Kheradmand and D. Corry, unpublished data). Overexpression of IL-13 using a doxicycline-induced CCSP driver in the adult murine lung increases transcription and translation of MMP2, MMP9, MMP12, MMP13, and MMP14, in a MMP12-dependent manner (135). These mice also exhibit respiratory failure due to increased lung volumes and compliance, mucus metaplasia, and spontaneous inflammation. Some of the IL-13-dependent lung damage can be prevented by administration of the MMP inhibitor GM6001 (318).
IL-17, a T-cell-specific cytokine, has been shown to promote degradation of collagen in vitro through a process that is dependent on upregulation of MMP1, MMP3, and MMP13 (129). Furthermore, when recombinant IL-17 is administered in the airways of mice, it results in an increase in the concentration and activity of MMP9 in the BAL (220). These findings point to the link between adaptive immune system and activation of MMPs in diseases that feature tissue destruction such as arthritis of the joints and emphysema in the lung.
4. Endogenous inflammatory intermediates
Reactive oxygen species (ROS), including singlet O2, hydroxyl radical, superoxide anion, and hydrogen peroxide, are created by the partial reduction of molecular O2 (71). ROS are found in high levels in cigarette smoke and are generated by mitochondria, peroxisomes, and the plasma membranes and vacuoles of activated leukocytes (72, 110). Imbalances in ROS have been implicated in a variety of lung pathologies: too little may impair host defense and too much may result in tissue damage, disease, or cell death (72).
ROS can affect MMP gene expression through activation of several transcription factors. H2O2 production by mitochondria can activate MMP1 gene expression through activation of AP-1 and Ets-1 transcription factors (185). In addition, ROS indirectly activate MMP expression via JNK and ERK signaling pathways (185).
Reactive nitrogen species (RNS) are also involved in inflammatory reactions. In a rat model of pulmonary embolism, MMP2 and MMP9 are upregulated, perhaps indirectly as a result of neutrophil and macrophage influx to the pulmonary artery or directly from oxidative stress associated with pulmonary embolism. l-Arginine, a known oxygen radical scavenger, is also a substrate for nitric oxide synthase (138). Treatment with l-arginine diminishes MMP expression, while inhibition of nitric oxide synthase abrogates the effect of l-arginine (253), suggesting that MMP production is driven by NO or one of its partially reduced forms, such as peroxynitrite. In support of this hypothesis, endogenous NO has been shown to increase MMP1 promoter activity in cultured cells (113).
5. Environmental factors
Ample studies have linked mRNA expression of several proteinases in lungs to cigarette smoke exposure (42, 43, 305). Short (few hours) and prolonged (6 mo) exposure to smoke significantly increases expression of matrix metalloproteinase MMP9 and MMP12 mRNA in lung and antigen presenting cell populations (27). Lungs from mice exposed to cigarette smoke show increased volume fractions of alveolar septa and airspaces by morphometrical analysis and have increased MMP12 in lung macrophages compared with sham-treated mice, pathological changes consistent with those seen in human emphysema (281). These effects may be mediated by ROS, since as mentioned above, cigarette smoke contains high concentrations of ROS (110).
Air pollution may also contain high levels of ROS, but also particulate matter composed of heavy metals. Urban air pollutants have been shown to increase production of MMP2, MMP7, and MMP9 (261, 262). However, since air particulates are composite mixtures, the actual causative agent remains unclear. Interestingly, the MMP activity profile varies depending on the type or fraction of urban pollution administered (2), indicating that the response to pollution is complex and may be driven by several factors.
6. Pathogen-derived mediators
Destruction of lung by cavity-forming bacterial pathogens has been well documented. Most recently, conditioned media from Mycobacterium tuberculosis (MTb)-infected monocytes, but not direct infection with the organism, was observed to upregulate epithelial cell MMP1 promoter activity, gene expression, and secretion. These effects are dependent on p38 mitogen-activated protein kinase (MAPK) phosphorylation. These data reveal that MTb regulates the expression of MMP1, which can drive lung tissue destruction (64). In contrast in cultured human monocytic cell line (THP-1), infection with both MTb and its cell wall glycolipid stimulate expression and activity of MMP9 (227). The cellular mechanism involved in this process is dependent on signaling through mannose receptors, the activation of protein kinases and transcriptional activation by AP-1 (226). In addition to MTb, other bacteria, including Staphylococcus, can cause similar types of lung damage. Perhaps this example of pathogen-mediated increase in MMP expression is a general response of the host innate immune system to the invading organism (more detail in sect. VII).
7. Mechanical stress
In addition to all of the known factors mentioned above, expression of MMPs in the lung is also modulated by the mechanical stimuli that continuously change airway pressure as part of the dynamic breathing cycle. During pathological conditions that cause bronchial constriction, or during mechanical ventilation, lung pressure fluctuates greatly (99, 270, 276, 303). Under experimental conditions, exposure of rats to 4 h of high-volume ventilation results in upregulation of MMP2, MMP9, and MMP14 in the lung, by a mechanism that depends on the upregulation CD147 (74, 100). In cultured cells and in isolated mouse lungs that are stimulated with methacholine, a potent bronchial smooth muscle stimulant, mechanical stress is communicated from stressed (bronchoalveolar) to unstressed (fibroblasts) cells. This elicits increased production of MMP9 and TIMP1, through shedding of cell surface receptors and activation of EGFR (266, 274, 275).
The role of shear stress in upregulation of MMPs is not unique to the lung, since stimulation of cultured synovial cells by shear stress also elevates the mRNA level of MMP1, MMP3, and MMP13 and the three transcription factors c-Fos, Ets-1, and Ets-2, suggesting a new mechanism for tissue degradation in rheumatic joints (263). These effects of mechanical stress may ultimately be caused by production of ROS, since mechanical stress increases ROS in vascular smooth muscle cells. In that case, the ROS generated are responsible for increased gene expression of MMP2 and increased release of pro-MMP2 (86).
B. Posttranslational Regulation
Posttranslational modification of MMPs in the lung is a crucial step in regulating MMP action in vivo and is mediated through activation of latent MMPs. Several MMPs have soluble and cell surface forms, providing another level of regulation through compartmentalization/localization (201, 203). In addition, two endogenous families of inhibitory proteins, TIMP1−4 and α2-macroglobulin, are known to regulate MMPs at the posttranslational level and are briefly discussed in section XI.
1. Activation of MMPs
Secreted pro-MMPs remain inactive due to the interaction of the unpaired cysteine sulfhydryl group (cysteine switch) in the propeptide domain with the active site containing the zinc ion (256). Of the diverse means of activating MMPs, one common thread is thought to be thiol (sulfhydryl) reactivity, which disrupts the binding of the cysteine switch and exposes the active site of the enzyme (185). Removal of the propeptide containing the cysteine group by enzyme cleavage or disruption of the zinc-cysteine interaction can result in activation of the latent enzyme (256). Several MMPs are exceptions to this rule, including MMP11, MMP28, and all of the transmembrane MMPs (MT1-MT6-MMP), which are activated in the secretory pathway via subtilisin-like furin family serine proteinases (216). Organic mercury compounds, SDS, heat, acidic pH, and ROS have all been shown to activate MMPs in vitro (304). For instance, Pro-MMP1 is activated by H2O2, HOCl, and OH radicals (230). Pro-MMP1 becomes partially activated when ROS oxidate the sulfhydryl group, making an unstable pro-MMP1. At that point, the unstable pro-MMP1 is susceptible to autocatalytic cleavage of the propeptide resulting in active enzyme (185).
Activation of MMP2 on the cell surface critically depends on the binding of pro-MMP2 to MMP14 and TIMP2. This complex, which tethers the zymogen in place, allows a second active MMP14 to cleave the prodomain and release the activated MMP2 (260). This type of multistep, pericellular activation is now also shown to play a role in the activation of pro-MMP-7 where this soluble zymogen is captured and activated on the cell membrane through interaction with CD151, a member of the transmembrane 4 superfamily (248). Furthermore, MMP7 activity is blocked by antibodies to CD151 (248).
Several other proteinases regulate activation of MMPs, including trypsin and cathepsin G (304). In vivo, pro-MMP2 and MMP9 are activated by a chymase proteinase. Mice lacking the mast cell chymase proteinase 4 (mMCP-4) fail to process pro-MMP9 and pro-MMP2 to their active forms, resulting in increased collagen, ear thickness, and a higher content of hydroxyproline in the ear tissue compared with wild-type mice (271).
Plasmin and its activator urokinase plasminogen activator (uPA) can activate an array of proteinases, including many MMPs (304). This system is intricately bound to the MMP system, as the zymogen form of plasmin, plasminogen, is a known substrate for several MMPs (256). Adding further complexity to this system, plasmin not only activates MMPs, it can inactivate the tissue inhibitors of metalloproteinases (TIMPs) (131).
Activation of MMPs in vivo under physiological or pathological conditions, such as sepsis, requires multiple steps, but the exact mechanisms remain unknown. In experimental endotoxemia, lipopolysaccharide (LPS), TNF-α, granulocyte colony stimulating factor (G-CSF), and IL-8 result in release of pro-MMP9 in the blood, while activated MMP9 and MMP2 are not detected (224). However, whole blood analysis of MMPs in patients with gram-negative sepsis shows activated forms of MMP2 and MMP9, the levels of which correlate with the severity of sepsis. This indicates that while proinflammatory mediators such as LPS can acutely induce the inactive form of MMPs, the activation process in response to bacterial infection is more complex and regulated differently in vivo (224).
Activity of MMPs can be altered in several ways. The relative fluxes of nitric oxide and superoxide at sites of inflammation are shown to differentially modulate MMP2 activity, because superoxide-derived metabolites increase activity of MMP2 in vitro, while peroxynitrite, which is formed from the interaction between superoxide and nitric oxide, inhibits MMP2 activity (204). MMP activity may also be altered by environmental pollutants. Rats exposed to a combination of particulate matter and ozone experience increased MMP2 activity in bronchoalveolar lavage (BAL) (272).
2. Localization of MMPs
Compartmentalization of MMPs in inflammatory cells is the most obvious example of this type of regulation. For example, pro-MMP8, pro-MMP9, and pro-MMP25 are packaged into peroxidase-negative granules within neutrophils to be released upon leukocyte activation (67). Peroxidase-positive azurophil granules contain both activators of MMPs, such as serine proteinases and ROS-generating enzymes, and inactivators of MMPs, such as thrombospondin (67). MMP9 and MMP12 can be released from macrophages in response to activation.
In addition, chemokines have been shown to regulate MMP activation by altering their localization. MMP14 is highly expressed on lung endothelial cells, and in its absence, there is decreased angiogenesis induced by the chemokines CCL2 and CXCL8 (6, 78, 320). In human endothelial cells, CCL2 and CXCL8 increase cell surface expression and clustering of MMP14 (78). The cell surface expression pattern of MMP14 is one potential regulator of MMP2 activity, since MMP2 activation would then be dependent on colocalization with MMP14 (93).
Another level of regulation is that MMPs can be either free in the cytosol or bound to the surface of a cell. The cell surface-bound forms of at least two MMPs are thought to enhance inflammatory cell functions. For example, MMP9 is secreted but also has a cell surface-bound form. This active, cell surface form of MMP9 is expressed on neutrophils in response to proinflammatory mediators and can cleave gelatin, type IV collagen, elastin, and α1-proteinase inhibitor in vitro (202). However, in contrast to soluble MMP9, membrane-bound MMP9 is substantially resistant to inhibition by TIMPs and may be responsible for the increase in pericellular proteolytic activity necessary for the proper action of neutrophils in vivo, although this hypothesis has not been formally tested (202). Similarly, neutrophils exposed to proinflammatory cytokines increase cell surface expression of active MMP8. Pericellular collagenase activity from activated neutrophils is attributed to membrane-bound MMP8, and its localization on the cell surface confers resistance to TIMP inhibition (203). MMP8 may be anti-inflammatory during acute lung injury, because MMP8 null mice given intratracheal LPS have significantly greater accumulation of neutrophils in the alveolar space than wild-type mice (203). Together with the cell surface activation of MMP2 and MMP7, these experiments suggest that localization of MMPs to the cell surface is a general means of regulating MMP activity. Additionally, MMP1, MMP2, and MMP9 have been found to bind to specific cell surface proteins such as αvβ3-integrin, α2β1-integrin, and CD44, respectively (30, 259, 314; for a recent review, see Ref. 213).
IV. SPATIOTEMPORAL EXPRESSION OF MATRIX METALLOPROTEINASES IN LUNG: BRANCHING MORPHOGENESIS AND ALVEOLAR DEVELOPMENT
Lung development is a multistep process, beginning with a group of endodermal cells, which interact to form the lung primordium, and this in turn forms the trachea. The differentiation of the cells is directed by cell-cell interactions and regulated by the genes Gli, Shh, and Nkx2.1 (57). From this point, the lung grows through branching morphogenesis, angiogenesis, and alveolar generation [for detailed reviews on the molecular regulation of lung morphogenesis, see Cardoso (37) and the ATS official workshop document (1)].
Very little is known about regeneration of lung after injury/destruction, but it is likely that repair events mimic those during development (317). Thus elucidating the mechanisms involved in alveologenesis will yield information that may facilitate understanding the repair process. In this regard, MMPs represent excellent targets, since these enzymes are critical for alveolar development and often, in lung injury, many of the same MMPs, such as MMP7, MMP8, and MMP9, reappear (303). In many instances, it is unclear whether the presence of MMPs is destructive or beneficial. Knowing how these enzymes function during development will help us to further our understanding of lung repair, possibly providing insight on how to repair emphysematous lungs and lungs damaged from chronic inflammation or injury.
Various MMPs participate in the development of lung from the very beginnings of lung bud formation throughout alveolarization, where some MMPs are more prominent players than others. For instance, in the developing mouse lung, there is no detectable MMP3, MMP9, MMP10, or TIMP1 at any stage (122). Expression of mRNA for MMP15 and MMP20 in fetal and adult lungs shows no significant changes with developmental stage, whereas MMP2 and MMP14 decrease with age (229). MMP21 is expressed transiently during embryonic development with expression peaking around days 11−13.5 as detected by PCR and may be associated with neuronal cells (3, 160). Amidst all the variation, two key MMPs stand out as being of major importance during development: MMP2 and MMP14 (MT1-MMP; Fig. 2).
FIG. 2.

MMP expression in mouse lung development. MMP2, MMP14, and an MMP inducer, CD147, are constitutively expressed in all five distinct stages of lung development: primary budding (E9.5−11.5), pseudoglandular (E12−16.5), canalicular (E17−18), saccular (E19-P5), and alveolar (P5-P21). Expression of these MMPs and their inducer tapers off with the completion of lung development. Expression of most other MMPs occurs following the postnatal period and in adult lungs. In inflammatory lung diseases, cells of the hematopoetic origin (e.g., macrophages, neutrophils, eosinophils, lymphocytes) home to the lung and express MMP8, MMP9, and MMP12. Alternatively, many MMPs are induced in the alveolar epithelial cells in response to exposure to environmental agents. For example, alveolar and bronchial epithelial cells express MMP7 and MMP9 induced by exposure to pathogens or toxins, respectively (see Refs. 3, 33, 34, 66, 122, 160, 165, 229).
MMP2 is constitutively expressed throughout lung development (33; rats, Ref. 142; humans, Ref. 165). MMP9 is upregulated after birth (34). MMP upregulation may be mediated by CD147, which has been shown to be highly expressed in lung on embryonic days 13.5 and 15.5 in mice (66), but is minimal in adult lung (39).
A. Branching Morphogenesis
Branching morphogenesis of bronchiole structures takes place during the pseuodoglandular stage, which is characterized by dichotomous branching of epithelial tubes (229). This stage occurs from 7−16 wk gestational age in humans and embryonic days 9.5 −16.5 in the mouse. In the mouse, MMP2, MMP14, and TIMP2 are expressed on days 11.5 and 13.5 (122). In humans, MMP1, MMP9, TIMP1, TIMP2, and TIMP3 are detected by immunohistochemistry in fetal epithelium, while MMP2, MMP1, TIMP2, and TIMP3 are expressed only in pulmonary vascular endothelium and media (165).
Branching morphogenesis can be inhibited with high concentrations of the broad spectrum metalloproteinase inhibitor GM6001 (81, 122). Yet, a lower concentration of GM6001 enhances branching (81). One model of branching morphogenesis suggests that cleft formation is driven by accumulation of TGF-β, which stimulates ECM deposition and directs branching to either side of the built-up ECM (37, 103). This process is facilitated by TGF-β-mediated inhibition of MMPs. In support of this hypothesis, TGF-β1 and TGF-β3 inhibit expression of MMP1 and upregulate expression of TIMP1 in fibroblasts (62). However, TGF-β family members do not regulate MMP2, which is highly upregulated during lung development. This relationship points to other, as yet unexplored, and complex interactions between MMP-ECM and growth factors.
Another model for branching morphogenesis suggests that low tissue PO2 could inhibit MMP degradation of tenascin C (TN-C), allowing for deposition of this important ECM component. In support of this model, low tissue PO2 (3%) increases branching in lung, increases expression of TN-C, and also decreases activity of MMP2 (but not its mRNA synthesis), possibly through decreased availability of ROS (79). Inhibition of MMPs with GM6001 results in decreased deposition of TN-C (79), confirming the role of MMPs in lung branching in explants. Potentially, TGF-β inhibition of MMPs may promote TN-C deposition, by working in concert with low PO2 to promote branching, since TN-C stimulates cell proliferation in human lung fibroblasts, and this response is amplified in the presence of TGF-β (236).
MMPs are shown to stimulate cellular migration, which is another plausible mechanism for their role in branching morphogenesis. MMP14 is expressed in lung tissue early in embryogenesis. MMP14 null embryos after embryonic day 16.5 have a subtle defect in peripheral bronchiolization. However, the role of MMP14 in branching is minimal and redundant (193). Interestingly, lung endothelial cells from the MMP14 null mice show reduced migration (193), possibly due to altered processing of laminin γ2-chain. MMP14 and MMP2 cleavage of laminin γ2-chain generates chemotactic fragments containing EGF-like motifs that are present in tissues undergoing remodeling (80) and can attract epithelial cells (128). This process suggests another mechanism for MMP14's role in the developing lung. MMP-mediated release of cryptic, bioactive fragments and neoepitopes from ECM macromolecules is detailed in a recent review (181).
B. Angiogenesis
The canalicular stage is characterized by extensive angiogenesis and the linking of bronchiolar structures with their capillary interface. In humans this stage occurs from 16 to 24 wk and in mice from embryonic days 16.5 to 17.5. In the human lung, MMP1 and MMP9 are detected throughout all stages. The role of MMPs in angiogenesis is nicely addressed in a recent review by Brauer (28), and a comprehensive review on the role of MMPs in intimal thickening was recently published by Newby (188); thus we will only provide highlights relating to lung development.
Fourteen of the 25 vertebrate MMPs are expressed in vascular smooth muscle cells and endothelial cells, indicating the importance of MMPs in vascular biology (188). During angiogenesis, fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), and their receptors are important for proper formation of blood vessels; these bioactive molecules can be released from ECM by MMP cleavage (28). Conversely, FGFs and VEGFs can increase expression of MMPs (5, 134). Mice lacking MMP9 have delayed angiogenesis that is likely corrected through compensation by other MMPs, resulting in normal adult mice (286). The dysregulation of angiogenesis is hypothesized to be lack of a critical growth factor in the absence of MMP9. Alternatively, MMP activity can abrogate angiogenesis. Degradation of several ECM components, including plasminogen, thrombospondin, and collagens IV, XV, and XVIII, can generate fragments that are inhibitors of angiogenesis, through MMP-dependent mechanisms, giving MMPs regulatory power over angiogenesis (217).
