

SUMMARY
Periodontal disease is a chronic inflammatory condition induced by tooth-associated microbial biofilms that induce a host immune response. Therapeutic control of progressive tissue destruction in high-risk patients is a significant challenge in therapy. Soluble protein delivery of antagonists to tumor necrosis factor alpha (TNF-α) inhibits alveolar bone resorption due to periodontitis. However, protein therapy raises several concerns, such as recurrence of disease activity after treatment cessation and repeated dosing regimens. In this study, we used pseudotyped adeno-associated virus vector based on serotype 1 (AAV2/1) to deliver the TNF receptor-immunoglobulin Fc (TNFR:Fc) fusion gene to rats subjected to experimental Porphyromonas gingivalis (Pg)-lipopolysaccharide (LPS)-mediated bone loss. Animals received Pg-LPS delivered to the gingivae thrice weekly for 8 weeks, vehicle alone, Pg-LPS and intramuscular delivery of pseudotyped AAV2/1-TNFR:Fc vector (1×1011 DNase I-resistant particles) or AAV2/1-TNFR:Fc vector delivered to naïve animals. AAV2/1-TNFR:Fc therapy led to sustained therapeutic levels of serum TNFR protein and protected against Pg-LPS-mediated loss of bone volume and density. Furthermore, AAV2/1-TNFR:Fc administration reduced local levels of multiple pro-inflammatory cytokines and osteoclast-like cells at the periodontal lesions. These findings suggest that delivery of AAV2/1-TNFR:Fc may be a viable approach to modulate periodontal disease progression.
Keywords: bone resorption, host modulation, periodontitis, tumor necrosis factor, gene therapy, gene transfer
INTRODUCTION
Periodontitis is a common inflammatory bone destructive disease that is the leading cause of tooth loss in adults. Periodontal disease has also been associated with increased risk of systemic diseases, such as coronary heart disease, bacterial pneumonia and stroke1,2. Although triggered by bacterial biofilms on the tooth surface, tissue destruction and disease progression are driven primarily by mediators of the host immune-inflammatory response to persistent gram-negative pathogens1. Activation of pathogen recognition receptors (e.g. toll-like receptors - TLRs) by bacterial LPS and other pathogen-associated molecular patterns (PAMPs) stimulate the production of pro-inflammatory cytokines [Interleukin (IL)-1, IL-6 and tumor necrosis factor (TNF)-α] and chemokines (IL-8) followed by a cascade of immune-inflammatory events3. Thus, therapeutic approaches that target the host response to local oral infection offer significant potential for periodontal disease management4,5.
TNF-α is a multifunctional cytokine that plays a crucial role in the pathogenesis of periodontitis6. TNF-α is released by activated monocytes, macrophages and T lymphocytes and contributes to both innate and adaptive immune responses, regulating growth, differentiation, survival and physiological function of a variety of different cells and further production of other cytokines, inflammatory mediators and enzymes6-10. TNF-α is a potent bone resorption inducer that stimulates osteoclast differentiation and activation7,11,12. TNF-α functions are mediated by two receptors, p55 and p75 TNF receptor (TNFR), which, respectively, up-regulate and dampen inflammation13,14. P75 TNFR knockout mice display defects in osteoclastogenesis and inflammation-mediated periodontal tissue destruction15,16.
Host-modulation therapies using TNF-α blockers have been successfully used in humans for the treatment of rheumatoid arthritis (RA)17-19. One class of drugs consists of monoclonal anti-TNF-α antibodies or fusion proteins containing p75 TNFR linked to the Fc portion of human IgG1. In both cases, the drug binds to TNF-α with high affinity and prevents it from interacting with its cognate cell surface receptors20. TNF-α blockers significantly improve clinical outcome by reducing erosive bone damage21, through inhibition of the receptor activator of nuclear factor kappa B ligand (RANKL) expression in the synovial tissue22. In periodontitis management, protein delivery of antagonists to IL-1β and TNF-α lead to reduction in periodontal tissue destruction in vivo23-27. However, the use of recombinant protein raises several concerns, such as recurrence of disease activity after cessation of therapy and repeated dosing regimens28. Recently, delivery of an adeno-associated virus (AAV) vector encoding the p75-TNFR:Fc fusion gene has been evaluated as an alternative approach for TNF-α blockade in animal models of RA and demonstrated long-term disease suppression29,30. The main advantages of gene therapy are maintenance of sustained and therapeutically relevant concentration of the protein29, eliminating the previously described limitations of recombinant protein delivery.