C. Alveolarization
The final stage, alveolarization, is long lasting (from 36 wk to 3 yr of age in humans) and is characterized by alveolar development and maturation of the capillary network. In mice this stage lasts from postnatal day 5 to day 30 and peaks at day 14 (229). This process requires coordination of ECM remodeling with epithelial morphogenesis and capillary growth. MMP14 (MT1-MMP) is required for alveolarization, because mice lacking this enzyme have decreased alveolar surface area and enlarged airspaces, attributes which could be caused by failure of capillary network formation and defective distal airspace branching (193).
However, the bronchioles of these mice branch properly, although defects in alveoli are apparent from the late saccular stage (8, 193). This led to the hypothesis that the abnormal alveolization results from problems with saccular development and septation (193). Interestingly, these are exactly the same stages impacted by altering TGF-β and angiogenesis (163). In addition, treatment with doxicycline, which had MMP inhibitory activity, during postnatal alveolar development (day 4 to day 13) in rats results in enlarged alveolar spaces compared with control rats (107), confirming the role of MMPs in alveolar development.
V. RESURGENCE OF MATRIX METALLOPROTEINASES IN LUNG DISEASES: ACUTE AND CHRONIC
In animal models of inflammatory lung diseases, the association between the onset of inflammation and the acute upregulation of MMPs is well established. For example, in models of acute allergic lung disease, MMP upregulation occurs within the lung parenchyma, and in the alveolar fluid, and is required for resolution of lung inflammation (48, 49, 318). However, in cases of chronic inflammatory lung diseases, the role of MMPs remains to be determined. Although very few animal models have been developed that mimic the chronic inflammation observed in human disease, transgenic models of MMP overexpression have provided evidence that chronic elevation of these enzymes in the lung can cause tissue destruction similar to human emphysema (56). Constitutive expression of human MMP1, driven by a haptoglobin promoter, causes disrupted alveolar wall formation in mice, indicating that the imbalance of this collagenase, which has no known elastin degrading properties and limited expression in human lung, can destroy normal murine lung architecture (56).
In a mouse model of hyperoxia-induced lung disease, direct O2 toxicity can result in alveolar type I cell injury and death. Lung damage is mediated by the synthesis and release of MMP9 and MMP12 by neutrophils and alveolar macrophages. Overexpression of STAT3C in alveolar epithelial cells results in increased survival, decreased capillary leakage, and suppressed neutrophil infiltration. These deleterious effects were prevented through STAT3C-mediated inhibition of MMP9 and MMP12 (146).
Other indirect evidence for the role of chronic exposure to MMPs in structural lung damage comes from transgenic overexpression models of IFN-γ, the canonical Th1 cytokine, and IL-13, a key Th2 cytokine. Inducible and targeted expression of IFN-γ in the lung results in increased alveolar space, lung volume, and pulmonary compliance and is associated with excessive expression of MMP12 and MMP9 (244, 291). The role of IFN-γ in modulation of MMPs in vivo is most likely an indirect one, because prior studies show that IFN-γ downregulates MMPs but CXCL10 (IFN-inducible protein of 10 kDa, IP10) upregulated MMP12 in alveolar macrophages (87, 108). Similarly, inducible and targeted expression of IL-13 in mice is associated with increased MMP2, MMP9, and MMP12 expression in the lung, and these mice also develop alveolar and lung enlargement, compliance variations, and respiratory failure, ultimately leading to death, which is prevented when they are crossed with MMP9- or MMP12-deficient mice. This result strongly suggests that the alveolar damage is mediated by MMPs (135).
MMP12 null mice exposed to long-term cigarette smoke fail to recruit macrophages into their lung and do not develop emphysema compared with wild-type litter-mates (102). These findings are one of the most definitive studies to show that chronic exposure to cigarette smoke induces MMP12 expression in macrophages, which is required for the development of emphysema in mice.
MMP7, an epithelial specific MMP, is constitutively expressed at low levels in the airway epithelia, but its expression is upregulated under a variety of pathological conditions in the lung (60, 211). Short exposure of lung epithelial cells to Pseudomonas aeruginosa or its flagellin results in a strong upregulation of MMP7 that is essential in eradicating this organism, because mice deficient in MMP7 fail to clear the pathogen efficiently (302). However, under chronic conditions, the resurgence of MMP7 may create a more ominous effect. For instance, in cystic fibrosis, MMP7 expression is increased in airway epithelial cells and induced in alveolar type II cells (60). Furthermore, osteopontin, an ECM molecule that distinguishes fibrotic from normal lungs in microarray analysis, upregulates MMP7 expression in lung epithelial cells and may exert a profibrotic effect in idiopathic pulmonary fibrosis (208). These results highlight the potentially different roles of MMPs from acute to chronic conditions.
Pulmonary fibrosis is a chronic condition that results in loss of elastic recoil of the lung. Although evidence for the role of MMPs in the pathogenic processes associated with pulmonary fibrosis and injury to basement membranes is indirect, nonetheless a few studies highlight the potential function of MMPs in this condition. For example, MMP9 expression is increased in the lung in response to intratracheal instillation of bleomycin; however, initiation or evolution of the inflammatory and fibrotic changes that occur are independent of MMP9, because MMP9 null mice have inflammatory cell infiltration and fibrosis equivalent to wild type (19). In addition, MMP9 null mice have disrupted bronchiolization after bleomycin-induced injury, suggesting that MMP9 may be important for repair and may play a more serious role in the long-term recovery from lung injury (19). In addition, in the chronic phase of bleomycin-induced pulmonary fibrosis, attenuation of MMP2 and MMP9 expression and activity in macrophages from mice lacking the C-C chemokine receptor 2 (CCR2−/−) is accompanied by reduced lung remodeling and hydroxyproline content of lung tissues (198), suggesting a role for MCP1 and macrophage infiltration in fibrotic disease.
Another instance of MMP resurgence in chronic inflammation is via CD147. As we discussed earlier in section IV, CD147 is present in the lung during development, but its expression drops to low levels in normal adult lung (39). CD147 has been shown to increase the expression of MMP1, MMP2, MMP3, MMP9, and MMP14 in the lung either directly or through stimulation of the cells adjacent to the tumor mass (90, 264, 265). Not surprisingly, early increases of CD147 have been found in several models of lung damage, such as ventilator-associated acute lung injury (74), LPS (7), and bleomycin-induced lung injury (20).
VI. MATRIX METALLOPROTEINASES: REDUNDANT VERSUS COMPENSATORY MECHANISM OF ACTION
The concept that MMPs are redundant in action originates from two lines of evidence. First, researchers have found enormous substrate overlap from in vitro cleavage assays, and the classification of MMPs based on their biochemical similarities suggests functional redundancy. Additional evidence for redundant behavior of MMPs comes from the observation that, in mice, single null mutations of MMPs do not generally result in embryonic lethality (8, 112, 252). With the notable exceptions of MMP14 knockout mice, which die by 3 wk of age from unknown cause with severe skeletal abnormalities and lung defects (193), and MMP2 knockout mice, which have less-severe nonlethal bone and alveolar defects (122), mice lacking other MMPs appear fairly normal, suggesting that there is compensation from other MMPs. Indeed, MMP9 null mice have transient developmental problems with angiogenesis at the growth plate, but the defect is reversed by ∼3 wk of age resulting in normal adult mice (286). It is unclear which MMP may be providing the compensatory action.
Despite the superficial appearance of redundancy, the differential tissue distribution of these enzymes suggests that they have distinct and nonoverlapping functions in vivo (3, 48, 122, 165). Even more importantly, the apparent compensatory and/or redundant action by other members of the MMP family during normal conditions falls apart when the same null mutations are tested under pathological conditions (48, 49, 144, 166, 170, 302).
Several disease models support the hypothesis that MMPs do not have redundant functions, at least in immune response. First, in an asthma model (Fig. 3), decreased BAL inflammatory cells are observed in MMP2−/− mice; however, the decrease is entirely accounted for by fewer eosinophils (48). Thus observed decreases in neutrophil and eosinophil counts in the BAL of mice deficient in MMP9 reveal similar but nonoverlapping specificity of MMP9 and MMP2 (48).
FIG. 3.

Role of MMPs in adaptive immunity. In response to inhaled allergens, Th2 inflammatory cells home to the lung and initiate allergic lung disease that can manifest in many pathological features such as accumulation of eosiniphils, basophils, and neutrophils; increases in mucus production; goblet cell metaplasia; and narrowing of the airways. Increases in concentration of interleukin (IL)-4 and IL-13, the canonical Th2 cytokines, also orchestrate upregulation of MMPs in the lung mesenchymal cells (MMP2, MMP3), hematopoietic cells (MMP9, MMP12), and epithelial cells (MMP7). In experimental models where MMPs are inhibited or in mice deficient in MMP2, MMP9, or MMP2/MMP9 there is an accumulation of inflammatory cells in the lung parenchyma and that predisposes mice to death from asphyxiation (see Refs. 48, 49, 88, 170, 186).
MMP2 and MMP9 also play nonredundant roles in the pathogenesis of obliterative airway disease (OAD), a known complication of chronic lung allograft rejection. In the murine model of OAD, MMP2 and MMP9 are upregulated, and researchers find a significant survival advantage for MMP9 null, but not MMP2 null mice. The mechanism for this protection is related to the enhanced T-cell alloreactivity and dendritic cell stimulatory capacity in MMP9 null mice (68). Thus inhibition of MMP9, but not MMP2, may represent a target for the therapeutic intervention of chronic lung allograft rejection.
VII. MATRIX METALLOPROTEINASES IN INNATE AND ADAPTIVE IMMUNE FUNCTION
A. Evidence for Host Defense
MMPs are now being recognized as one of the critical mediators for host defense. In recent years, mammalian defensins have been shown to function as effectors of antimicrobial innate immunity. Defensins are small peptides expressed in the leukocytes, mucosal surfaces, and epithelia as prepropeptides (241) from the Paneth cells and require activation by a convertase. In the case of murine α-defensin, the convertase is MMP7, an epithelialspecific MMP (301, 302). Mice deficient in MMP7 die more rapidly when exposed to gram-negative bacterial pathogens, and the mechanism for this increased susceptibility is lack of the active form of α-defensin in the gut (302). Whether other members of the MMP family act as convertases for other defensins is not known, but based on emerging evidence from animal models of bacterial infection, MMP7 is strongly upregulated as part of mucosal immunity in response to infection (153, 213). Studies in mice have shown that MMP2 and MMP9 are upregulated in response to MTb infection and may be critical in clearance of this organism in the lung (65, 227). Mice infected with MTb and treated with a BB-94, a general inhibitor of MMPs, develop an aberrant Th2 immune response and also develop increased IL-4. This treatment resulted in rapid progression of the disease (105).
Other evidence for the role of MMPs in lung innate immune system originates from the report that pulmonary surfactant proteins regulate the expression of several MMPs in alveolar macrophages. Lung surfactant, which is primarily composed of 90% lipids and 10% proteins, coats the distal airspaces to reduce the alveolar surface tension, a process that is essential in prevention of atelectasis, and in host defense against environmental pathogens (92). Of the four surfactant-associated proteins that have been cloned in mammals, two with hydrophilic properties, surfactant protein (SP)-A and SP-D, play critical roles in innate host defense and adaptive immunity (156, 306). Recombinant SP-D upregulates expression of MMP1, MMP3, and MMP12 in human alveolar macrophages (273). SP-D-mediated upregulation of MMPs is independent of the production of TNF-α or IL-1β, two potent inducers of MMPs (273). One hypothesis is that specific upregulation of MMPs in alveolar macrophages by SP-D enhances their antimicrobial function in vivo by directly activating macrophages and stimulating the production of more MMPs, which could result in formation of bioactive proteins (Fig. 4).
FIG. 4.
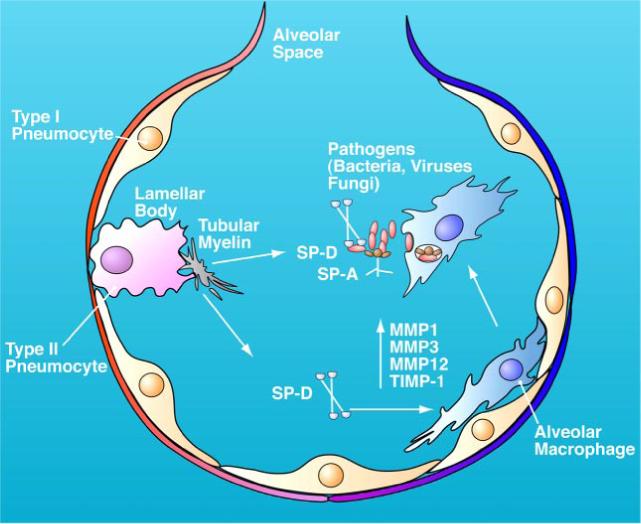
Role of MMPs in innate immunity: clearance of lung pathogens. Schematic diagram of the interaction between alveolar epithelial type II cells, macrophage, MMPs, and surfactant proteins in the alveoli. Alveoli are the gas exchange unit in the lung and are lined with pulmonary surfactant, a dynamic and lipid-rich fluid that is associated with four proteins, two of which (SP-A, SP-D) play important roles in bacterial clearance. SP-D is an in vivo target of cleavage by MMPs; induces expression of MMP1, MMP3, and MMP12; and activates alveolar macrophages. SP-A and SP-D can bind the offending pathogens that enter the airway and enhance their clearance by activated macrophages in the lung. Whether induction of MMPs in alveolar macrophages directly affects bacterial killing is unknown (see Refs. 156, 273, 296, 306).
SP-D and SP-A, when not associated with lipids, can bind bacteria, fungi, and viral particles that enhance opsonization of the microorganisms (306). Lung surfactant levels decrease during acute pulmonary inflammation due to degradation by inflammatory cells, including neutrophils and macrophages (157). Additionally, SP-A and SP-D are putative in vivo substrates for MMP2 and MMP9, but whether the cleaved products of these proteins could alter the antimicrobial function of the surfactant proteins is unknown (83a). Although these findings are quite intriguing, the validity and/or functional significance of these observations has not been explored in vivo.
SP-D-induced upregulation of MMPs in vitro appears to be in sharp contrast to multiple reports that indicate the absence of SP-D in mice results in accumulation of activated macrophages and proinflammatory enzymes including MMP2, MMP9, and MMP12 in the lung (194, 296). The phenotype of SP-D null (SP-D −/−) mice is quite complex, but these mice show marked inflammation and histological changes in the lung that have similarities with human emphysema (296). However, it is not clear if the absence of SP-D results in inefficient turnover of surfactant proteins or increases susceptibility to infection, which could contribute to the inflammatory changes seen in the lung of SP-D −/− mice. Consistent with this possibility, SP-D −/− mice fare worse during bacterial infection due to decreased cell-surface expression of CD14, a pattern recognition receptor present on many innate immune cells (242). Macrophages without CD14 presumably are unable to form the complex receptor (with TLR4 and MD-2) that mediates the cellular responses to pathogens. However, soluble CD14 increases in BAL, likely through increased shedding by MMP9 and MMP12. Because alveolar macrophages from SP-D null mice have increased MMP production and also have 10-fold higher H2O2 production, it is possible that SP-D regulates MMPs by acting as an antioxidant in macrophages. In support of this hypothesis, treating SP-D −/− mice with antioxidants inhibits MMP production (296, 313). Finally, a balance between SP-D and MMPs expression is critical in host defense, since overexpression of SP-D in an immunocompromised mouse infected with Pneumocystis resulted in a worse outcome when compared with wild-type mice (287).
Another pulmonary surfactant family member, SP-C, has also been found to play a role in immunity. SP-C −/− mice have alveolar defects that predispose them to pulmonary fibrosis and pneumonitis through increased macrophage infiltration and MMP2 and MMP9 production (82). When SP-C −/− mice are subjected to bleomycin-induced lung injury, they show increased mortality, increased neutrophil influx, and significantly delayed tissue repair compared with wild-type mice, suggesting that SP-C plays a major role in leukocyte trafficking and lung remodeling (139).
Microbial proteinase activation of MMPs may be an essential part of the immune response for elimination of bacterial infections in vivo (see Table 2 in Ref. 213 for summary). Some bacterial exopeptidases have been shown to activate zymogens of MMPs through proteolytic processing of their autoinhibitory domains and by formation of ROS/RNS (activation of MMPs by ROS is discussed in section IIIB1). Thus proper expression/activation of MMPs may be of potential therapeutic value for various infections and inflammatory lung diseases (197).
MMP2 and MMP9 are also implicated in host defense against Angiostrongylus cantonensis, a parasitic worm infestation that causes granulomatous fibrosis reactions in the lung (109). Gelatin zymography revealed that the active forms of MMP2 and MMP9 during granulomatous fibrosis are significantly higher than at other times during a 90-day infestation in rats (109). It remains unclear whether the presence of these gelatinases is critical to the confinement of the worm infestation, which would provide a mechanism for natural defense against the infection.
A more clear-cut role for MMPs in resistance against gram-negative pathogens was recently shown using single deletion of MMPs or their inhibitor TIMP1 in mice. TIMP1 null mice showed increased resistance to Pseudomonas aeruginosa infection in both cornea and lung, suggesting that activity of certain MMPs makes mice more susceptible to these infections. To identify which MMP is responsible for the increased resistance, researchers created double null mice by crossing TIMP1 null mice with mice deficient in MMPs known to be inhibited by TIMP1 in vivo, including MMP2, MMP3, MMP7, MMP9, and MMP12. MMP9/TIMP1 double null mice completely lose their resistance to infection, and MMP3/TIMP1 and MMP7/TIMP1 mice have decreased resistance, while MMP2 and MMP12 do not play a role (140). Although these studies did not directly examine the mechanism involved in the protective effect of MMPs in infection with Pseudomonas, other studies have shown that shedding of syndecan1 from airway epithelial cells, which is dependent on cleavage by MMP7, is critical for defense against this organism (169, 210).
B. MMPs as Acute Inflammatory Mediators
In addition to their complex roles in innate immunity, MMPs are being implicated in adaptive immunity as well. For example, using a mouse model of asthma, recruitment of allergic inflammatory cells and AHR is independent of MMP9 and MMP2 (Fig. 5). However, pathological examination of lungs in the same mice indicates that, relative to wild-type mice, lack of MMP9 or MMP9 and MMP2 increases inflammatory cells in lung parenchyma (48). Differential cell counts of the BAL cells reveal a significant reduction in total number of eosinophils and neutrophils in the alveolar lining fluid of MMP9 null and MMP2/MMP9 double null mice. Thus several features of the asthma phenotype are preserved in the absence of MMP2 and MMP9, but lack of either or both enzymes disrupts trafficking of inflammatory cells into the airway, and the excess inflammatory cells that accumulate abnormally around bronchovascular bundles in the lung parenchyma produce IL-4 and IL-13 (48).
FIG. 5.

Proteolytic effector function: chemokine gradient formation. The schematic diagrams depict the migration (extravasation, intraparenchymal homing, and transepithelial egression) of allergic inflammatory cells recruited to the lungs under normal conditions (top) or in the absence of MMPs (bottom). Cellular migration is shown progressing from right to left, with recently extravasated cells (including T cells, monocytes, eosinophils, and mast cells) traversing the pulmonary interstitium and the airway epithelium to enter the airway lumen, where they are cleared in the wild-type (WT) mice (top) and less so in MMP null mice (bottom). Interstitial inflammatory cells are recruited to the lumen by establishing a transepithelial chemotactic gradient in which chemokines (CCL) are strongly expressed in the lumen and on the apical surface of epithelial cells relative to the interstitium. Lack of MMPs disrupts the formation of this chemokine gradient and impairs migration of cells at the points marked “X”.