In this study, AAV2/1-TNFR:Fc was delivered for the prevention of periodontal disease induced by local, chronic, oral exposure of Porphyromonas gingivalis lipopolysaccharide (Pg-LPS) in vivo. The results of this study show that AAV2/1-TNFR:Fc led to potent inhibition of periodontal disease progression.
RESULTS
Single administration of AAV2/1-TNFR:Fc results in sustained serum rat TNFR:Fc protein levels
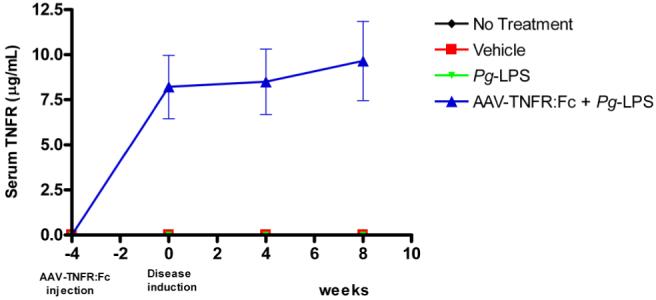
The circulating serum levels of rat TNFR:Fc protein were monitored by enzyme linked immunosorbent assay (ELISA) during the study period to confirm the transduction efficiency of AAV2/1-TNFR:Fc after a single IM administration of 1011 DNase-resistant particles (DRP). The injections were performed four weeks prior to the Pg-LPS delivery to allow for therapeutic levels at the time of disease induction. The administrations resulted in mean TNFR serum levels of 8.2±1.8 μg/mL, by 4 weeks (time of disease induction). These levels were sustained during the 8 week experimental period (Fig.S1). In the other groups not treated with the AAV2/1-TNFR:Fc, including Pg-LPS, Vehicle only (Veh) or no treatment group (NT), TNFR:Fc serum levels were significantly lower than AAV2/1-TNFR:Fc treated animals with results at or below the detection level. The result is in agreement with a previous report, which demonstrated a long-term and sustained secretion of TNFR after a single IM injection of AAV2/1-TNFR:Fc30
AAV2/1-TNFR:Fc inhibits local inflammatory cell infiltrates induced by Pg-LPS in vivo
Animals were sacrificed at four and eight weeks after Pg-LPS disease induction. Stereometric histological analysis using hematoxylin and eosin (H/E) staining was performed to evaluate the inflammatory cell infiltrate induced by an 8-week course of local Pg-LPS administration with or without IM AAV2/1-TNFR:Fc gene therapy. At both timepoints, an intense inflammatory cell infiltrate was consistently observed in the subepithelial connective tissue and surrounding alveolar bone of the periodontia of Pg-LPS-treated animals (Fig. 1b,e). In contrast, a significantly less intense inflammatory reaction was observed in the AAV2/1-TNFR:Fc + Pg-LPS-treated animal (Fig. 1c, f). Control animals (Veh, NT and vector only) did not display evidence of significant inflammatory cell infiltrates (Fig. 1a, d). Statistical analysis demonstrated higher number of inflammatory cells for diseased group (Pg-LPS) in both analyzed areas at 4 weeks timepoint and in subepithelial area at 8 weeks timepoint when compared to treated animals (AAV2/1-TNFR:Fc + Pg-LPS) (Fig. 1). These results demonstrate the anti-inflammatory effect of TNFR:Fc in the face of continued Pg-LPS challenge.
Figure 1.