In a mouse model of acute, bleomycin-induced lung injury, neutrophils accumulate in the interstitium of the lung in MMP7 null, but not wild-type mice (145). The mechanism for the lack of neutrophil migration is aberrant shedding of syndecan-1 that binds KC, the mouse homolog of human chemokine gro-α (CXCL-1). Thus MMP7-mediated shedding of syndecan-1/KC complexes from the airway epithelium facilitates neutrophil migration to sites of injury (145). MMP8 and MMP12 also play a role in leukocyte trafficking and survival. Tracheal instillation of MMP-12 results in a transient increase in Th1 chemokines and cytokines such as IL-6, TNF-α, macrophage inflammatory protein-1α (MIP-1α), monocyte chemoattractant protein (MCP-1), and KC. This treatment also results in acute recruitment of neutrophils and macrophages to the lung, as determined by cell counts in BAL fluid (186). One explanation is that MMP12 releases bioactive peptides through degradation of the ECM protein components.
In response to allergen challenge, MMP8 −/− mice develop airway inflammation characterized by an increased neutrophilic and eosinophilic infiltration in the lung, indicating that MMP8 is not critical for development of asthma (88). Similar to the findings in MMP2 and MMP9 null mice, MMP8 deficiency in a mouse model of asthma is also associated with increased levels of IL-4 and anti-ovalbumin IgE and IgG1. But, unlike the finding in MMP2 and MMP9 null mice, inflammatory cell apoptosis decreases in the lungs from MMP8 −/− mice (88). However, the mechanism of MMP8-mediated inflammatory cell survival is unclear. These findings demonstrate the importance of MMPs in inflammatory cell function and highlight the lack of information regarding their mechanism of action (Fig. 6).
FIG. 6.
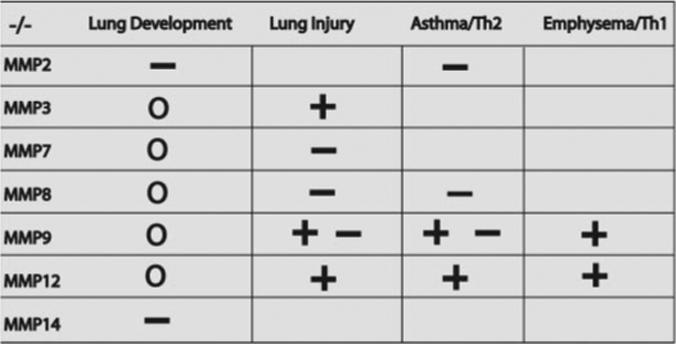
Effect of MMP deletion in lung development and in animal models of lung diseases. In MMP14 and a small subset of MMP2 null mice (back-crossed to C57BL/6), developmental lung defects (−; disadvantageous) have been reported (8, 112, 122, 193). Gene deletion in all other MMPs (0; no effect) has not been shown to result in abnormal lung development. Absence of MMP3 (144), MMP9 (68), and MMP12 (186) has been shown to be advantageous (+; beneficial) in a mouse model of acute lung injury. Whereas others have shown that absence of MMP7 (302), MMP8 (203), and MMP9 (19) is disadvantageous in a mouse model of acute lung injury (−). In the absence of MMP2 (49), MMP8 (88), and MMP9 (48, 170), mice develop exaggerated allergic inflammation in an acute model of asthma that is consistent with a disadvantage (−); however, inhibition of MMP9 showed a beneficial (+) effect in other studies (68). In the absence of MMP9 and MMP12, there was a consistent beneficial (+) effect in a mouse model of smoking-related lung disease. Blank area indicates that mice deficient in the MMPs have not been tested or the results of the studies have not been published to date.
VIII. PROTEOLYTIC EFFECTOR FUNCTION: MECHANISM OF ACTION IN INFLAMMATION
Several physiological substrates for MMPs have been described using a variety of screening methods. Many of these substrates are chemokines or their receptors (Table 1). For instance, the MMP2 COOH-terminal domain used as bait in a yeast two-hybrid screen identified MCP-3 as an MMP2 substrate (171, 172). Furthermore, using oriented peptide libraries to identify potential MMP cleavage sites, researchers have identified several integrins and proteoglycans as MMP substrates (279).
TABLE 1.
Partial list of chemokines and their receptors that are either cleaved by MMPs or their interactions can activate/upregulate MMPs
| Chemokine | Alternate Name | Receptor | MMP Substrate | Regulation of MMP by Chemokines/Receptor Interaction |
|---|---|---|---|---|
| CCL2 | MCP-1, JE | CCR2, 10 | Y(173) | ↑ MMP1, MMP2 |
| ↑ Clustering of MMP14 (78) | ||||
| CCL3 | MIP-1α | CCR1, 5 | U | |
| CCL4 | MIP-1β, HC21 | CCR5, 8 | U | |
| CCL5 | RANTES | CCR1, 3, 5 | U | ↑ MMP9 (126) |
| CCL6 | C10, MPR-1 | CCR1 | U | ↑ MMP2, 9 (282, 283) |
| CCL7 | MCP-3, MARC | CCR1, 2, 3 | Y(173) | ↑ MMP9 (126) |
| CCL8 | MCP-2 | CCR1, 2, 3, 5 | Y(173) | |
| CCL9/10 | MIP-1γ | CCR1 | U | |
| CCL11 | Eotaxin-1 | CCR3 | Y(83a) | ↑ MMP2 (127) |
| CCL13 | MCP-4, CKβ10 | CCR2, 3 | Y(173) | |
| CCL17 | TARC | CCR4 | Y(83a) | |
| CCL23 | MPIF, CKβ8 | CCR1 | U | ↑ MMP2 (251) |
| CCL25 | TECK | CCR9 | U | ↑ MMP1, 2, 9, 10, 11, 13 (249) |
| CXCL5 | ENA-78 | CXCR2 | Y(11, 283) | |
| CXCL6 | GCP-2 | CXCR1, 2 | Y(283) | |
| CXCL7 | NAP-2, CTAP-III | CXCR2 | Y | MMPs activate (130) |
| CXCL8 | IL-8, NAP-1, MIP-2, MDNCF | CXCR1, 2 | Y | ↑ Clustering of MMP14 (78) |
| CXCL9 | MIG | CXCR3 | U | ↑ MMP12 (87) |
| CXCL10 | IP-10, CRG-2 | CXCR3 | U | ↑ MMP12 (87) |
| CXCL11 | I-TAC, beta-R1, H174, IP-9 | CXCR3 | U | ↑ MMP12 (87) |
| CXCL12 | SDF-1, PBSF | CXCR4 | Y(171) | ↑ MMP2 (127) |
| ↑ MMP9 (126) | ||||
| CXCL15 | Lungkine | ? | Y(83a) |
MMP, matrix metalloproteinase; Y, yes; U, unknown. Reference numbers are given in parentheses.
Using a novel functional proteomics approach, we identified several proteins in the BAL fluid that are cleaved by MMP2 and MMP9 and are essential for regulating inflammatory pathways in experimental asthma, including S100A8, S100A9, and Ym1. These proteins all have chemotactic properties and are upregulated in allergic lung inflammation. In addition, using function-blocking antibodies to S100A8 and S100A9, inflammatory cell egression into the alveolar space is reduced, pointing to the importance of these proteins in resolution of allergic inflammation (83a). Searching for other in vivo substrates for MMPs that function as bioactive molecules can aid in detection of new biochemical pathways that regulate allergic lung disease and provide novel insight into the function of proteinases in allergic lung disease (Fig. 5).
Generation and inactivation of bioactive molecules by MMPs during lung inflammation could alter cellular function and impact lung function. For instance, MMPs can cleave many serine class proteinases, such as plasminogen and u-PA that are potent mitogenic factors for lung fibroblasts (31). Similarly, MMP2, MMP7, MMP9, and MMP12 can cleave α1-proteinase inhibitor, the major endogenous serine class proteinase inhibitor and the first gene whose lack of function has been implicated in smoke-induced COPD and emphysema (148, 243). MMP cleavage inactivates the α1-proteinase inhibitor and also generates a new biologically active fragment that is a powerful neutrophil chemoattractant factor (12). Since α1-proteinase inhibitor is a potent endogenous inhibitor of neutrophil elastase, its degradation by MMP9 and MMP12 may indirectly activate neutrophil elastase, resulting in elastin degradation common in smoke-induced emphysema (15, 115). Thus interfering with inhibitory molecules may be a plausible mechanism by which the prominent presence of MMP9 may participate in tissue remodeling.
MMP7 is highly expressed in lungs of patients with pulmonary fibrosis and mediates shedding of E-cadherin ectodomain from bleomycin-injured lung epithelium, a process that was shown to be critical for epithelial repair (168). Most recently, researchers have identified a peptide fragment containing the neutrophil chemoattractant motif N-acetyl Pro-Gly-Pro (PGP). The PGP peptide is released from ECM presumably by cleavage from MMP1 or MMP9. This peptide resembles the chemokine IL-8 and activates its receptor CXCR2, prolonging the influx of neutrophils to the lung (292).
MMP2 is upregulated during exposure to air pollution. Also, during this exposure, mRNA of endothelin-1 (ET-1) increases (272). ET-1 is a known substrate for MMP2, where its cleavage results in ET1−21, a vasodilator (69, 70). However, in this case the authors found no increase in ET-11−21 and hypothesize that MMP2 may cleave ET-1 to another form (ET-11−32), which is a potent vasoconstrictor (272). These data suggest that MMP2 could be responsible for cardiovascular changes in response to inhaled pollution. Furthermore, it is interesting to speculate how variable cleaving of one protein may be regulated.
IX. HUMAN GENETIC VARIATION OF MATRIX METALLOPROTEINASES
Genetic models of MMP deficiency in mice, with the exception of MMP2 and MMP14, result in little phenotypic morphology or shorter life span (8, 122). Although a systematic study of natural occurrence of all of the MMP mutations in humans has not been performed, few genetic studies have pointed to the role of several MMPs in human diseases.
A. MMP Deficiency
The most deeply investigated type of MMP deficiency in humans is that of MMP2, where a homozygous recessive mutation in MMP2 allele results in no MMP2 production and severe bone deformations. There are two disorders that have been associated with this genotype. The first identified three families in Saudi Arabia of which many members have a disorder referred to as Nodulosis-Arthropathy-Osteolysis syndrome, or multicentric osteolysis (162). The afflicted family members have no detectable MMP2, resulting from a missense mutation in the MMP2 allele (162). Recently, another osteolytic disorder, Winchester syndrome, is also linked to a homozygous recessive mutation in the MMP2 active site found in a patient in Southern Italy who has generalized osteoporosis and severe osteolytic changes, including brachydactyly of the hands and feet (315). It has also been hypothesized that Torg syndrome, a less severe osteolytic disease, may also be related to MMP2 mutations; however, this has not been tested.
Other MMP deficiencies that have been implicated in human disease include MMP13 deficiency resulting from a homozygous dominant mutation that is the underlying cause of Missouri-type spondyloepimetaphyseal dysplasia (SEMD) that was identified in a large kindred in Missouri, United States (214). This mutation in MMP13 results in production of enzyme that is either degraded intracellularly or is of low molecular weight and proteolytically inactive (118). MMP20 deficiency from homozygous recessive mutation in the zinc catalytic site renders the enzyme inactive and results in hypomaturation amelogenesis imperfecta (205). The Italian patient with MMP2 mutation also had discolored and easily chipped teeth, possibly due to altered MMP20 (315). The only apparent disturbances in these patients was with bone structures; however, it would be interesting to know of their immune system function or the presence of any lung dysfunction.
B. MMP Polymorphisms
Several MMP alleles found in human population have functional polymorphisms, including MMP1, MMP3, MMP8, MMP9, and MMP12. For MMP3, researchers have identified variation in the promoter region (having either 5A or 6A) that alters expression of MMP3 protein (312). Relative to heterozygotes, individuals homozygous for 5A have increased MMP3 gene expression and individuals homozygous for 6A have decreased gene expression (174). The 6A genotype has been correlated with increased risk for coronary disease and progression of atherosclerosis (312), oral submucosal fibrosis (277), and celiac disease in men (179). For MMP8, an association between a minor allele haplotype and preterm premature rupture of membranes has been identified (289).
Several MMP polymorphisms have been identified in lung-related disease susceptibility. In asthma, a polymorphism in TIMP1 is associated with asthma in women, but not in men (154). Possession of at least one copy of the −1607GG allele (1 of 5 polymorphisms identified) for MMP1 correlates with rapid decline of lung function regardless of smoking history, age, or sex (116). Having the −1607GG allele in combination with a copy of MMP12 that contains a certain substitution also correlates highly with declining lung function (116). Of the numerous polymorphisms known for MMP9, the C to T mutation at position −1562 appears to be most important. Despite its prevalence in asthma, MMP9 polymorphisms, including the functional polymorphism C-1562T, have not been found to correlate with presence of the disease (106, 154). Initial research found no relationship between MMP9 polymorphisms and decline of lung function, but more recent studies have found that this mutation appears more frequently among smokers with COPD compared with healthy smokers (319); however, the functional significance of this remains unclear. Development of COPD diagnosed by lung function is not associated with the C-1562T polymorphism. However, the presence of the T allele correlates with developing upper lung dominant emphysema in patients who were diagnosed with COPD (114).
X. MATRIX METALLOPROTEINASES IN HUMAN INFLAMMATORY LUNG DISEASES
As we have discussed above, MMPs are important effectors of both innate and adaptive immunity and have been extensively studied in animal models of human lung diseases. Because MMPs are critical for lung development and tissue repair processes, it is not surprising that over the past decade a large effort has been devoted to determining their role in various inflammatory disease of the lung. The use of genetic approaches has increased our understanding of the contribution of MMPs in animal models of disease; however, much less is known about how MMPs may behave in the pathogenesis of human lung disease. Due to the complexity of human lung inflammatory conditions, the fact that most human studies are performed on lung diseases in different stages of their progression, and the heterogeneous nature of the human population, a clear role for the MMPs in the pathogenesis of lung diseases is difficult to conclude. Nonetheless, the resurgence of MMPs in the lung during inflammation has made their manipulation an appealing therapeutic strategy. Below, we review major classes of human inflammatory lung diseases and the part that MMPs are known to play.
A. Bronchopulmonary Dysplasia and Chronic Lung Disease of Prematurity
Infants born prematurely (before 36 wk gestation) lack adequate concentration of surfactant (discussed in sect. VII) in their distal airspace and have immature alveoli; this condition is referred to as hyaline membrane disease and puts infants at risk for later development of bronchopulmonary dysplasia (BPD). The development of BPD can be divided into four stages: acute (2−4 days), regenerative (4−8 days), transitional (8−16 days), and chronic (>16 days) (8). A hallmark of this lung injury is reepithelialization of denuded alveoli followed by the increased presence of fibroblasts and major areas of fibrosis. The cause of BPD is thought to be both the hyaline membrane disease and possible lung injury related to prolonged treatment with oxygen and mechanical ventilation (55).
One study found that increased amounts of MMP2 and a higher ratio of MMP9 to TIMP1 during the first 3 days of life correlates with poor outcome that was measured by increased dependency and the use of mechanical ventilation in infants with BPD. However, the infants who developed chronic lung disease (CLD) were born significantly earlier than those who did not. Thus the increase in airway MMPs may present an association rather than the cause in this case (63). Another study found that among preterm infants, amount of MMP2 at birth successfully predicted the outcome of BPD; however, this was confounded by the finding that birth weight was significantly correlated with amount of MMP2, again suggesting that the biggest factor in whether an infant develops BPD or CLD is the stage of development at birth (55).
Perhaps premature infants have a higher concentration of MMP2 (Fig. 5), and lower surfactant concentrations, which results in a lack of adequate protection from the oxidative damage that occurs from high oxygen treatment and higher alveolar pressures induced by mechanical ventilation. In the same study, serial BAL sampling on days 3 and 5 from infants on mechanical ventilation showed a negative correlation between MMP9 activation and TIMP1 concentration and development of BPD (55). By day 5, infants with BPD had a higher amount of TIMP1 compared with those without BPD, suggesting that the shift from acute to regenerative stage is altered in the more premature infants. A study of the pathology of BPD using immunohistochemical detection of MMPs and TIMP from lung sections of preterm infants in various stages of BPD found changes in the concentration of TIMP1 from acute to chronic stages, which was mostly associated with the occurrence of fibroblasts (59). The interactions between the surfactant proteins and MMPs and TIMPs in the alveolar space most likely play an important role in the lung maturation process, but more information is needed to identify the exact nature of these interactions in infants (273).
B. Interstitial Lung Disease and Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF), one of the most common forms of interstitial lung diseases, is a progressive fibrotic lung condition of unknown etiology (22). The diagnosis of IPF is made by surgical lung biopsy, and the histopathological features include the presence of patchy inflammatory cells, foci of proliferating fibroblasts and myofibroblasts, and collagen deposition (238, 123). Development of IPF has been linked to mutations in the SP-C gene in humans. The SP-C profile in patients with the mutation shows aberrant migration of pro-SP-C on SDS gels to a lower molecular weight than in normal patients and also shows decreased processing to mature SP-C (191, 192). Interestingly, in an effort to identify a genetic fingerprint of this disease, microarray analysis of the lung samples obtained from patients with IPF showed not only upregulation of ECM genes including collagens and tenascin N, but increases in proteinase expression including two members of ADAMTS family, MMP1 and MMP7 (240, 322). These findings highlight the complex interaction of the ECM and proteinases in IPF, but the significance of increased proteinases in this fibrotic lung disease is not known.
One aspect of IPF that correlates with rapid progression of fibrosis is the presence of a large number of fibrotic foci, which results in enhanced ECM (123). TGF-β plays a key role in stimulation of fibroblast proliferation and has been implicated in progression of IPF (299). In addition, TGF-β stimulation increases MMP2, but not MMP9 activity in a dose-dependent manner in A549 cells treated for 48 h, suggesting that fibrosis is due to an aberration in the remodeling process (117). Recent work suggests that patients with IPF may be predisposed to developing the disease due to an imbalance between chemokine/chemokine receptors, as they found increased CCR4 expression on CD4-positive T lymphocytes in BAL from patients with IPF (219). CCR4 is the receptor for CCL17 (TARC), which is also found in more patients with IPF compared with either non IPF-diseased or normal subjects. One possibility is that the MMPs that normally regulate these chemokines through cleavage are not functioning properly. In support of this, fibrosis resulting from systemic sclerosis may be due to development of function-blocking antibodies to MMP1 and MMP3, allowing deposition of ECM (190).
C. Asthma
Asthma and related atopic syndromes have emerged as major public health concerns, and studies from around the world suggest that the incidence and prevalence of asthma began to rise more than two decades ago with no signs that these disturbing trends may be reversing (120). Asthma is characterized by episodic dyspnea, lung inflammation, and in some patients, progressive irreversible airway dysfunction (16). Clinically, patients with asthma display AHR in response to methacholine and can become short of breath or develop asphyxia during an acute exacerbation of their disease. Histological hallmarks of asthma include homing of Th2 inflammatory cells into the lung parenchyma, eosinophilia, and increased mucus metaplasia (47). Expression of several MMPs has been associated with asthma; increases in MMP1, MMP2, MMP3, MMP8, and MMP9 have all been found in sputum and BAL from patients with asthma (58). In a special subset of patients with severe asthma, there is a greater increase in MMP9 activity in BAL relative to that from patients with mild asthma or control patients. Wenzel et al. (293) found an increase in MMP9 activity in the subepithelial basement membrane that is accompanied by higher TGF-β. These studies suggest that in patients with severe asthma, neutrophils play a key role in lung remodeling because they express both MMP9 and TGF-β, which are involved in breakdown and repair of tissue, respectively.
In addition to the MMP9-mediated action of neutrophils, data from human disease support the hypothesis that MMP9 plays a role in clearing eosinophils from lung after inflammation. In asthma, MMP9 is expressed in bronchial epithelium and submucosa, where its abundance correlates with tissue eosinophil number (96). MMP9 is also produced by eosinophils, macrophages, and neutrophils (35, 196). In sputum, concentration of MMP9 is positively correlated with neutrophil number (26) and also with the cumulative macrophage, neutrophil, and eosinophil count (141) during asthma. In addition, the number of MMP positive cells inversely correlates with forced expiratory volume (FEV1); this association may indicate that under acute exacerbations, the influx of inflammatory cells interferes with normal gas exchange (26).
BAL fluid from normal and asthmatic patients, when added to human airway smooth muscle cells in culture, increases cell count and thymidine incorporation indicating increased proliferation. However, BAL taken from asthmatic patients after allergen challenge increases the mitogenic indices even more than a normal patient's BAL (184). This phenomenon could contribute to the airway remodeling observed in patients with chronic asthma; however, the pathway underlying the proliferation remains unclear. EGFR, TGF-β, or platelet-derived growth factor are all candidates based on their presence in lung during disease and their relationship to various MMPs.
D. COPD and Emphysema
COPD, characterized by irreversible reduction of maximum expiratory flow, is currently the fourth leading cause of death in the United States (National Center for Health Statistics; http://www.cdc.gov/nchs/about.htm) and the fourth leading cause of death worldwide (http://www.who.int/respiratory/copd/en/). Exposure to tobacco smoke accounts for the majority of clinically significant COPD. Classification of COPD based on lung function studies and symptoms identify four stages of the disease: mild, moderate, severe, and very severe (215).
Elastin fibers form the mechanical stress-resistant structure of the lung by maintaining elasticity and recoil properties. It is not surprising then that destruction of these fibers can directly result in reduction of maximum airflow, one of the physiological hallmarks of this disease (14). As discussed in section V, animal models of emphysema using purified papain or elastase support the hypothesis that elastic fiber repair is associated with formation of secondary inflammation that leads to secretory cell metaplasia comparable to chronic bronchitis (243, 254). Although destruction of elastin fibers is known to occur in patients with emphysema, few clinical studies have linked human COPD and emphysema to dysregulated expression of MMPs. In one such study alveolar lining fluid of subjects with COPD and emphysema show a higher concentration of MMP8 and MMP9 compared with nonsmoker, age-matched controls (73). Furthermore, during the macrophage influx to lung, both the number of MMP12-expressing macrophages and the amount of MMP12 they express, as shown by immunohistochemistry and Western blotting, is higher in BAL samples from COPD patients than in control subjects. In addition, human lung tissue from COPD patients shows a larger number of macrophages expressing MMP12 than control subjects (87, 177).
The most well-studied MMPs in human COPD and emphysema are MMP1, MMP8, MMP9, and MMP12, which have all been implicated in tissue destruction in human COPD and emphysema (177). The lymphocytes present in emphysematous lungs have a strong Th1 bias, expressing higher levels of the CCR5 and CXCR3, and their ligands CXCL10 (IP-10) and CXCL9 (MIG). CXCL9 can upregulate expression of MMP12 in pulmonary macrophages (87), providing a mechanism for chronic and progressive destruction of lung parenchyma that is seen in emphysema.
In addition to direct upregulation of MMP9 and MMP12 by cigarette smoke in human lung, MMPs can be upregulated indirectly through activation of CD147 that is a known inducer of MMPs (see sect. IV). BAL from current and former smokers compared with those who had never smoked show an increase in CD147 expression, although a positive correlation with emphysema was not established in these studies (20, 21). Furthermore, using immunohistochemistry, CD147 expression is found in lung tissue from smokers, where it is localized to bronchiolar epithelium and alveolar macrophages (21). While these studies point to the role of MMPs and some of the inducers of this family in tobacco-induced lung diseases, in general most human-based studies can only provide an association or lack thereof between MMPs and destructive lung diseases such as COPD and emphysema.
E. Cystic Fibrosis
Cystic fibrosis (CF) is a common, inheritable genetic disorder that results in malfunctioning of the chloride channels of many exocrine epithelial linings including the airway epithelia (77). Failure of the chloride ion channel to function properly is thought to contribute to the morbidity and mortality observed in CF patients who suffer from chronic bacterial infections and inflammation. The exact mechanism or pathophysiology of progressive lung inflammation in patients with CF is not clear, but patients with CF have increased levels of MMP2, MMP8, and MMP9 in their BAL (232). In a study of 23 children with CF, BAL fluid concentrations of MMP8 and MMP9 were higher in untreated children and lower in those who were treated with DNase (225). In children with stable CF, there is a significant inverse relationship between MMP9 and lung function, as measured by FEV1. Furthermore, levels of MMP9 are higher in sputum of asymptomatic children with CF compared with controls, suggesting that MMP9 and total neutrophil count may be useful markers of airway injury and airflow obstruction in persons with CF (233). As discussed in section VII, MMPs play a critical role in the host defense against pathogens in the lung. Patients with CF have chronic gram-negative organisms such as Pseudomonas in their distal airways and upregulated expression of MMP7 in the epithelium of upper airway and type II pneumocytes (60).
F. Bronchiolitis Obliterans
Survival of lung transplant recipients is limited by the development of bronchiolitis obliterans (BO), an irreversible condition that responds poorly to therapy and results in airway remodeling with obstructive and restrictive features (187). In the past few years, the role of MMPs in BO-induced lung remodeling has been examined. In a study looking at the biological markers in the lungs of patients with lung transplant, despite a similar number of inflammatory cells, the BAL, but not serum, concentrations of MMP8, MMP9, and TIMP1 are significantly elevated in subjects with advanced BO (267). Similar evidence from a pilot study comparing BAL from patients with and without BO identified a higher ratio of MMP9 to TIMP1 in patients with BO (111). The source of MMP9 in these studies is shown to be neutrophils recruited into the lung as the result of inflammation; however, much like other clinical studies, it is unknown whether the presence of MMP9 results in the progression of BO. Consistent with the findings in the above-mentioned human studies, in murine heterotopic tracheal transplant model of BO, selective induction of MMP3, MMP9, MMP12, and TIMP1 was detected in tracheal allografts compared with isografts (40).
G. Pulmonary Alveolar Proteinosis
Pulmonary alveolar proteinosis (PAP) is a rare disease that is characterized by the presence of immature alveolar macrophages and an accumulation of surfactant in the lung (51). This condition can occur as an autoimmune disorder in which autoantibodies to granulocyte macrophage colony stimulating factor (GM-CSF) can inhibit normal maturation and function of alveolar macrophages (23). Patients with PAP have been shown to have chronic inflammation in the lung with elevated levels of monocyte chemotactic proteins (MCP-1, MCP-2, MCP-3, IL-8, IL-10, and M-CSF). However, despite increases in chemotactic mediators, there is no increase in the number of inflammatory cells in the BAL (25) or evidence of tissue remodeling and fibrosis (83, 221). Interestingly, MMP2 and MMP9 mRNA and protein are increased in BAL from PAP patients (24). MMP2 is shown to cleave CCR2 (the receptor for many MCPs), and binding of the cleaved form of this protein acts as an antagonist that prevents chemotaxis in vitro (173; see Table 1). Thus MMP processing of chemokines in addition to decreased receptor function could contribute to the lack of cellular migration to lungs. The mechanism for increased MMPs is unclear, but one hypothesis is that increased surfactant could be responsible. Alternatively, elevated M-CSF has been found to increase MMPs in cultured cells (24).
XI. MATRIX METALLOPROTEINASES: TO INHIBIT OR NOT INHIBIT?
In the lung, one of the most important factors in recovering from disease or injury is clearing of inflammatory cells. Failure to do so can lead to tissue destruction and alveolar blockage. Researchers now must determine the transcriptional and translational regulation of the cytokines and chemokines necessary for proper functioning of the inflammatory pathway that ultimately leads to clearance of the recruited inflammatory cells from the lung. Because MMPs are increased in so many disease states and are correlated with tissue damage, inhibition of MMPs would seem an attractive therapy. However, before we can use MMP targets for therapy, we must know that they “unambiguously contribute to disease initiation or progression” (199).
At present, researchers have knocked out at least 11 of the 25 MMPs in mice (213). During pathological experimental conditions, some MMPs confer protective effects (Fig. 6). For example, MMP3 has a protective effect against cancer in mice (166). Yet, other MMPs contribute to worsening of the disease or injury (Fig. 6). Mice lacking MMP7 are protected from bleomycin-induced lung injury (322) and have decreased tumor incidence (300). In addition, a knockout mouse may fare better in one treatment and worse in another, further confounding the contribution of MMPs (213). In an acute model of pulmonary embolism in rats, within an hour MMP-9 activity increases by 43%, but interestingly, despite known expression of MMP2 by endothelial cells that are affected by embolization, no significant increase is observed in MMP2 activity. Pretreatment of rats with doxicycline attenuated the physiological impairments seen in pulmonary embolism and blunted the increase in lung MMP9 activity (207). This study exemplifies how pharmacological strategies targeting specific MMPs with selective inhibitors are necessary in treatment of disease.
A. Natural Inhibitors
MMPs have several naturally occurring inhibitors (213). A general inhibitor that also inhibits MMPs, α2-macroglobulin, is an abundant plasma protein that irreversibly inhibits MMPs. Four TIMPs have been identified and characterized in mammals. Each TIMP inhibits specific MMPs, and the molar ratio of MMP to TIMP correlates significantly with disease outcome and/or severity in several model systems and human diseases. In addition, several other proteins, including reversion-inducing cysteine-rich protein with Kazal motifs (RECK; Ref. 195), tissue-factor pathway inhibitor-2 (TFPI2; Ref. 104), pro-collagen COOH-terminal proteinase enhancer (PCPE; Ref. 180), fragments from certain collagens (218), and agrin (257), have been shown to inhibit MMPs in vivo (199).
B. Synthetic Inhibitors
Among synthetic inhibitors, there are broad-spectrum inhibitors and specific inhibitors. Broad-spectrum MMP inhibitors interfere with numerous MMPs and may have deleterious effects due to the multiplicity of the MMP system. In addition, their effects are not easy to predict, making them poor candidates for therapeutic use. For example, one general inhibitor, Batimastat, was used in mice during bleomycin-induced lung injury, where it had a protective effect and was thought to be a potential target for preventing pulmonary fibrosis (44). However, when the same inhibitor was tested in a model of LPS infection, it had no effect on inflammatory cells and no change in MMP activity in BAL, underlining the differential effects of MMPs during various pathologies (45). In a clinical trial, researchers reported serious musculoskeletal side effects in cancer patients that were given Marimastat. In addition, other clinical trials were stopped due to decreased survival or no survival benefit detected (53). However, in a small clinical study of mild asthma, Marimastat reduced bronchial hyperresponsiveness to inhaled allergen and resulted in a nonsignificant drop in inflammatory cell egression to sputum (32).
On the other hand, more promising, specific MMP inhibitors are in preliminary stages of research. A specific inhibitor with affinity for MMP2 and MMP9, ONO-4817, has been shown to decrease neutrophil migration to BAL and oxygen radical formation in response to ischemia-reperfusion-induced lung injury (247).
XII. SUMMARY
The MMP family regulates a multitude of biological responses in the lung that range from normal development to destructive and fibrotic changes. Although members of the MMPs family share variable degrees of sequence homology and in vitro show overlapping substrate specificity, it should be emphasized that different cell types express MMPs under dissimilar conditions. As such, it is not surprising that MMP genes have diverse and, at times, opposite transcriptional regulatory mechanisms. MMPs can act directly by degrading lung ECM and/or other secreted proteins; alternatively, they function indirectly by changing the properties of the cleaved proteins in the alveolar space. Future efforts to identify their in vivo substrates will most likely yield the necessary information that will pave the way for elucidating their mechanism of action under normal or diseased states. Animal models of human lung diseases and clinical studies have provided ample evidence for the involvement of MMPs in the development or progression of a number of common lung diseases such as COPD, emphysema, and asthma. However, merely detecting increases in MMPs expression in various lung inflammatory conditions may not provide sufficient information to understand the contribution of MMPs in human lung diseases. On one hand, it appears that MMPs are upregulated in a variety of inflammatory and fibrotic lung diseases; however, inhibition of MMPs in models of human disease shows unexpected deleterious effects. Thus, while excess production of proteinases during inflammation occurs, this may prove to be a necessary phase of the repair process in the lung. Genetic models that result in over- or underexpression of MMPs applied to models of lung disease will provide invaluable information into MMP function, information which is required to determine the utility of proteinases and proteinase inhibitors in the clinical management of inflammatory lung diseases.
ACKNOWLEDGMENTS
GRANTS
This work was supported by National Institutes of Health Grants T32-HL-07747 (to K. J. Greenlee), R01-HL-72419 and K02-HL-72062 (to F. Kheradmand), and P01-AI-53194 and R01-HL-75602 (to Z. Werb).
REFERENCES
- 1.Mechanisms and limits of induced postnatal lung growth. Am J Respir Crit Care Med. 2004;170:319–343. doi: 10.1164/rccm.200209-1062ST. [DOI] [PubMed] [Google Scholar]
- 2.Adamson IYR, Vincent R, Bakowska J. Differential production of metalloproteinases after instilling various urban air particle samples to rat lung. Exp Lung Res. 2003;29:375–388. doi: 10.1080/01902140303753. [DOI] [PubMed] [Google Scholar]
- 3.Ahokas K, Lohi J, Illman SA, Llano E, Elomaa O, Impola U, Karjalainen-Lindsberg ML, Saarialho-Kere U. Matrix metalloproteinase-21 is expressed epithelially during development and in cancer and is up-regulated by transforming growth factor-beta 1 in keratinocytes. Lab Invest. 2003;83:1887–1899. doi: 10.1097/01.lab.0000106721.86126.39. [DOI] [PubMed] [Google Scholar]
- 4.Al-Aqeel AI. Al-Aqeel Sewairi syndrome, a new autosomal recessive disorder with multicentric osteolysis, nodulosis, and arthropathy. The first genetic defect of matrix metalloproteinase 2 gene. Saudi Med J. 2005;26:24–30. [PubMed] [Google Scholar]
- 5.Anteby EY, Greenfield C, Natanson-Yaron S, Goldman-Wohl D, Hamani Y, Khudyak V, Ariel I, Yagel S. Vascular endothelial growth factor, epidermal growth factor and fibroblast growth factor-4 and -10 stimulate trophoblast plasminogen activator system and metalloproteinase-9. Mol Hum Reprod. 2004;10:229–235. doi: 10.1093/molehr/gah031. [DOI] [PubMed] [Google Scholar]
- 6.Apte SS, Fukai N, Beier DR, Olsen BR. The matrix metalloproteinase-14 (MMP-14) gene is structurally distinct from other MMP genes and is co-expressed with the TIMP-2 gene during mouse embryogenesis. J Biol Chem. 1997;272:25511–25517. doi: 10.1074/jbc.272.41.25511. [DOI] [PubMed] [Google Scholar]
- 7.Arora K, Gwinn WM, Bower MA, Watson A, Okwumabua I, MacDonald HR, Bukrinsky MI, Constant SL. Extracellular cyclophilins contribute to the regulation of inflammatory responses. J Immunol. 2005;175:517–522. doi: 10.4049/jimmunol.175.1.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atkinson JJ, Holmbeck K, Yamada S, Birkedal-Hansen H, Parks WC, Senior RM. Membrane-type 1 matrix metalloproteinase is required for normal alveolar development. Dev Dyn. 2005;232:1079–1090. doi: 10.1002/dvdy.20267. [DOI] [PubMed] [Google Scholar]
- 9.Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol. 2003;28:12–24. doi: 10.1165/rcmb.2002-0166TR. [DOI] [PubMed] [Google Scholar]
- 10.Bai S, Thummel R, Godwin AR, Nagase H, Itoh Y, Li L, Evans R, McDermott J, Seiki M, Sarras JMP. Matrix metalloproteinase expression and function during fin regeneration in zebrafish: analysis of MT1-MMP, MMP2 and TIMP2. Matrix Biol. 2005;24:247–260. doi: 10.1016/j.matbio.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 11.Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, Lopez-Otin C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nature Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 12.Banda MJ, Rice AG, Griffin GL, Senior RM. The inhibitory complex of human alpha 1-proteinase inhibitor and human leukocyte elastase is a neutrophil chemoattractant. J Exp Med. 1988;167:1608–1615. doi: 10.1084/jem.167.5.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banda MJ, Werb Z. Mouse macrophage elastase. Biochem J. 1981;193:589–605. doi: 10.1042/bj1930589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269–280. doi: 10.1056/NEJM200007273430407. [DOI] [PubMed] [Google Scholar]
- 15.Barnes PJ. Small airways in COPD. N Engl J Med. 2004;350:2635–2637. doi: 10.1056/NEJMp048102. [DOI] [PubMed] [Google Scholar]
- 16.Barrios RJ, Kheradmand F, Batts L, Corry DB. Asthma: pathology and pathophysiology. Arch Pathol Lab Med. 2006;130:447–451. doi: 10.5858/2006-130-447-APAP. [DOI] [PubMed] [Google Scholar]
- 17.Bartlett JD, Simmer JP, Xue J, Margolis HC, Moreno EC. Molecular cloning and mRNA tissue distribution of a novel matrix metalloproteinase isolated from porcine enamel organ. Gene. 1996;183:123–128. doi: 10.1016/s0378-1119(96)00525-2. [DOI] [PubMed] [Google Scholar]