Histological sections (H/E, 200X) of subepithelial (upper panels) and bone crest (lower panels) levels of periodontal tissues around the maxillary second molar of Vehicle (a and d), Pg-LPS (b and e) and AAV2/1-TNFR:Fc + Pg-LPS (c and f) treated animals, at 4 weeks. The number of inflammatory cells was determined by stereometric analysis using a point-counting technique. A 500 μm2 rectangular-lattice grid with 50 intersection points was constructed and the type of structure found on the intersection of the grid lines was counted on a optical microscope. (One-way ANOVA and Tukey’s post hoc tests).
AAV2/1-TNFR:Fc prevents alveolar bone loss and preserves bone mineral density in Pg-LPS exposed animals
To investigate the preventive effect of AAV2/1-TNFR:Fc on alveolar bone loss induced by an 8-week course Pg-LPS delivery, bi-dimensional (2-D) and three-dimensional (3-D) microcomputed tomography (μCT) images of maxillae and associated teeth were evaluated by linear and volumetric measurements. Linear bone loss was measured from the cementum enamel junction (CEJ) to the bone crest (BC) at the palatal surface of the maxillary second molar teeth, which were the sites most directly affected by Pg-LPS local delivery. Significant alveolar bone destruction was observed in the Pg-LPS group continuously over the course of eight weeks, demonstrating a progressive loss of tooth-supporting bone (Fig. 2, Pg-LPS).Administration of the AAV2/1-TNFR:Fc vector prevented linear bone resorption during the entire study observation period, represented by significant preservation of tooth-supporting bone during Pg-LPS challenge, at both timepoints (Fig. 2, AAV2/1-TNFR:Fc + Pg-LPS).
Figure 2.

Preventive effect of AAV2/1-TNFR:Fc administration on alveolar bone loss induced by Pg-LPS injections. Linear vertical bone loss was measured in digitalized micro CT 2D images in the palatal roots of the maxillary second molars at 4 and 8 weeks after Pg-LPS-disease induction. Data expressed as means ± SD (n=5/group/timepoint).* Statistically different from all other groups (One-way ANOVA and Tukey’s post hoc tests).
This preservation corresponded, on average, to 61.2% and 48.5% of bone loss reduction at 4 and 8 week timepoints, respectively, compared with the Pg-LPS only-treated group that was normalized to Veh-treated animals. Interestingly, a single administration of AAV2/1-TNFR:Fc vector to a group of healthy animals also resulted in preservation of alveolar bone level compared with the vehicle. This bone-sparing effect was significantly greater than that of the vehicle-treated healthy controls, demonstrating a preventive effect in the normal physiologic alveolar bone loss observed in rats (Fig. 2). These results clearly show the beneficial effect of the TNF-α blockade on alveolar bone preservation.
Volumetric bone loss was measured in a specific region of interest (ROI) captured from the 3-D images, based on fixed reference points. ROI involved furcation and interproximal alveolar bone around the maxillary second molars and detected the extension of the destructive effect of local Pg-LPS delivery. The results were comparable to linear measurements, showing more severe bone loss around molar teeth of the Pg-LPS group and a preventive effect for the AAV2/1-TNFR:Fc vector-treated groups. This response was more pronounced after longer periods of disease progression (8 weeks) (Table 1). Bone mineral density measures also demonstrated that the AAV2/1-TNFR:Fc vector preserved bone quality in Pg-LPS exposed animals, after longer periods of disease induction. Vector alone-treated animals displayed bone density values similar to that observed in healthy animals and superior to diseased Pg-LPS treated animals (Table 1).
TABLE 1.