- 18.Basset P, Bellocq JP, Wolf C, Stoll I, Hutin P, Limacher JM, Podhajcer OL, Chenard MP, Rio MC, Chambon P. A novel metalloproteinase gene specifically expressed in stromal cells of breast carcinomas. Nature. 1990;348:699–704. doi: 10.1038/348699a0. [DOI] [PubMed] [Google Scholar]
- 19.Betsuyaku T, Fukuda Y, Parks WC, Shipley JM, Senior RM. Gelatinase B is required for alveolar bronchiolization after intratracheal bleomycin. Am J Pathol. 2000;157:525–535. doi: 10.1016/S0002-9440(10)64563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Betsuyaku T, Kadomatsu K, Griffin GL, Muramatsu T, Senior RM. Increased basigin in bleomycin-induced lung injury. Am J Respir Cell Mol Biol. 2003;28:600–606. doi: 10.1165/rcmb.2002-0059OC. [DOI] [PubMed] [Google Scholar]
- 21.Betsuyaku T, Tanino M, Nagai K, Nasuhara Y, Nishimura M, Senior RM. Extracellular matrix metalloproteinase inducer is increased in smokers’ bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 2003;168:222–227. doi: 10.1164/rccm.200301-103OC. [DOI] [PubMed] [Google Scholar]
- 22.Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157:199–203. doi: 10.1164/ajrccm.157.1.9704130. [DOI] [PubMed] [Google Scholar]
- 23.Blic JD. Pulmonary alveolar proteinosis in children. Pediatr Respir Rev. 2004;5:316–322. doi: 10.1016/j.prrv.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Bonfield TL, Swaisgood CM, Barna BP, Farver CF, Kavuru MS, Thomassen MJ. Elevated gelatinase activity in pulmonary alveolar proteinosis: role of macrophage-colony stimulating factor. J Leukoc Biol. 2006;79:133–139. doi: 10.1189/jlb.0805447. [DOI] [PubMed] [Google Scholar]
- 25.Bonfield TL, Swaisgood CM, Barna BP, Farver CF, Kavuru MS, Thomassen MJ. Elevated gelatinase activity in pulmonary alveolar proteinosis: role of macrophage-colony stimulating factor. J Leukoc Biol. 2006;79:133–139. doi: 10.1189/jlb.0805447. [DOI] [PubMed] [Google Scholar]
- 26.Boulay ME, Prince P, Deschesnes F, Chakir J, Boulet LP. Metalloproteinase-9 in induced sputum correlates with the severity of the late allergen-induced asthmatic response. Respiration. 2004;71:216–224. doi: 10.1159/000077418. [DOI] [PubMed] [Google Scholar]
- 27.Bracke K, Cataldo D, Maes T, Gueders M, Noel A, Foidart JM, Brusselle G, Pauwels RA. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int Arch Allergy Immunol. 2005;138:169–179. doi: 10.1159/000088439. [DOI] [PubMed] [Google Scholar]
- 28.Brauer PR. MMPs: role in cardiovascular development and disease. Front Biosci. 2006;11:447–478. doi: 10.2741/1810. [DOI] [PubMed] [Google Scholar]
- 29.Breathnach R, Matrisian LM, Gesnel MC, Staub A, Leroy P. Sequences coding for part of oncogene-induced transin are highly conserved in a related rat gene. Nucleic Acids Res. 1987;15:1139–1151. doi: 10.1093/nar/15.3.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks PC, Stromblad S, Sanders LC, vonSchalscha TL, Aimes RT, StetlerStevenson WG, Quigley JP, Cheresh DA. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- 31.Brown JK, Tyler CL, Jones CA, Ruoss SJ, Hartmann T, Caughey GH. Tryptase, the dominant secretory granular protein in human mast cells, is a potent mitogen for cultured dog tracheal smooth muscle cells. Am J Respir Cell Mol Biol. 1995;13:227–236. doi: 10.1165/ajrcmb.13.2.7626290. [DOI] [PubMed] [Google Scholar]
- 32.Bruce C, Thomas PS. The effect of marimastat, a metalloprotease inhibitor, on allergen-induced asthmatic hyper-reactivity. Toxicol Appl Pharmacol. 2005;205:126–132. doi: 10.1016/j.taap.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Buckley S, Driscoll B, Shi W, Anderson K, Warburton D. Migration and gelatinases in cultured fetal, adult, and hyperoxic alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L427–L434. doi: 10.1152/ajplung.2001.281.2.L427. [DOI] [PubMed] [Google Scholar]
- 34.Buckley S, Warburton D. Dynamics of metalloproteinase-2 and -9, TGF-beta, and uPA activities during normoxic vs. hyperoxic alveolarization. Am J Physiol Lung Cell Mol Physiol. 2002;283:L747–L754. doi: 10.1152/ajplung.00415.2001. [DOI] [PubMed] [Google Scholar]
- 35.Campbell EJ, Cury JD, Shapiro SD, Goldberg GI, Welgus HG. Neutral proteinases of human mononuclear phagocytes: cellular differentiation markedly alters cell phenotype for serine proteinases, metalloproteinases, and tissue inhibitor of metalloproteinases. J Immunol. 1991;146:1286–1293. [PubMed] [Google Scholar]
- 36.Campbell LG, Ramachandran S, Liu W, Shipley JM, Itohara S, Rogers JG, Moazami N, Senior RM, Jaramillo A. Different roles for matrix metalloproteinase-2 and matrix metalloproteinase-9 in the pathogenesis of cardiac allograft rejection. Am J Transplant. 2005;5:517–528. doi: 10.1111/j.1600-6143.2005.00744.x. [DOI] [PubMed] [Google Scholar]
- 37.Cardoso WV. Molecular regulation of lung development. Annu Rev Physiol. 2001;63:471–494. doi: 10.1146/annurev.physiol.63.1.471. [DOI] [PubMed] [Google Scholar]
- 38.Carinato ME, Walter BE, Henry JJ. Xenopus laevis gelatinase B (Xmmp-9): development, regeneration, and wound healing. Dev Dyn. 2000;217:377–387. doi: 10.1002/(SICI)1097-0177(200004)217:4<377::AID-DVDY5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 39.Caudroy S, Polette M, Tournier JM, Burlet H, Toole B, Zucker S, Birembaut P. Expression of the extracellular matrix metalloproteinase inducer (EMMPRIN) and the matrix metalloproteinase-2 in bronchopulmonary and breast lesions. J Histochem Cytochem. 1999;47:1575–1580. doi: 10.1177/002215549904701209. [DOI] [PubMed] [Google Scholar]
- 40.Chen P, Farivar AS, Mulligan MS, Madtes DK. Tissue inhibitor of metalloproteinase-1 deficiency abrogates obliterative airway disease after heterotopic tracheal transplantation. Am J Respir Cell Mol Biol. 2006;34:464–472. doi: 10.1165/rcmb.2005-0344OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi J, Choi K, Benveniste EN, Hong YS, Lee JH, Kim J, Park K. Bcl-2 promotes invasion and lung metastasis by inducing matrix metalloproteinase-2. Cancer Res. 2005;65:5554–5560. doi: 10.1158/0008-5472.CAN-04-4570. [DOI] [PubMed] [Google Scholar]
- 42.Churg A, Brauer M, Avila-Casado Mdel C, Fortoul TI, Wright JL. Chronic exposure to high levels of particulate air pollution and small airway remodeling. Environ Health Perspect. 2003;111:714–718. doi: 10.1289/ehp.6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, Shapiro SD, Wright JL. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med. 2003;167:1083–1089. doi: 10.1164/rccm.200212-1396OC. [DOI] [PubMed] [Google Scholar]
- 44.Corbel M, Caulet-Maugendre S, Germain N, Molet S, Lagente V, Boichot E. Inhibition of bleomycin-induced pulmonary fibrosis in mice by the matrix metalloproteinase inhibitor batimastat. J Pathol. 2001;193:538–545. doi: 10.1002/path.826. [DOI] [PubMed] [Google Scholar]
- 45.Corbel M, Lanchou J, Germain N, Malledant Y, Boichot E, Lagente V. Modulation of airway remodeling-associated mediators by the antifibrotic compound, pirfenidone, and the matrix metalloproteinase inhibitor, batimastat, during acute lung injury in mice. Eur J Pharmacol. 2001;426:113–121. doi: 10.1016/s0014-2999(01)01209-2. [DOI] [PubMed] [Google Scholar]
- 46.Corry DB, Kheradmand F. Biology and therapeutic potential of the interleukin-4/interleukin-13 signaling pathway in asthma. Am J Respir Med. 2002;1:185–193. doi: 10.1007/BF03256608. [DOI] [PubMed] [Google Scholar]
- 47.Corry DB, Kheradmand F. Control of allergic airway inflammation through immunomodulation. J Allergy Clin Immunol. 2006;117:S461–S464. doi: 10.1016/j.jaci.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Corry DB, Kiss A, Song LZ, Song L, Xu J, Lee SH, Werb Z, Kheradmand F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corry DB, Rishi K, Kanellis J, Kiss A, Song Lz LZ, Xu J, Feng L, Werb Z, Kheradmand F. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cossins J, Dudgeon TJ, Catlin G, Gearing AJH, Clements JM. Identification of MMP-18, a putative novel human matrix metalloproteinase. Biochem Biophys Res Commun. 1996;228:494–498. doi: 10.1006/bbrc.1996.1688. [DOI] [PubMed] [Google Scholar]
- 51.Costabel U, Guzman J. Pulmonary alveolar proteinosis: a new autoimmune disease. Sarcoidosis Vasculitis Diffuse Lung Dis. 2005;22:S67–S73. [PubMed] [Google Scholar]
- 52.Coussens LM, Shapiro SD, Soloway PD, Werb Z. Models for gain-of-function and loss-of-function of MMPs. Transgenic and gene targeted mice. Methods Mol Biol. 2001;151:149–179. doi: 10.1385/1-59259-046-2:149. [DOI] [PubMed] [Google Scholar]
- 53.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crawford BD, Pilgrim DB. Ontogeny and regulation of matrix metalloproteinase activity in the zebrafish embryo by in vitro and in vivo zymography. Dev Biol. 2005;286:405–414. doi: 10.1016/j.ydbio.2005.06.035. [DOI] [PubMed] [Google Scholar]
- 55.Danan C, Jarreau PH, Franco ML, Dassieu G, Grillon C, Abd Alsamad I, Lafuma C, Harf A, Delacourt C. Gelatinase activities in the airways of premature infants and development of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1086–L1093. doi: 10.1152/ajplung.00066.2002. [DOI] [PubMed] [Google Scholar]
- 56.D'Armiento J, Dalal SS, Okada Y, Berg RA, Chada K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell. 1992;71:955–961. doi: 10.1016/0092-8674(92)90391-o. [DOI] [PubMed] [Google Scholar]
- 57.Demayo F, Minoo P, Plopper CG, Schuger L, Shannon J, Torday JS. Mesenchymal-epithelial interactions in lung development and repair: are modeling and remodeling the same process? Am J Physiol Lung Cell Mol Physiol. 2002;283:L510–L517. doi: 10.1152/ajplung.00144.2002. [DOI] [PubMed] [Google Scholar]
- 58.Demedts IK, Brusselle GG, Bracke KR, Vermaelen KY, Pauwels RA. Matrix metalloproteinases in asthma and COPD. Curr Opin Pharmacol. 2005;5:257–263. doi: 10.1016/j.coph.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 59.Dik WA, De Krijger RR, Bonekamp L, Naber BAE, Zimmermann LJI, Versnel MA. Localization and potential role of matrix metalloproteinase-1 and tissue inhibitors of metalloproteinase-1 and -2 in different phases of bronchopulmonary dysplasia. Pediatr Res. 2001;50:761–766. doi: 10.1203/00006450-200112000-00022. [DOI] [PubMed] [Google Scholar]
- 60.Dunsmore SE, Saarialho-Kere UK, Roby JD, Wilson CL, Matrisian LM, Welgus HG, Parks WC. Matrilysin expression and function in airway epithelium. J Clin Invest. 1998;102:1321–1331. doi: 10.1172/JCI1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 62.Eickelberg O, Kohler E, Reichenberger F, Bertschin S, Woodtli T, Erne P, Perruchoud AP, Roth M. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-beta1 and TGF-beta3. Am J Physiol Lung Cell Mol Physiol. 1999;276:L814–L824. doi: 10.1152/ajplung.1999.276.5.L814. [DOI] [PubMed] [Google Scholar]
- 63.Ekekezie II, Thibeault DW, Simon SD, Norberg M, Merrill JD, Ballard RA, Ballard PL, Truog WE. Low levels of tissue inhibitors of metalloproteinases with a high matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio are present in tracheal aspirate fluids of infants who develop chronic lung disease. Pediatrics. 2004;113:1709–1714. doi: 10.1542/peds.113.6.1709. [DOI] [PubMed] [Google Scholar]
- 64.Elkington PTG, Emerson JE, Lopez-Pascua LDC, O'Kane CM, Horncastle DE, Boyle JJ, Friedland JS. Mycobacterium tuberculosis up-regulates matrix metalloproteinase-1 secretion from human airway epithelial cells via a p38 MAPK switch. J Immunol. 2005;175:5333–5340. doi: 10.4049/jimmunol.175.8.5333. [DOI] [PubMed] [Google Scholar]
- 65.Elkington PTG, Friedland JS. Matrix metalloproteinases in destructive pulmonary pathology. Thorax. 2006;61:259–266. doi: 10.1136/thx.2005.051979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fan QW, Kadomatsu K, Uchimura K, Muramatsu T. Embigin/basigin subgroup of the immunoglobulin superfamily: different modes of expression during mouse embryogenesis and correlated expression with carbohydrate antigenic markers. Development Growth Differentiation. 1998;40:277–286. doi: 10.1046/j.1440-169x.1998.t01-1-00003.x. [DOI] [PubMed] [Google Scholar]
- 67.Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infection. 2003;5:1317–1327. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 68.Fernandez FG, Campbell LG, Liu W, Shipley JM, Itohara S, Patterson GA, Senior RM, Mohanakumar T, Jaramillo A. Inhibition of obliterative airway disease development in murine tracheal allografts by matrix metalloproteinase-9 deficiency. Am J Transplant. 2005;5:671–683. doi: 10.1111/j.1600-6143.2005.00751.x. [DOI] [PubMed] [Google Scholar]
- 69.Fernandez-Patron C, Radomski MW, Davidge ST. Vascular matrix metalloproteinase-2 cleaves big endothelin-1 yielding a novel vasoconstrictor. Circ Res. 1999;85:906–911. doi: 10.1161/01.res.85.10.906. [DOI] [PubMed] [Google Scholar]
- 70.Fernandez-Patron C, Zouki C, Whittal R, Chan JSD, Davidge ST, Filep JG. Matrix metalloproteinases regulate neutrophil-endothelial cell adhesion through generation of endothelin-1[1–32]. FASEB J. 2001;15:2230–2240. doi: 10.1096/fj.01-0178com. [DOI] [PubMed] [Google Scholar]
- 71.Fink MP. Role of reactive oxygen and nitrogen species in acute respiratory distress syndrome. Curr Opin Crit Care. 2002;8:6–11. doi: 10.1097/00075198-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 72.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 73.Finlay GA, Russell KJ, McMahon KJ, Darcy EM, Masterson JB, FitzGerald MZ, O'Connor CM. Elevated levels of matrix metalloproteinases in bronchoalveolar lavage fluid of emphysematous patients. Thorax. 1997;52:502–506. doi: 10.1136/thx.52.6.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Foda HD, Rollo EE, Drews M, Conner C, Appelt K, Shalinsky DR, Zucker S. Ventilator-induced lung injury upregulates and activates gelatinases and EMMPRIN: attenuation by the synthetic matrix metalloproteinase inhibitor, Prinomastat (AG3340). Am J Respir Cell Mol Biol. 2001;25:717–724. doi: 10.1165/ajrcmb.25.6.4558f. [DOI] [PubMed] [Google Scholar]
- 75.Franco AA, Mundy LM, Trucksis M, Wu SG, Kaper JB, Sears CL. Cloning and characterization of the Bacteroides fragilis metalloprotease toxin gene. Infect Immunity. 1997;65:1007–1013. doi: 10.1128/iai.65.3.1007-1013.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Freije JMP, Diezitza I, Balbin M, Sanchez LM, Blasco R, Tolivia J, Lopezotin C. Molecular-cloning and expression of collagenase-3, a novel human matrix metalloproteinase produced by breast carcinomas. J Biol Chem. 1994;269:16766–16773. [PubMed] [Google Scholar]
- 77.Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Galvez BG, Genis L, Matias-Roman S, Oblander SA, Tryggvason K, Apte SS, Arroyo AG. Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoattractant protein-1/ccl2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J Biol Chem. 2005;280:1292–1298. doi: 10.1074/jbc.M408673200. [DOI] [PubMed] [Google Scholar]
- 79.Gebb SA, Fox K, Vaughn J, McKean D, Jones PL. Fetal oxygen tension promotes tenascin-C-dependent lung branching morphogenesis. Dev Dyn. 2005;234:1–10. doi: 10.1002/dvdy.20500. [DOI] [PubMed] [Google Scholar]
- 80.Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–228. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- 81.Gill SE, Pape MC, Khokha R, Watson AJ, Leco KJ. A null mutation for tissue inhibitor of metalloproteinases-3 (Timp-3) impairs murine bronchiole branching morphogenesis. Dev Biol. 2003;261:313–323. doi: 10.1016/s0012-1606(03)00318-x. [DOI] [PubMed] [Google Scholar]
- 82.Glasser SW, Detmer EA, Ikegami M, Na CL, Stahlman MT, Whitsett JA. Pneumonitis and emphysema in sp-C gene targeted mice. J Biol Chem. 2003;278:14291–14298. doi: 10.1074/jbc.M210909200. [DOI] [PubMed] [Google Scholar]
- 83.Godwin JD, Muller NL, Takasugi JE. Pulmonary alveolar proteinosis—CT findings. Radiology. 1988;169:609–613. doi: 10.1148/radiology.169.3.3186983. [DOI] [PubMed] [Google Scholar]
- 83a.Greenlee KJ, Corry DB, Engler DA, Matsunami RK, Tessier P, Cook RG, Werb Z, Kheradmand F. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J Immunol. doi: 10.4049/jimmunol.177.10.7312. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gross J. How tadpoles lose their tails: path to discovery of the first matrix metalloproteinase. Matrix Biol. 2004;23:3–13. doi: 10.1016/j.matbio.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 85.Gross J, Lapiere CM. Collagenolytic activity in amphibian tissues: a tissue culture assay. Proc Natl Acad Sci USA. 1962;48:1014–1022. doi: 10.1073/pnas.48.6.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grote K, Flach I, Bavendiek U, Hilfiker-Kleiner D, Schieffer B. Mechanical stretch promotes pro-angiogenic mediators via mechanosensitive transcription factors. Circulation. 2003;108:170–170. [Google Scholar]
- 87.Grumelli S, Corry D, Song L, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis D, Kheradmand F. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. Public Library Sci Med. 2004;1:74–83. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gueders MM, Balbin M, Rocks N, Foidart JM, Gosset P, Louis R, Shapiro S, Lopez-Otin C, Noel A, Cataldo DD. Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. J Immunol. 2005;175:2589–2597. doi: 10.4049/jimmunol.175.4.2589. [DOI] [PubMed] [Google Scholar]
- 89.Gueguen Y, Cadoret JP, Flament D, Barreau-Roumiguiere C, Girardot AL, Garnier J, Hoareau A, Bachere E, Escoubas JM. Immune gene discovery by expressed sequence tags generated from hemocytes of the bacteria-challenged oyster, Crassostrea gigas. Gene. 2003;303:139–145. doi: 10.1016/s0378-1119(02)01149-6. [DOI] [PubMed] [Google Scholar]
- 90.Guo HM, Zucker S, Gordon MK, Toole BP, Biswas C. Stimulation of matrix metalloproteinase production by recombinant extracellular matrix metalloproteinase inducer from transfected Chinese hamster ovary cells. J Biol Chem. 1997;272:24–27. [PubMed] [Google Scholar]
- 91.Gururajan R, Grenet J, Lahti JM, Kidd VJ. Isolation and characterization of two novel metalloproteinase genes linked to the Cdc2L locus on human chromosome 1p36.3. Genomics. 1998;52:101–106. doi: 10.1006/geno.1998.5401. [DOI] [PubMed] [Google Scholar]
- 92.Haagsman HP, Diemel RV. Surfactant-associated proteins: functions and structural variation. Comp Biochem Physiol A Mol Integr Physiol. 2001;129:91–108. doi: 10.1016/s1095-6433(01)00308-7. [DOI] [PubMed] [Google Scholar]
- 93.Haas TL. Endothelial cell regulation of matrix metalloproteinases. Can J Physiol Pharmacol. 2005;83:1–7. doi: 10.1139/y04-120. [DOI] [PubMed] [Google Scholar]
- 94.Haas TL, Stitelman D, Davis SJ, Apte SS, Madri JA. Egr-1 mediates extracellular matrix-driven transcription of membrane type 1 matrix metalloproteinase in endothelium. J Biol Chem. 1999;274:22679–22685. doi: 10.1074/jbc.274.32.22679. [DOI] [PubMed] [Google Scholar]
- 95.Han X, Boyd PJ, Colgan S, Madri JA, Haas TL. Transcriptional up-regulation of endothelial cell matrix metalloproteinase-2 in response to extracellular cues involves GATA-2. J Biol Chem. 2003;278:47785–47791. doi: 10.1074/jbc.M309482200. [DOI] [PubMed] [Google Scholar]
- 96.Han Z, Jun XU, Zhong N. Expression of matrix metalloproteinases MMP-9 within the airways in asthma. Respir Med. 2003;97:563–567. doi: 10.1053/rmed.2001.1162. [DOI] [PubMed] [Google Scholar]
- 97.Harris EDJ, Krane SM. An endopeptidase from rheumatoid synovial tissue culture. Biochim Biophys Acta. 1972;258:566–576. doi: 10.1016/0005-2744(72)90249-5. [DOI] [PubMed] [Google Scholar]
- 98.Harrison M, Abu-Elmagd M, Grocott T, Yates C, Gavrilovic J, Wheeler GN. Matrix metalloproteinase genes in Xenopus development. Dev Dyn. 2004;231:214–220. doi: 10.1002/dvdy.20113. [DOI] [PubMed] [Google Scholar]
- 99.Hasaneen NA, Zucker S, Cao J, Chiarelli C, Panettieri RA, Foda HD. Cyclic mechanical strain-induced proliferation and migration of human airway smooth muscle cells: role of EMMPRIN and MMPs. FASEB J. 2005;19:1507–1509. doi: 10.1096/fj.04-3350fje. [DOI] [PubMed] [Google Scholar]
- 100.Haseneen NA, Vaday GG, Zucker S, Foda HD. Mechanical stretch induces MMP-2 release and activation in lung endothelium: role of EMMPRIN. Am J Physiol Lung Cell Mol Physiol. 2003;284:L541–L547. doi: 10.1152/ajplung.00290.2002. [DOI] [PubMed] [Google Scholar]
- 101.Hasty KA, Pourmotabbed TF, Goldberg GI, Thompson JP, Spinella DG, Stevens RM, Mainardi CL. Human neutrophil collagenase: a distinct gene-product with homology to other matrix metalloproteinases. J Biol Chem. 1990;265:11421–11424. [PubMed] [Google Scholar]
- 102.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 103.Heine UI, Munoz EF, Flanders KC, Roberts AB, Sporn MB. Colocalization of TGF-beta 1 and collagen I and III, fibronectin and glycosaminoglycans during lung branching morphogenesis. Development. 1990;109:29–36. doi: 10.1242/dev.109.1.29. [DOI] [PubMed] [Google Scholar]
- 104.Herman MP, Sukhova GK, Kisiel W, Foster D, Kehry MR, Libby P, Schonbeck U. Tissue factor pathway inhibitor-2 is a novel inhibitor of matrix metalloproteinases with implications for atherosclerosis. J Clin Invest. 2001;107:1117–1126. doi: 10.1172/JCI10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hernandez-Pando R, Orozco H, Arriaga K, Pavon L, Rook G. Treatment with BB-94, a broad spectrum inhibitor of zinc-dependent metalloproteinases, causes deviation of the cytokine profile towards Type-2 in experimental pulmonary tuberculosis in Balb/c mice. Int J Exp Pathol. 2000;81:199–209. doi: 10.1046/j.1365-2613.2000.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Holla LI, Vasku A, Stejskalova A, Znojil V. Functional polymorphism in the gelatinase B gene and asthma. Allergy. 2000;55:900–901. doi: 10.1034/j.1398-9995.2000.00736.x. [DOI] [PubMed] [Google Scholar]
- 107.Hosford GE, Fang XIN, Olson DM. Hyperoxia decreases matrix metalloproteinase-9 and increases tissue inhibitor of matrix metalloproteinase-1 protein in the newborn rat lung: association with arrested alveolarization. Pediatr Res. 2004;56:26–34. doi: 10.1203/01.PDR.0000130658.45564.1F. [DOI] [PubMed] [Google Scholar]
- 108.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116:753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hsu LS, Lee HH, Chen KM, Chou HL, Lai SC. Matrix metalloproteinase-2 and -9 in the granulomatous fibrosis of rats infected with Angiostrongylus cantonensis. Ann Trop Med Parasitol. 2005;99:61–70. doi: 10.1179/136485905X19919. [DOI] [PubMed] [Google Scholar]
- 110.Huang MF, Lin WL, Ma YC. A study of reactive oxygen species in mainstream of cigarette. Indoor Air. 2005;15:135–140. doi: 10.1111/j.1600-0668.2005.00330.x. [DOI] [PubMed] [Google Scholar]
- 111.Hubner RH, Meffert S, Mundt U, Bottcher H, Freitag S, El Mokhtari NE, Pufe T, Hirt S, Folsch UR, Bewig B. Matrix metalloproteinase-9 in bronchiolitis obliterans syndrome after lung transplantation. Eur Respir J. 2005;25:494–501. doi: 10.1183/09031936.05.00091804. [DOI] [PubMed] [Google Scholar]
- 112.Irie K, Komori K, Seiki M, Tsuruga E, Sakakura Y, Kaku T, Yajima T. Impaired alveolization in mice deficient in membrane-type matrix metalloproteinase 1 (MT1-MMP). Med Mol Morphol. 2005;38:43–46. doi: 10.1007/s00795-004-0277-9. [DOI] [PubMed] [Google Scholar]
- 113.Ishii Y, Ogura T, Tatemichi M, Fujisawa H, Otsuka F, Esumi H. Induction of matrix metalloproteinase gene transcription by nitric oxide and mechanisms of MMP-1 gene induction in human melanoma cell lines. Int J Cancer. 2003;103:161–168. doi: 10.1002/ijc.10808. [DOI] [PubMed] [Google Scholar]
- 114.Ito I, Nagai S, Handa T, Muro S, Hirai T, Tsukino M, Mishima M. Matrix metalloproteinase-9 promoter polymorphism associated with upper lung dominant emphysema. Am J Respir Crit Care Med. 2005;172:1378–1382. doi: 10.1164/rccm.200506-953OC. [DOI] [PubMed] [Google Scholar]
- 115.Jeffery P. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164:S28–S38. doi: 10.1164/ajrccm.164.supplement_2.2106061. [DOI] [PubMed] [Google Scholar]
- 116.Joos L, He JQ, Shepherdson MB, Connett JE, Anthonisen NR, Pare PD, Sandford AJ. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Hum Mol Genet. 2002;11:569–576. doi: 10.1093/hmg/11.5.569. [DOI] [PubMed] [Google Scholar]
- 117.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta 1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kennedy AM, Inada M, Krane SM, Christie PT, Harding B, Lopez-Otin C, Sanchez LM, Pannett AAJ, Dearlove A, Hartley C, Byrne MH, Reed AAC, Nesbit MA, Whyte MP, Thakker RV. MMP13 mutation causes spondyloepimetaphyseal dysplasia, Missouri type (SEMDMO). J Clin Invest. 2005;115:2832–2842. doi: 10.1172/JCI22900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kheradmand F, Rishi K. The Role of Proteinases in Airway Remodeling. Dekker; New York: 2003. pp. 749–765. [Google Scholar]
- 120.Kheradmand F, Rishi K, Corry DB. Environmental contributions to the allergic asthma epidemic. Environ Health Perspect. 2002;110:553–556. doi: 10.1289/ehp.02110s4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kheradmand F, Werner E, Tremble P, Symons M, Werb Z. Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science. 1998;280:898–902. doi: 10.1126/science.280.5365.898. [DOI] [PubMed] [Google Scholar]
- 122.Kheradmand P, Rishi K, Werb Z. Signaling through the EGF receptor controls lung morphogenesis in part by regulating MT1-MMP-mediated activation of gelatinase A/MMP2. J Cell Sci. 2002;115:839–848. doi: 10.1242/jcs.115.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.King TEJ, Schwarz MI, Brown K, Tooze JA, Colby TV, Waldron JAJ, Flint A, Thurlbeck W, Cherniack RM. Idiopathic pulmonary fibrosis. Relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001;164:1025–1032. doi: 10.1164/ajrccm.164.6.2001056. [DOI] [PubMed] [Google Scholar]
- 124.Kinoh H, Hayashita H, Kajita M, Okada A, Seiki M. Assignment of the genes for membrane-type-4 matrix metalloproteinase (Mmp17, MMP17) to mouse chromosome 5, human chromosome band 12q24.3 and membrane-type-5 matrix metalloproteinase (MMP24, MMP24) to mouse chromosome 2 and human chromosome band 20q11 2 → q12, respectively, by radiation hybrid and in situ hybridization. Cytogenet Cell Genet. 1999;87:97–98. doi: 10.1159/000015402. [DOI] [PubMed] [Google Scholar]
- 125.Kinoshita T, Fukuzawa H, Shimada T, Saito T, Matsuda Y. Primary structure and expression of a gamete lytic enzyme in Chlamydomonas-Reinhardtii: similarity of functional domains to matrix metalloproteases. Proc Natl Acad Sci USA. 1992;89:4693–4697. doi: 10.1073/pnas.89.10.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Klier CM, Nelson EL, Cohen CD, Horuk R, Schlondorff D, Nelson PJ. Chemokine-induced secretion of gelatinase B in primary human monocytes. Biol Chem. 2001;382:1405–1410. doi: 10.1515/BC.2001.173. [DOI] [PubMed] [Google Scholar]
- 127.Kodali R, Hajjou M, Berman AB, Bansal MB, Zhang SH, Pan JJ, Schecter AD. Chemokines induce matrix metalloproteinase-2 through activation of epidermal growth factor receptor in arterial smooth muscle cells. Cardiovasc Res. 2006;69:706–715. doi: 10.1016/j.cardiores.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 128.Koshikawa N, Minegishi T, Sharabi A, Quaranta V, Seiki M. Membrane-type matrix metalloproteinase-1 (MT1-MMP) is a processing enzyme for human laminin gamma 2 chain. J Biol Chem. 2005;280:88–93. doi: 10.1074/jbc.M411824200. [DOI] [PubMed] [Google Scholar]
- 129.Koshy PJ, Henderson N, Logan C, Life PF, Cawston TE, Rowan AD. Interleukin 17 induces cartilage collagen breakdown: novel synergistic effects in combination with proinflammatory cytokines. Ann Rheum Dis. 2002;61:704–713. doi: 10.1136/ard.61.8.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kruidenier L, MacDonald TT, Collins JE, Pender SLF, Sanderson IR. Myofibroblast matrix metalloproteinases activate the neutrophil chemoattractant CXCL7 from intestinal epithelial cells. Gastroenterology. 2006;130:127–136. doi: 10.1053/j.gastro.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 131.Kucharewicz I, Kowal K, Buczko W, Bodzenta-Lukaszyk A. The plasmin system in airway remodeling. Thromb Res. 2003;112:1–7. doi: 10.1016/j.thromres.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 132.Kusano K, Miyaura C, Inada M, Tamura T, Ito A, Nagase H, Kamoi K, Suda T. Regulation of matrix metalloproteinases (MMP-2, -3, -9, and -13) by interleukin-1 and interleukin-6 in mouse calvaria: association of MMP induction with bone resorption. Endocrinology. 1998;139:1338–1345. doi: 10.1210/endo.139.3.5818. [DOI] [PubMed] [Google Scholar]
- 133.Lagente V, Manoury B, Nenan S, Le Quement C, Martin-Chouly C, Boichot E. Role of matrix metalloproteinases in the development of airway inflammation and remodeling. Braz J Med Biol Res. 2005;38:1521–1530. doi: 10.1590/s0100-879x2005001000009. [DOI] [PubMed] [Google Scholar]
- 134.Lamoreaux WJ, Fitzgerald MEC, Reiner A, Hasty KA, Charles ST. Vascular endothelial growth factor increases release of gelatinase a and decreases release of tissue inhibitor of metalloproteinases by microvascular endothelial cells in vitro. Microvasc Res. 1998;55:29–42. doi: 10.1006/mvre.1997.2056. [DOI] [PubMed] [Google Scholar]
- 135.Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, Chen Q, Homer RJ, Wang J, Rabach LA, Rabach ME, Shipley JM, Shapiro SD, Senior RM, Elias JA. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest. 2002;110:463–474. doi: 10.1172/JCI14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lapiere CM. Tadpole collagenase, the single parent of such a large family. Biochimie. 2005;87:243–247. doi: 10.1016/j.biochi.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 137.Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 138.Lass A, Suessenbacher A, Wolkart G, Mayer B, Brunner F. Functional and analytical evidence for scavenging of oxygen radicals by L-arginine. Mol Pharmacol. 2002;61:1081–1088. doi: 10.1124/mol.61.5.1081. [DOI] [PubMed] [Google Scholar]
- 139.Lawson WE, Polosukhin VV, Stathopoulos GT, Zoia O, Han W, Lane KB, Li B, Donnelly EF, Holburn GE, Lewis KG, Collins RD, Hull WM, Glasser SW, Whitsett JA, Blackwell TS. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol. 2005;167:1267–1277. doi: 10.1016/S0002-9440(10)61214-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee MM, Yoon BJ, Osiewicz K, Preston M, Bundy B, van Heeckeren AM, Werb Z, Soloway PD. Tissue inhibitor of metalloproteinase 1 regulates resistance to infection. Infect Immun. 2005;73:661–665. doi: 10.1128/IAI.73.1.661-665.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lee YC, Lee HB, Rhee YK, Song CH. The involvement of matrix metalloproteinase-9 in airway inflammation of patients with acute asthma. Clin Exp Allergy. 2001;31:1623–1630. doi: 10.1046/j.1365-2222.2001.01211.x. [DOI] [PubMed] [Google Scholar]
- 142.Lemke RP, Zhang W, Balcerazak D, Kobayashi K, Schwingshackl A, Cheung PY, Dixon WT, Baracos VE, Greer JJ. Expression and activity of matrix metalloproteinases 2 and 9 and their inhibitors in rat lungs during the perinatal period and in diaphragmatic hernia. Exp Lung Res. 2003;29:261–276. doi: 10.1080/01902140303783. [DOI] [PubMed] [Google Scholar]
- 143.Leontovich AA, Zhang JS, Shimokawa K, Nagase H, Sarras MP. A novel hydra matrix metalloproteinase (HMMP) functions in extracellular matrix degradation, morphogenesis and the maintenance of differentiated cells in the foot process. Development. 2000;127:907–920. doi: 10.1242/dev.127.4.907. [DOI] [PubMed] [Google Scholar]
- 144.Li CKF, Pender SLF, Pickard KM, Chance V, Holloway JA, Huett A, Goncalves NS, Mudgett JS, Dougan G, Frankel G, MacDonald TT. Impaired immunity to intestinal bacterial infection in stromelysin-1 (matrix metalloproteinase-3)-deficient mice. J Immunol. 2004;173:5171–5179. doi: 10.4049/jimmunol.173.8.5171. [DOI] [PubMed] [Google Scholar]
- 145.Li QL, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 146.Lian X, Qin Y, Hossain SA, Yang L, White A, Xu H, Shipley JM, Li T, Senior RM, Du H, Yan C. Overexpression of Stat3C in pulmonary epithelium protects against hyperoxic lung injury. J Immunol. 2005;174:7250–7256. doi: 10.4049/jimmunol.174.11.7250. [DOI] [PubMed] [Google Scholar]
- 147.Lim S, Roche N, Oliver BG, Mattos W, Barnes PJ, Fan Chung K. Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10. Am J Respir Crit Care Med. 2000;162:1355–1360. doi: 10.1164/ajrccm.162.4.9910097. [DOI] [PubMed] [Google Scholar]
- 148.Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz LA, Senior RM, Werb Z. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102:647–655. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 149.Llano E, Adam G, Pendas AM, Quesada V, Sanchez LM, Santamaria I, Noselli S, Lopez-Otin C. Structural and enzymatic characterization of Drosophila Dm2-MMP, a membrane-bound matrix metalloproteinase with tissue-specific expression. J Biol Chem. 2002;277:23321–23329. doi: 10.1074/jbc.M200121200. [DOI] [PubMed] [Google Scholar]
- 150.Llano E, Pendas AM, Aza-Blanc P, Kornberg TB, Lopez-Otin C. Dm1-MMP, a matrix metalloproteinase from Drosophila with a potential role in extracellular matrix remodeling during neural development. J Biol Chem. 2000;275:35978–35985. doi: 10.1074/jbc.M006045200. [DOI] [PubMed] [Google Scholar]
- 151.Llano P, Pendas AM, Freije JP, Nakano A, Knauper V, Murphy G, Lopez-Otin C. Identification and characterization of human MT5-MMP, a new membrane-bound activator of progelatinase A overexpressed in brain tumors. Cancer Res. 1999;59:2570–2576. [PubMed] [Google Scholar]
- 152.Lohi J, Wilson CL, Roby JD, Parks WC. Epilysin, a novel human matrix metalloproteinase (MMP-28) expressed in testis and keratinocytes and in response to injury. J Biol Chem. 2001;276:10134–10144. doi: 10.1074/jbc.M001599200. [DOI] [PubMed] [Google Scholar]
- 153.Lopez-Boado YS, Wilson CL, Parks WC. Regulation of matrilysin expression in airway epithelial cells by Pseudomonas aeruginosa flagellin. J Biol Chem. 2001;276:41417–41423. doi: 10.1074/jbc.M107121200. [DOI] [PubMed] [Google Scholar]
- 154.Lose F, Thompson PJ, Duffy D, Stewart GA, Kedda MA. A novel tissue inhibitor of metalloproteinase-1 (TIMP-1) polymorphism associated with asthma in Australian women. Thorax. 2005;60:623–628. doi: 10.1136/thx.2004.026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Maidment JM, Moore D, Murphy GP, Murphy G, Clark IM. Matrix metalloproteinase homologues from Arabidopsis thaliana: expression and activity. J Biol Chem. 1999;274:34706–34710. doi: 10.1074/jbc.274.49.34706. [DOI] [PubMed] [Google Scholar]
- 156.Malherbe DC, Erpenbeck VJ, Abraham SN, Crouch EC, Hohlfeld JM, Wright JR. Surfactant protein D decreases pollen-induced IgE-dependent mast cell degranulation. Am J Physiol Lung Cell Mol Physiol. 2005;289:L856–L866. doi: 10.1152/ajplung.00009.2005. [DOI] [PubMed] [Google Scholar]
- 157.Malloy JL, Wright JR. In vivo clearance of surfactant lipids during acute pulmonary inflammation. Respir Res. 2004;5:8. doi: 10.1186/1465-9921-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mannello F, Canesi L, Faimali M, Piazza V, Gallo G, Geraci S. Characterization of metalloproteinase-like activities in barnacle (Balanus amphitrite) nauplii. Comp Biochem Physiol B Biochem Mol Biol. 2003;135:17–24. doi: 10.1016/s1096-4959(03)00049-6. [DOI] [PubMed] [Google Scholar]
- 159.Mannello F, Canesi L, Gazzanelli G, Gallo G. Biochemical properties of metalloproteinases from the hemolymph of the mussel Mytilus galloprovincialis Lam. Comp Biochem Physiol B Biochem Mol Biol. 2001;128:507–515. doi: 10.1016/s1096-4959(00)00352-3. [DOI] [PubMed] [Google Scholar]
- 160.Marchenko GN, Marchenko ND, Strongin AY. The structure and regulation of the human and mouse matrix metalloproteinase-21 gene and protein. Biochem J. 2003;372:503–515. doi: 10.1042/BJ20030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Marchenko GN, Strongin AY. MMP-28, a new human matrix metalloproteinase with an unusual cysteine-switch sequence is widely expressed in tumors. Gene. 2001;265:87–93. doi: 10.1016/s0378-1119(01)00360-2. [DOI] [PubMed] [Google Scholar]
- 162.Martignetti JA, Al Aqeel A, Al Sewain W, Glucksman CE, Bahabri S, Meyer BF, Desnick RJ. The first matrix metalloproteinase disease: MMP-2 deficiency results in a multicentric osteolysis syndrome. Am J Hum Genet. 2001;69:189–189. [Google Scholar]
- 163.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 164.Massova I, Kotra LP, Fridman R, Mobashery S. Matrix metalloproteinases: structures, evolution, and diversification. FASEB J. 1998;12:1075–1095. [PubMed] [Google Scholar]
- 165.Masumoto K, de Rooij JD, Suita S, Rottier R, Tibboel D, de Krijger RR. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases during normal human pulmonary development. Histopathology. 2005;47:410–419. doi: 10.1111/j.1365-2559.2005.02228.x. [DOI] [PubMed] [Google Scholar]