Bone Volumetric Fraction (BVF) and Bone Mineral Density (BMD), according to measures 4 and 8 weeks after experimental periodontal disease induction
| Group | Bone Volumetric Fraction | Bone Mineral Density | ||
|---|---|---|---|---|
| Mean ± SD 4 weeks | Mean ± SD 8 weeks | Mean ± SD 4 weeks | Mean ± SD 8 weeks | |
| Vehicle | 0.617 ± 0.038 1 | 0.596 ± 0.082 4 | 725.67 ± 28.49 8 | 664.95 ± 18.47 9 |
| AAV2/1-TNFR:Fc | 0.680 ± 0.024 2,3 | 0.682± 0.029 5,6 | 673.43 ± 25.38 | 680.85 ± 31.52 10,11 |
| AAV2/1-TNFR:Fc + Pg-LPS | 0.567 ± 0.099 2 | 0.509 ± 0.079 5,6 | 650.65 ± 90.65 | 555.50 ± 103.12 10 |
| Pg-LPS | 0.515 ± 0.032 1,3 | 0.373 ± 0.082 4,6,7 | 587.46 ±105.47 8 | 489.75 ±112.68 9,11 |
1 - 11 - designate mean statistically significant difference between groups, at the same timepoint (1 way-ANOVA and Tukey’s post hoc tests).
BVF and BMD were measured in specific ROIs drawn on μCT 3-D images, using the Microview Analysis software (GE healthcare).
Taken together, these results strongly suggest that anti-inflammatory TNFα-blocking therapy prevents disease-induced bone loss and preserves bone density in the periodontium.
AAV2/1-TNFR:Fc suppresses inflammatory and bone resorptive cytokine expression in periodontal tissues exposed to Pg-LPS
To better understand the mechanisms by which TNF-α blockade, in AAV2/1-TNFR:Fc-treated animals, suppress the progression of Pg-LPS-mediated tissue destruction, the expression of relevant inflammatory cytokines (IL-1 β, TNF-α, IL-6, IL-10) and bone-related molecules [receptor activator of nuclear factor kB ligand (RANKL) and osteoprotegerin (OPG)] involved in periodontal disease progression were evaluated in the gingival tissues, at the RNA level by real-time PCR. Significantly higher RNA expression of IL-6, IL-10, RANKL and OPG was observed at four weeks in Pg-LPS-exposed animals, but not in animals previously treated with the AAV2/1-TNRF:Fc vector (Fig. 3), suggesting an inhibition of the Pg-LPS-mediated inflammatory and bone destructive effects. IL-1β and TNF-α expression levels were not affected by continuous Pg-LPS administration and were similar for all the groups at four and eight weeks. These results corroborate previous in vitro31 and in vivo16 studies showing that long term, repeated administration of Pg-LPS induces cytokine tolerance to Pg-LPS. Based on these observations, a time course experiment was performed to determine IL-1β and TNF-α gene expression over a short interval (12-72h). This parallel study demonstrated significant up-regulation of IL-1β and TNF-α expression, 24 to 48h after a single injection of Pg-LPS when compared to NT or vehicle injected animals (Fig. 4). Animals previously treated with AAV2/1-TNFR:Fc presented higher expression of both cytokines than NT or vehicle-injected animals, yet significantly lower than Pg-LPS only animals. Taken together, these results suggest that AAV2/1-TNFR:Fc gene therapy has an inhibitory effect on inflammatory and bone resorptive cytokines such as IL-1 and TNF during the short-term and to a lesser degree long-term.
Figure 3.

Real time PCR results for cytokines (IL-1β, IL-6, TNF-α, IL-10), RANKL and OPG expressions in the palatal gingival tissue, at 4 and 8 weeks timepoints, for groups: AAV2/1:Fc-TNFR + Pg-LPS, Pg-LPS only and Veh injection. Data expressed as means ± SD (n=5/group/timepoint). (One-way ANOVA and Tukey’s post hoc tests).
Figure 4.

Short-term quantitative real time PCR results for TNF-α and IL-1β cytokines expression in a time course experiment after single Pg-LPS injection. Data expressed as means ± SD (n=5/group/timepoit). (One-way ANOVA and Tukey’s post hoc tests).