- 166.McCawley LJ, Crawford HC, King LE, Jr, Mudgett J, Matrisian LM. A protective role for matrix metalloproteinase-3 in squamous cell carcinoma. Cancer Res. 2004;64:6965–6972. doi: 10.1158/0008-5472.CAN-04-0910. [DOI] [PubMed] [Google Scholar]
- 167.McGeehan G, Burkhart W, Anderegg R, Becherer JD, Gillikin JW, Graham JS. Sequencing and characterization of the soybean leaf metalloproteinase: structural and functional similarity to the matrix metalloproteinase family. Plant Physiol. 1992;99:1179–1183. doi: 10.1104/pp.99.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McGuire JK, Li Q, Parks WC. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am J Pathol. 2003;162:1831–1843. doi: 10.1016/S0002-9440(10)64318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.McGuire JK, Li QL, Parks WC. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am J Pathol. 2003;162:1831–1843. doi: 10.1016/S0002-9440(10)64318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.McMillan SJ, Kearley J, Campbell JD, Zhu XW, Larbi KY, Shipley JM, Senior RM, Nourshargh S, Lloyd CM. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol. 2004;172:2586–2594. doi: 10.4049/jimmunol.172.4.2586. [DOI] [PubMed] [Google Scholar]
- 171.McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, Overall CM. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276:43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- 172.McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–1206. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- 173.McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–1167. [PubMed] [Google Scholar]
- 174.Medley TL, Kingwell BA, Gatzka CD, Pillay P, Cole TJ. Matrix metalloproteinase-3 genotype contributes to age-related aortic stiffening through modulation of gene and protein expression. Circ Res. 2003;92:1254–1261. doi: 10.1161/01.RES.0000076891.24317.CA. [DOI] [PubMed] [Google Scholar]
- 175.Mirastschijski U, Johannesson K, Jeppsson B, Agren MS. Effect of a matrix metalloproteinase activity and TNF-alpha converting enzyme inhibitor on intra-abdominal adhesions. Eur Surg Res. 2005;37:68–75. doi: 10.1159/000083150. [DOI] [PubMed] [Google Scholar]
- 176.Mizuno S, Matsumoto K, Li MY, Nakamura T. HGF reduces advancing lung fibrosis in mice: a potential role for MMP-dependent myofibroblast apoptosis. FASEB J. 2005;19:580–582. doi: 10.1096/fj.04-1535fje. [DOI] [PubMed] [Google Scholar]
- 177.Molet S, Belleguic C, Lena H, Germain N, Bertrand CP, Shapiro SD, Planquois JM, Delaval P, Lagente V. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflammation Res. 2005;54:31–36. doi: 10.1007/s00011-004-1319-4. [DOI] [PubMed] [Google Scholar]
- 178.Montagnani C, Le Roux F, Berthe F, Escoubas JM. Cg-TIMP, an inducible tissue inhibitor of metalloproteinase from the Pacific oyster Crassostrea gigas with a potential role in wound healing and defense mechanisms. FEBS Lett. 2001;500:64–70. doi: 10.1016/s0014-5793(01)02559-5. [DOI] [PubMed] [Google Scholar]
- 179.Mora B, Bonamico M, Ferri M, Megiorni F, Osborn J, Pizzuti A, Mazzilli MC. Association of the matrix metalloproteinase-3 (MMP-3) promoter polymorphism with Celiac disease in male subjects. Hum Immunol. 2005;66:716–720. doi: 10.1016/j.humimm.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 180.Mott JD, Thomas CL, Rosenbach MT, Takahara K, Greenspan DS, Banda MJ. Posttranslational proteolytic processing of procollagen C-terminal proteinase enhancer releases a metalloproteinase inhibitor. J Biol Chem. 2000;275:1384–1390. doi: 10.1074/jbc.275.2.1384. [DOI] [PubMed] [Google Scholar]
- 181.Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–564. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Muller D, Quantin B, Gesnel MC, Milloncollard R, Abecassis J, Breathnach R. The collagenase gene family in humans consists of at least 4 members. Biochem J. 1988;253:187–192. doi: 10.1042/bj2530187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Munoz P, Vance K, Gomez-Chiarri M. Protease activity in the plasma of American oysters, Crassostrea virginica, experimentally infected with the protozoan parasite Perkinsus marinus. J Parasitol. 2003;89:941–951. doi: 10.1645/GE-3126. [DOI] [PubMed] [Google Scholar]
- 184.Naureckas ET, Ndukwu IM, Halayko AJ, Maxwell C, Hershenson MB, Solway J. Bronchoalveolar lavage fluid from asthmatic subjects is mitogenic for human airway smooth muscle. Am J Respir Crit Care Med. 1999;160:2062–2066. doi: 10.1164/ajrccm.160.6.9903131. [DOI] [PubMed] [Google Scholar]
- 185.Nelson KK, Melendez JA. Mitochondrial redox control of matrix metalloproteinases. Free Radical Biol Med. 2004;37:768–784. doi: 10.1016/j.freeradbiomed.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 186.Nenan S, Planquois JM, Berna P, De Mendez I, Hitier S, Shapiro SD, Boichot E, Lagente V, Bertrand CP. Analysis of the inflammatory response induced by rhMMP-12 catalytic domain instilled in mouse airways. Int Immunopharmacol. 2005;5:511–524. doi: 10.1016/j.intimp.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 187.Neuringer IP, Chalermskulrat W, Aris R. Obliterative bronchiolitis or chronic lung allograft rejection: a basic science review. J Heart Lung Transplant. 2005;24:3–19. doi: 10.1016/j.healun.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 188.Newby AC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85:1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- 189.Ning W, Li CJ, Kaminski N, Feghali-Bostwick CA, Alber SM, Di YP, Otterbein SL, Song R, Hayashi S, Zhou Z, Pinsky DJ, Watkins SC, Pilewski JM, Sciurba FC, Peters DG, Hogg JC, Choi AM. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci USA. 2004;101:14895–14900. doi: 10.1073/pnas.0401168101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nishijima C, Hayakawa I, Matsushita T, Komura K, Hasegawa M, Takehara K, Sato S. Autoantibody against matrix metalloproteinase-3 in patients with systemic sclerosis. Clin Exp Immunol. 2004;138:357–363. doi: 10.1111/j.1365-2249.2004.02615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nogee LM, Dunbar AE, Wert S, Askin F, Hamvas A, Whitsett JA. Mutations in the surfactant protein C gene associated with interstitial lung disease. Chest. 2002;121:20S–21S. doi: 10.1378/chest.121.3_suppl.20s. [DOI] [PubMed] [Google Scholar]
- 192.Nogee LM, Dunbar AE, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344:573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 193.Oblander SA, Zhou ZJ, Galvez BG, Starcher B, Shannon JM, Durbeej M, Arroyo AG, Tryggvason K, Apte SS. Distinctive functions of membrane type 1 matrix-metalloprotease (MT1-MMP or MMP-14) in lung and submandibular gland development are independent of its role in pro-MMP-2 activation. Dev Biol. 2005;277:255–269. doi: 10.1016/j.ydbio.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 194.Ochs M, Knudsen L, Allen L, Stumbaugh A, Levitt S, Nyengaard JR, Hawgood S. GM-CSF mediates alveolar epithelial type II cell changes, but not emphysema-like pathology, in SP-D-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1333–L1341. doi: 10.1152/ajplung.00137.2004. [DOI] [PubMed] [Google Scholar]
- 195.Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, Ide C, Horan TP, Arakawa T, Yoshida H, Nishikawa SI, Itoh Y, Seiki M, Itohara S, Takahashi C, Noda M. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- 196.Ohno I, Ohtani H, Nitta Y, Suzuki J, Hoshi H, Honma M, Isoyama S, Tanno Y, Tamura G, Yamauchi K, Nagura H, Shirato K. Eosinophils as a source of matrix metalloproteinase-9 in asthmatic airway inflammation. Am J Respir Cell Mol Biol. 1997;16:212–219. doi: 10.1165/ajrcmb.16.3.9070604. [DOI] [PubMed] [Google Scholar]
- 197.Okamoto T, Akuta T, Tamura F, van Der Vliet A, Akaike T. Molecular mechanism for activation and regulation of matrix metalloproteinases during bacterial infections and respiratory inflammation. Biol Chem. 2004;385:997–1006. doi: 10.1515/BC.2004.130. [DOI] [PubMed] [Google Scholar]
- 198.Okuma T, Terasaki Y, Kaikita K, Kobayashi H, Kuziel WA, Kawasuji M, Takeya M. C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J Pathol. 2004;204:594–604. doi: 10.1002/path.1667. [DOI] [PubMed] [Google Scholar]
- 199.Overall CM, Kleifeld O. Tumour microenvironment - opinion -validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nature Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 200.Overall CM, Wrana JL, Sodek J. Transcriptional and posttranscriptional regulation of 72-kDa gelatinase type-IV collagenase by transforming growth factor-beta-1 in human fibroblasts: comparisons with collagenase and tissue inhibitor of matrix metalloproteinase gene-expression. J Biol Chem. 1991;266:14064–14071. [PubMed] [Google Scholar]
- 201.Owen CA. Proteinases and oxidants as targets in the treatment of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:373–385. doi: 10.1513/pats.200504-029SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Owen CA, Hu Z, Barrick B, Shapiro SD. Inducible expression of tissue inhibitor of metalloproteinases-resistant matrix metalloproteinase-9 on the cell surface of neutrophils. Am J Respir Cell Mol Biol. 2003;29:283–294. doi: 10.1165/rcmb.2003-0034OC. [DOI] [PubMed] [Google Scholar]
- 203.Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172:7791–7803. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- 204.Owens MW, Milligan SA, Jourd'heuil D, Grisham MB. Effects of reactive metabolites of oxygen and nitrogen on gelatinase A activity. Am J Physiol. 1997;273 doi: 10.1152/ajplung.1997.273.2.L445. [DOI] [PubMed] [Google Scholar]
- 205.Ozdemir D, Hart PS, Ryu OH, Choi SJ, Ozdemir-Karatas M, Firatli E, Piesco N, Hart TC. MMP20 active-site mutation in hypomaturation Amelogenesis Imperfecta. J Dent Res. 2005;84:1031–1035. doi: 10.1177/154405910508401112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Page-McCaw A, Serano J, Sante JM, Rubin GM. Drosophila matrix metalloproteinases are required for tissue remodeling, but not embryonic development. Dev Cell. 2003;4:95–106. doi: 10.1016/s1534-5807(02)00400-8. [DOI] [PubMed] [Google Scholar]
- 207.Palei AC, Zaneti RA, Fortuna GM, Gerlach RF, Tanus-Santos JE. Hemodynamic benefits of matrix metalloproteinase-9 inhibition by doxycycline during experimental acute pulmonary embolism. Angiology. 2005;56:611–617. doi: 10.1177/000331970505600513. [DOI] [PubMed] [Google Scholar]
- 208.Pardo A, Gibson K, Cisneros J, Richards TJ, Yang Y, Becerril C, Yousem S, Herrera I, Ruiz V, Selman M, Kaminski N. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Medicine/Public Library Sci. 2005 doi: 10.1371/journal.pmed.0020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Park HI, Ni J, Gerkema FE, Liu D, Belozerov VE, Sang QXA. Identification and characterization of human endometase (matrix metalloproteinase-26) from endometrial tumor. J Biol Chem. 2000;275:20540–20544. doi: 10.1074/jbc.M002349200. [DOI] [PubMed] [Google Scholar]
- 210.Park PW, Pier GB, Hinkes MT, Bernfield M. Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature. 2001;411:98–102. doi: 10.1038/35075100. [DOI] [PubMed] [Google Scholar]
- 211.Parks WC, Lopez-Boado YS, Wilson CL. Matrilysin in epithelial repair and defense. Chest. 2001;120:36S–41S. doi: 10.1378/chest.120.1_suppl.s36. [DOI] [PubMed] [Google Scholar]
- 212.Parks WC, Shapiro SD. Matrix metalloproteinases in lung biology. Respir Res. 2001;2:10–19. doi: 10.1186/rr33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 214.Patel AC, McAlister WH, Whyte MP. Spondyloepimetaphyseal dysplasia: clinical and radiologic investigation of a large kindred manifesting autosomal-dominant inheritance, and a review of the literature. Medicine. 1993;72:326–342. [PubMed] [Google Scholar]
- 215.Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163:1256–1276. doi: 10.1164/ajrccm.163.5.2101039. [DOI] [PubMed] [Google Scholar]
- 216.Pei D, Weiss SJ. Transmembrane-deletion mutants of the membrane-type matrix metalloproteinase-1 process progelatinase A and express intrinsic matrix-degrading activity. J Biol Chem. 1996;271:9135–9140. doi: 10.1074/jbc.271.15.9135. [DOI] [PubMed] [Google Scholar]
- 217.Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arteriosclerosis Thromb Vasc Biol. 2001;21:1104–1117. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- 218.Petitclerc E, Boutaud A, Prestayko A, Xu JS, Sado Y, Ninomiya Y, Sarras MP, Hudson BG, Brooks PC. New functions for non-collagenous domains of human collagen type IV: novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem. 2000;275:8051–8061. doi: 10.1074/jbc.275.11.8051. [DOI] [PubMed] [Google Scholar]
- 219.Pignatti P, Brunetti G, Moretto D, Yacoub MR, Fiori M, Balbi B, Balestrino A, Cervio G, Nava S, Moscato G. Role of the chemokine receptors CXCR3 and CCR4 in human pulmonary fibrosis. Am J Respir Crit Care Med. 2006;173:310–317. doi: 10.1164/rccm.200502-244OC. [DOI] [PubMed] [Google Scholar]
- 220.Prause O, Bozinovski S, Anderson GP, Linden A. Increased matrix metalloproteinase-9 concentration and activity after stimulation with interleukin-17 in mouse airways. Thorax. 2004;59:313–317. doi: 10.1136/thx.2003.008854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Presneill JJ, Nakata K, Inoue Y, Seymour JF. Pulmonary alveolar proteinosis. Clin Chest Med. 2004;25:593. doi: 10.1016/j.ccm.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 222.Puente XS, Pendas AM, Llano E, Velasco G, LopezOtin C. Molecular cloning of a novel membrane-type matrix metalloproteinase from a human breast carcinoma. Cancer Res. 1996;56:944–949. [PubMed] [Google Scholar]
- 223.Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med. 1999;27:304–312. doi: 10.1097/00003246-199902000-00036. [DOI] [PubMed] [Google Scholar]
- 224.Pugin J, Widmer MC, Kossodo S, Liang CM, Preas HLN, Suffredini AF. Human neutrophils secrete gelatinase B in vitro and in vivo in response to endotoxin and proinflammatory mediators. Am J Respir Cell Mol Biol. 1999;20:458–464. doi: 10.1165/ajrcmb.20.3.3311. [DOI] [PubMed] [Google Scholar]
- 225.Ratjen F, Hartog CM, Paul K, Wermelt J, Braun J. Matrix metalloproteases in BAL fluid of patients with cystic fibrosis and their modulation by treatment with dornase alpha. Thorax. 2002;57:930–934. doi: 10.1136/thorax.57.11.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Rivera-Marrero CA, Schuyler W, Roser S, Ritzenthaler JD, Newburn SA, Roman J. M-tuberculosis induction of matrix metalloproteinase-9: the role of mannose and receptor-mediated mechanisms. Am J Physiol Lung Cell Mol Physiol. 2002;282:L546–L555. doi: 10.1152/ajplung.00175.2001. [DOI] [PubMed] [Google Scholar]
- 227.Rivera-Marrero CA, Schuyler W, Roser S, Roman J. Induction of MMP-9 mediated gelatinolytic activity in human monocytic cells by cell wall components of Mycobacterium tuberculosis. Microbial Pathogenesis. 2000;29:231–244. doi: 10.1006/mpat.2000.0383. [DOI] [PubMed] [Google Scholar]
- 228.Robinson JJ. Characterization of a metalloproteinase: a late stage specific gelatinase activity in the sea urchin embryo. J Cell Biochem. 1997;66:337–345. [PubMed] [Google Scholar]
- 229.Ryu J, Vicencio AG, Yeager ME, Kashgarian M, Haddad GG, Eickelberg O. Differential expression of matrix metalloproteinases and their inhibitors in human and mouse lung development. Thrombosis Haemostasis. 2005;94:175–183. doi: 10.1160/TH04-10-0656. [DOI] [PubMed] [Google Scholar]
- 230.Saari H, Sorsa T, Lindy O, Suomalainen K, Halinen S, Konttinen YT. Reactive oxygen species as regulators of human neutrophil and fibroblast interstitial collagenases. Int J Tissue Reactions Exp Clin Aspects. 1992;14:113–120. [PubMed] [Google Scholar]
- 231.Saarialhokere UK, Welgus HG, Parks WC. Distinct mechanisms regulate interstitial collagenase and 92-kDa gelatinase expression in human monocytic-like cells exposed to bacterial-endotoxin. J Biol Chem. 1993;268:17354–17361. [PubMed] [Google Scholar]
- 232.Sagel SD, Kapsner RK, Osberg I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr Pulmonol. 2005;39:224–232. doi: 10.1002/ppul.20165. [DOI] [PubMed] [Google Scholar]
- 233.Sagel SD, Kapsner RK, Osberg I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr Pulmonol. 2005;39:224–232. doi: 10.1002/ppul.20165. [DOI] [PubMed] [Google Scholar]
- 234.Sapolsky AI, Howell DS, Woessner JF., Jr Neutral proteases and cathepsin D in human articular cartilage. J Clin Invest. 1974;53:1044–1053. doi: 10.1172/JCI107641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M. A matrix metalloproteinase expressed on the surface of invasive tumor-cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 236.Scaffidi AK, Petrovic N, Moodley YP, Fogel-Petrovic M, Kroeger KM, Seeber RM, Eidne KA, Thompson PJ, Knight DA. αvβ3 Integrin interacts with the transforming growth factor β (TGFβ) type II receptor to potentiate the proliferative effects of TGFβ1 in living human lung fibroblasts. J Biol Chem. 2004;279:37726–37733. doi: 10.1074/jbc.M403010200. [DOI] [PubMed] [Google Scholar]
- 237.Schonbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- 238.Schwartz DA, Van Fossen DS, Davis CS, Helmers RA, Dayton CS, Burmeister LF, Hunninghake GW. Determinants of progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1994;149:444–449. doi: 10.1164/ajrccm.149.2.8306043. [DOI] [PubMed] [Google Scholar]
- 239.Sellers A, Woessner JF. The extraction of a neutral metalloproteinase from the involuting rat uterus, and its action on cartilage proteoglycan. Biochem J. 1980;189:521–531. doi: 10.1042/bj1890521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, Aziz N, Kaminski N, Zlotnik A. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2006;173:188–198. doi: 10.1164/rccm.200504-644OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. 2005;6:551–557. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- 242.Senft AP, Korfhagen TR, Whitsett JA, Shapiro SD, LeVine AM. Surfactant protein-D regulates soluble CD14 through matrix metalloproteinase-12. J Immunol. 2005;174:4953–4959. doi: 10.4049/jimmunol.174.8.4953. [DOI] [PubMed] [Google Scholar]
- 243.Senior RM. Mechanisms of COPD: conference summary. Chest. 2000;117:320S–323S. doi: 10.1378/chest.117.5_suppl_1.320s-a. [DOI] [PubMed] [Google Scholar]
- 244.Shapiro SD, Campbell EJ, Kobayashi DK, Welgus HG. Immune modulation of metalloproteinase production in human macrophages. Selective pretranslational suppression of interstitial collagenase and stromelysin biosynthesis by interferon-gamma. J Clin Invest. 1990;86:1204–1210. doi: 10.1172/JCI114826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Shapiro SD, Griffin GL, Gilbert DJ, Jenkins NA, Copeland NG, Welgus HG, Senior RM, Ley TJ. Molecular-cloning, chromosomal localization, and bacterial expression of a murine macrophage metalloelastase. J Biol Chem. 1992;267:4664–4671. [PubMed] [Google Scholar]
- 246.Shapiro SD, Senior RM. Matrix metalloproteinases. Matrix degradation and more. Am J Respir Cell Mol Biol. 1999;20:1100–1102. doi: 10.1165/ajrcmb.20.6.f151. [DOI] [PubMed] [Google Scholar]
- 247.Shimoyama T, Tabuchi N, Chung JW, Koyama T, Sunamori M. Matrix metalloproteinase inhibitor (ONO-4817) attenuates ischemia-reperfusion injury in rat lung. Med Sci Monitor. 2006;12:BR51–BR56. [PubMed] [Google Scholar]
- 248.Shiomi T, Inoki I, Kataoka F, Ohtsuka T, Hashimoto G, Nemori R, Okada Y. Pericellular activation of proMMP-7 (promatrilysin-1) through interaction with CD151. Lab Invest. 2005;85:1489–1506. doi: 10.1038/labinvest.3700351. [DOI] [PubMed] [Google Scholar]
- 249.Singh S, Singh UP, Stiles JK, Grizzle WE, Lillard JW. Expression and functional role of CCR9 in prostate cancer cell migration and invasion. Clin Cancer Res. 2004;10:8743–8750. doi: 10.1158/1078-0432.CCR-04-0266. [DOI] [PubMed] [Google Scholar]
- 250.Sivakumar P, Sampath P, Chandrakasan G. Collagenolytic metalloprotease (gelatinase) from the hepatopancreas of the marine crab, Scylla serrata. Comp Biochem Physiol B Biochem Mol Biol. 1999;123:273–279. [Google Scholar]
- 251.Son KN, Hwang JS, Kwon BS, Kim JY. Human CC chemokine CCL23 enhances expression of matrix metalloproteinase-2 and invasion of vascular endothelial cells. Biochem Biophys Res Commun. 2006;340:498–504. doi: 10.1016/j.bbrc.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 252.Soref CM, Di YP, Hayden L, Zhao YH, Satre MA, Wu R. Characterization of a novel airway epithelial cell-specific short chain alcohol dehydrogenase/reductase gene whose expression is up-regulated by retinoids and is involved in the metabolism of retinol. J Biol Chem. 2001;276:24194–24202. doi: 10.1074/jbc.M100332200. [DOI] [PubMed] [Google Scholar]
- 253.Souza-Costa DC, Zerbini T, Palei AC, Gerlach RF, Tanus-Santos JE. l-Arginine attenuates acute pulmonary embolism-induced increases in lung matrix metalloproteinase-2 and matrix metalloproteinase-9. Chest. 2005;128:3705–3710. doi: 10.1378/chest.128.5.3705. [DOI] [PubMed] [Google Scholar]
- 254.Starcher B, Williams I. The beige mouse: role of neutrophil elastase in the development of pulmonary emphysema. Exp Lung Res. 1989;15:785–800. doi: 10.3109/01902148909062861. [DOI] [PubMed] [Google Scholar]
- 255.Sternlicht MD, Werb Z. ECM. Proteinases. In: Kreis T, Vale R, editors. Guidebook to the Extracellular Matrix and Adhesion Proteins. Oxford Univ. Press; New York: 1999. pp. 503–562. [Google Scholar]
- 256.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Stetefeld J, Jenny M, Schulthess T, Landwehr R, Schumacher B, Frank S, Ruegg MA, Engel J, Kammerer RA. The laminin-binding domain of agrin is structurally related to N-TIMP-1. Nature Struct Biol. 2001;8:705–709. doi: 10.1038/90422. [DOI] [PubMed] [Google Scholar]
- 258.Stolow MA, Bauzon DD, Li JW, Sedgwick T, Liang VCT, Sang QXA, Shi YB. Identification and characterization of a novel collagenase in Xenopus laevis: possible roles during frog development. Mol Biol Cell. 1996;7:1471–1483. doi: 10.1091/mbc.7.10.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Stricker TP, Dumin JA, Dickeson SK, Chung L, Nagase H, Parks WC, Santoro SA. Structural analysis of the alpha(2) integrin I domain/procollagenase-1 (matrix metalloproteinase-1) interaction. J Biol Chem. 2001;276:29375–29381. doi: 10.1074/jbc.M102217200. [DOI] [PubMed] [Google Scholar]
- 260.Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- 261.Su WY, Jaskot RH, Dreher KL. Particulate matter induction of pulmonary gelatinase A, gelatinase B, and tissue inhibitor of metalloproteinase expression. Inhalation Toxicol. 2000;12:105–119. doi: 10.1080/08958378.2000.11463203. [DOI] [PubMed] [Google Scholar]
- 262.Su WY, Jaskot RH, Richards J, Abramson SR, Woessner JF, Yu WH, Dreher KL. Induction of pulmonary matrilysin expression by combustion and ambient air particles. Am J Physiol Lung Cell Mol Physiol. 2000;279:L152–L160. doi: 10.1152/ajplung.2000.279.1.L152. [DOI] [PubMed] [Google Scholar]
- 263.Sun HB, Yokota H. Messenger-RNA expression of matrix metalloproteinases, tissue inhibitors of metalloproteinases, and transcription factors in rheumatic synovial cells under mechanical stimuli. Bone. 2001;28:303–309. doi: 10.1016/s8756-3282(00)00454-3. [DOI] [PubMed] [Google Scholar]
- 264.Sun JX, Hemler ME. Regulation of MMP-1 and MMP-2 production through CD147/extracellular matrix metalloproteinase inducer interactions. Cancer Res. 2001;61:2276–2281. [PubMed] [Google Scholar]
- 265.Suzuki S, Sato M, Senoo H, Ishikawa K. Direct cell-cell interaction enhances pro-MMP-2 production and activation in co-culture of laryngeal cancer cells and fibroblasts: involvement of EMMPRIN and MT1-MMP. Exp Cell Res. 2004;293:259–266. doi: 10.1016/j.yexcr.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 266.Swartz MA, Tschumperlin DJ, Kamm RD, Drazen JM. Mechanical stress is communicated between different cell types to elicit matrix remodeling. Proc Natl Acad Sci USA. 2001;98:6180–6185. doi: 10.1073/pnas.111133298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Taghavi S, Krenn K, Jaksch P, Klepetko W, Aharinejad S. Broncho-alveolar lavage matrix metalloproteases as a sensitive measure of bronchiolitis obliterans. Am J Transplant. 2005;5:1548–1552. doi: 10.1111/j.1600-6143.2005.00865.x. [DOI] [PubMed] [Google Scholar]
- 268.Takagi M, Kikko T, Hosoi M, Hayashi I, Toyohara H. CDNA cloning of oyster matrix metalloproteinase and its possible involvement in hypoxic adaptation. Fish Sci. 2004;70:682–687. [Google Scholar]
- 269.Takino T, Sato H, Shinagawa A, Seiki M. Identification of the 2nd membrane-type matrix metalloproteinase (Mt-Mmp-2) gene from a human placenta cDNA library - Mt-Mmps form a unique membrane-type subclass in the Mmp Family. J Biol Chem. 1995;270:23013–23020. doi: 10.1074/jbc.270.39.23013. [DOI] [PubMed] [Google Scholar]
- 270.Tambunting F, Beharry KD, Hartleroad J, Waltzman J, Stavitsky Y, Modanlou HD. Increased lung matrix metalloproteinase-9 levels in extremely premature baboons with bronchopulmonary dysplasia. Pediatr Pulmonol. 2005;39:5–14. doi: 10.1002/ppul.20135. [DOI] [PubMed] [Google Scholar]
- 271.Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. 2005:9291–9296. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 272.Thomson E, Kumarathasan P, Goegan P, Aubin RA, Vincent R. Differential regulation of the lung endothelin system by urban particulate matter and ozone. Toxicol Sci. 2005;88:103–113. doi: 10.1093/toxsci/kfi272. [DOI] [PubMed] [Google Scholar]
- 273.Trask BC, Malone MJ, Lum EH, Welgus HG, Crouch EC, Shapiro SD. Induction of macrophage matrix metalloproteinase biosynthesis by surfactant protein D. J Biol Chem. 2001;276:37846–37852. doi: 10.1074/jbc.M102524200. [DOI] [PubMed] [Google Scholar]
- 274.Tschumperlin DJ. EGFR autocrine signaling in a compliant interstitial space: mechanotransduction from the outside in. Cell Cycle. 2004;3:996–997. doi: 10.4161/cc.3.8.1062. [DOI] [PubMed] [Google Scholar]
- 275.Tschumperlin DJ, Dai G, Maly IV, Kikuchi T, Laiho LH, McVittie AK, Haley KJ, Lilly CM, So PT, Lauffenburger DA, Kamm RD, Drazen JM. Mechanotransduction through growth-factor shedding into the extracellular space. Nature. 2004;429:83–86. doi: 10.1038/nature02543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Tschumperlin DJ, Drazen JM. Chronic effects of mechanical force on airways. Annu Rev Physiol. 2005;30:30. doi: 10.1146/annurev.physiol.68.072304.113102. [DOI] [PubMed] [Google Scholar]
- 277.Tu HF, Liu CJ, Chang CS, Lui MT, Kao SY, Chang CP, Liu TY. The functional (−1171 5A → 6A) polymorphisms of matrix metalloproteinase 3 gene as a risk factor for oral submucous fibrosis among male areca users. J Oral Pathol Med. 2006;35:99–103. doi: 10.1111/j.1600-0714.2006.00370.x. [DOI] [PubMed] [Google Scholar]
- 278.Turk BE, Cantley LC. Using peptide libraries to identify optimal cleavage motifs for proteolytic enzymes. Methods. 2004;32:398–405. doi: 10.1016/j.ymeth.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 279.Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nature Biotechnol. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 280.Uparanukraw P, Morakote N, Harnnoi T, Dantrakool A. Molecular cloning of a gene encoding matrix metalloproteinase-like protein from Gnathostoma spinigerum. Parasitol Res. 2001;87:751–757. doi: 10.1007/s004360100440. [DOI] [PubMed] [Google Scholar]
- 281.Valenca SS, da Hora K, Castro P, Moraes VG, Carvalho L, Porto LC. Emphysema and metalloelastase expression in mouse lung induced by cigarette smoke. Toxicol Pathol. 2004;32:351–356. doi: 10.1080/01926230490431466. [DOI] [PubMed] [Google Scholar]
- 282.Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–2681. [PubMed] [Google Scholar]
- 283.Van den Steen PE, Wuyts A, Husson SJ, Proost P, Van Damme J, Opdenakker G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur J Biochem. 2003;270:3739–3749. doi: 10.1046/j.1432-1033.2003.03760.x. [DOI] [PubMed] [Google Scholar]
- 284.Venkayya R, Lam M, Willkom M, Grunig G, Corry DB, Erle DJ. The Th2 lymphocyte products IL-4 and IL-13 rapidly induce airway hyperresponsiveness through direct effects on resident airway cells. Am J Respir Cell Mol Biol. 2002;26:202–208. doi: 10.1165/ajrcmb.26.2.4600. [DOI] [PubMed] [Google Scholar]
- 285.Vinarsky V, Atkinson DL, Stevenson TJ, Keating MT, Odelberg SJ. Normal newt limb regeneration requires matrix metalloproteinase function. Dev Biol. 2005;279:86–98. doi: 10.1016/j.ydbio.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 286.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Vuk-Pavlovic Z, Mo EK, Icenhour CR, Standing JE, Fisher JH, Limper AH. Surfactant protein D enhances Pneumocystis infection in immune-suppressed mice. Am J Physiol Lung Cell Mol Physiol. 2006;290:L442–L449. doi: 10.1152/ajplung.00112.2005. [DOI] [PubMed] [Google Scholar]
- 288.Wada K, Sato H, Kinoh H, Kajita M, Yamamoto H, Seiki M. Cloning of three Caenorhabditis elegans genes potentially encoding novel matrix metalloproteinases. Gene. 1998;211:57–62. doi: 10.1016/s0378-1119(98)00076-6. [DOI] [PubMed] [Google Scholar]
- 289.Wang H, Parry S, Macones G, Sammel MD, Ferrand PE, Kuivaniemi H, Tromp G, Halder I, Shriver MD, Romero R, Strauss JF. Functionally significant SNP MMP8 promoter haplotypes and preterm premature rupture of membranes (PPROM). Hum Mol Genet. 2004;13:2659–2669. doi: 10.1093/hmg/ddh287. [DOI] [PubMed] [Google Scholar]
- 290.Wang X, Inoue S, Gu J, Miyoshi E, Noda K, Li W, Mizuno-Horikawa Y, Nakano M, Asahi M, Takahashi M, Uozumi N, Ihara S, Lee SH, Ikeda Y, Yamaguchi Y, Aze Y, Tomiyama Y, Fujii J, Suzuki K, Kondo A, Shapiro SD, Lopez-Otin C, Kuwaki T, Okabe M, Honke K, Taniguchi N. Dysregulation of TGF-beta1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc Natl Acad Sci USA. 2005;102:15791–15796. doi: 10.1073/pnas.0507375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA, Jr, Shapiro SD, Elias JA. Interferon gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med. 2000;192:1587–1600. doi: 10.1084/jem.192.11.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12:317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 293.Wenzel SE, Balzar S, Cundall M, Chu HW. Subepithelial basement membrane immunoreactivity for matrix metalloproteinase 9. Association with asthma severity, neutrophilic inflammation, and wound repair. J Allergy Clin Immunol. 2003;111:1345–1352. doi: 10.1067/mai.2003.1464. [DOI] [PubMed] [Google Scholar]
- 294.Werb Z, Reynolds JJ. Stimulation by endocytosis of the secretion of collagenase and neutral proteinase from rabbit synovial fibroblasts. J Exp Med. 1974;140:1482–1497. doi: 10.1084/jem.140.6.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Werb Z, Tremble PM, Behrendtsen O, Crowley E, Damsky CH. Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J Cell Biol. 1989;109:877–889. doi: 10.1083/jcb.109.2.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Wert SE, Yoshida M, LeVine AM, Ikegami M, Jones T, Ross GF, Fisher JH, Korfhagen TR, Whitsett JA. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc Natl Acad Sci USA. 2000;97:5972–5977. doi: 10.1073/pnas.100448997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Wilhelm SM, Collier IE, Marmer BL, Eisen AZ, Grant GA, Goldberg GI. SV40-transformed human-lung fibroblasts secrete a 92-kDa type-IV collagenase which is identical to that secreted by normal human macrophages. J Biol Chem. 1989;264:17213–17221. [PubMed] [Google Scholar]
- 298.Will H, Hinzmann B. cDNA sequence and messenger-RNA tissue distribution of a novel human matrix metalloproteinase with a potential transmembrane segment. Eur J Biochem. 1995;231:602–608. doi: 10.1111/j.1432-1033.1995.tb20738.x. [DOI] [PubMed] [Google Scholar]
- 299.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-ss 1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Wilson CL, Heppner KJ, Labosky PA, Hogan BLM, Matrisian LM. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci USA. 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Wilson CL, Heppner KJ, Rudolph LA, Matrisian LM. The metalloproteinase matrilysin is preferentially expressed by epithelial cells in a tissue-restricted pattern in the mouse. Mol Biol Cell. 1995;6:851–869. doi: 10.1091/mbc.6.7.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 303.Winkler MK, Foldes JK, Bunn RC, Fowlkes JL. Implications for matrix metalloproteinases as modulators of pediatric lung disease. Am J Physiol Lung Cell Mol Physiol. 2003;284:L557–L565. doi: 10.1152/ajplung.00195.2002. [DOI] [PubMed] [Google Scholar]
- 304.Woessner F, Nagase H. Matrix Metalloproteinases and TIMPs. Oxford Univ. Press; Oxford, UK: 2000. p. 223. [Google Scholar]
- 305.Wright JL, Churg A. Animal models of cigarette smoke-induced COPD. Chest. 2002;122:301S–306S. doi: 10.1378/chest.122.6_suppl.301s. [DOI] [PubMed] [Google Scholar]
- 306.Wright JR. Immunoregulatory functions of surfactant proteins. Nature Rev Immunol. 2005;5:58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 307.Wu SG, Lim KC, Huang J, Saidi RF, Sears CL. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc Natl Acad Sci USA. 1998;95:14979–14984. doi: 10.1073/pnas.95.25.14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Yamamoto T, Eckes B, Mauch C, Hartmann K, Krieg T. Monocyte chemoattractant protein-1 enhances gene expression and synthesis of matrix metalloproteinase-1 in human fibroblasts by an autocrine IL-1α loop. J Immunol. 2000;164:6174–6179. doi: 10.4049/jimmunol.164.12.6174. [DOI] [PubMed] [Google Scholar]
- 309.Yang MZ, Kurkinen M. Cloning and characterization of a novel matrix metalloproteinase (MMP), CMMP, from chicken embryo fibroblasts: CMMP, Xenopus XMMP, and human MMP19 have a conserved unique cysteine in the catalytic domain. J Biol Chem. 1998;273:17893–17900. doi: 10.1074/jbc.273.28.17893. [DOI] [PubMed] [Google Scholar]
- 310.Yang MZ, Murray MT, Kurkinen M. A novel matrix metalloproteinase gene (XMMP) encoding vitronectin-like motifs is transiently expressed in Xenopus early development. Matrix Biol. 1997;16:80–80. doi: 10.1074/jbc.272.21.13527. [DOI] [PubMed] [Google Scholar]
- 311.Yao PM, Maitre B, Delacourt C, Buhler JM, Harf A, Lafuma C. Divergent regulation of 92-kDa gelatinase and TIMP-1 by HBECs in response to IL-1 beta and TNF-alpha. Am J Physiol Lung Cell Mol Physiol. 1997;17:L866–L874. doi: 10.1152/ajplung.1997.273.4.L866. [DOI] [PubMed] [Google Scholar]
- 312.Ye S, Eriksson P, Hamsten A, Kurkinen M, Humphries SE, Henney AM. Progression of coronary atherosclerosis is associated with a common genetic variant of the human stromelysin-1 promoter which results in reduced gene expression. J Biol Chem. 1996;271:13055–13060. doi: 10.1074/jbc.271.22.13055. [DOI] [PubMed] [Google Scholar]
- 313.Yoshida M, Korfhagen TR, Whitsett JA. Surfactant protein D regulates NF-kappa B and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol. 2001;166:7514–7519. doi: 10.4049/jimmunol.166.12.7514. [DOI] [PubMed] [Google Scholar]
- 314.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 315.Zankl A, Bonafe L, Calcaterra V, Di Rocco M, Superti-Furga A. Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin Genet. 2005;67:261–266. doi: 10.1111/j.1399-0004.2004.00402.x. [DOI] [PubMed] [Google Scholar]
- 316.Zhang JS, Bai S, Zhang XM, Nagase H, Sarras MP. The expression of gelatinase A (MMP-2) is required for normal development of zebrafish embryos. Dev Genes Evol. 2003;213:456–463. doi: 10.1007/s00427-003-0346-4. [DOI] [PubMed] [Google Scholar]
- 317.Zhao L, Leung JK, Yamamoto H, Goswami S, Kheradmand F, Vu TH. Identification of P311 as a potential gene regulating alveolar generation. Am J Respir Cell Mol Biol. doi: 10.1165/rcmb.2005-0475OC. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Chapman HA, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Zhou M, Huang SG, Wan HY, Li B, Deng WW, Li M. Genetic polymorphism in matrix metalloproteinase-9 and the susceptibility to chronic obstructive pulmonary disease in Han population of south China. Chinese Med J. 2004;117:1481–1484. [PubMed] [Google Scholar]
- 320.Zhou Z, Apte SS, Soininen R, Cao R, Baaklini GY, Rauser RW, Wang J, Cao Y, Tryggvason K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci USA. 2000;97:4052–4057. doi: 10.1073/pnas.060037197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ziegler G, Paynter K, Fisher D. Matrix metalloproteinase-like activity from hemocytes of the eastern oyster, Crassostrea virginica. Comp Biochem Physiol B Biochem Mol Biol. 2002;131:361–370. doi: 10.1016/s1095-6433(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 322.Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, Lollini L, Morris D, Kim Y, DeLustro B, Sheppard D, Pardo A, Selman M, Heller RA. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA. 2002;99:6292–6297. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]