AAV2/1-TNFR:Fc IM administration reduces TRAP+ osteoclasts-like cells associated with the alveolar crest in Pg-LPS-treated animals
To evaluate the effect of AAV2/1-TNFR:Fc administration on osteoclast differentiation at the alveolar crest in Pg-LPS-treated animals, histological sections were immunostained and osteoclast-like cells enumerated by image analysis for tartrate-resistant acid phosphatase (TRAP), an enzyme highly expressed in osteoclasts and activated macrophages. Pg-LPS-treated animals displayed significantly higher numbers of TRAP+ osteoclasts-like cells on the palatal bone surface compared with all other groups (Fig.5). Administration of AAV2/1-TNFR:Fc vector reduced osteoclast number in animals challenged with Pg-LPS yielding results statistically similar to the other control groups. The same results were observed when the linear cumulative distance of the palatal bone surface in contact with multinucleated osteoclast-like cells was measured in the coronal 1 mm of the bone crest. From these results we conclude that AAV2/1-TNFR:Fc gene therapy reduces active osteoclast presence at the alveolar bone crest level and, consequently, prevents alveolar bone resorption.
Figure 5.

TRAP immunohistochemistry for detection of osteoclasts-like cells in the bone surface of the alveolar bone crest in the palatal side of the maxillary molars of animals from the vehicle, Pg-LPS only and AAV2/1:Fc-TNFR + Pg-LPS groups, at 4 and 8 weeks after experimental disease induction.
DISCUSSION
Host modulation therapies are rising as promising alternatives for the treatment of periodontal diseases.4 These new strategies are based on advances in the knowledge of periodontal disease pathogenesis, which indicates the host response against pathogenic bacteria as the major driver of periodontal tissue destruction. However, similar to other chronic diseases, periodontal disease management require long-term treatment that can be affected by patient compliance. In developed countries, the level of adherence to long-term treatment of chronic diseases is 50%, and the problem is significantly worse in developing countries32, suggesting the necessity of new therapies to overcome this problem. In this study, we evaluated the application of gene therapy technology, using an AAV vector encoding the TNFR:Fc transgene for modulation of the host immune-inflammatory response in periodontal disease. This new approach has been successfully evaluated for the treatment of other chronic diseases, such as neurological, cardiovascular and autoimmune diseases33. The AAV2/1-TNFR:Fc vector used in the present study led to high level transduction efficiency, prolonged gene expression and therapeutic efficacy in a RA model, encouraging this new application29,30. Both periodontal disease and RA are chronic inflammatory diseases with complex etiology and clinically characterized by sustained over-expression of pro-inflammatory cytokines culminating in progressive bone destruction34. In this study, AAV2/1-TNFR:Fc administration resulted in a significant prevention of experimental periodontal disease progression, illustrating another approach for blocking cytokine production, one driven by pathogenic LPS.
The results of this study found that gene delivery achieved systemically therapeutic circulating levels of soluble TNFR protein, which were maintained during the entire observation period, consistent with our previous studies in RA30. To investigate the potential efficacy of a single delivery of AAV2/1-TNFR:Fc in prevention of alveolar bone loss and inflammation, we used a Pg-LPS-induced experimental periodontal bone resorption model. This model has the advantage to simulate the complex disease process occurring in human periodontitis. Pg-LPS was extracted from a highly pathogenic strain of Porphyromonas gingivalis, W-83, which is an important putative periodontal pathogen35. This bacterium stimulates the destructive innate host response through releasing high level LPS containing vesicles into the periodontal tissues. Pg-LPS is recognized by host cells transmembrane receptors TLR-2 and TLR-4 that activate key intracellular pathways (e.g., mitogen-activated protein kinases) and, thereafter, target genes involved in the activation of the host defense, especially pro-inflammatory cytokine production36. In this study, the Pg-LPS-induced disease model stimulated expression of key inflammatory and bone-related cytokines, detected at the mRNA level by real time PCR at short timepoints, 24-48h after single Pg-LPS administration (TNF-α and IL-1), or at longer intervals, 4 weeks after multiple Pg-LPS injections (IL-6, IL-10, RANKL and OPG). The time course experiment demonstrated an initial stimulatory effect of Pg-LPS on TNF-α and IL-1 expression, not observed after multiple Pg-LPS administration, suggesting an endotoxin tolerance process. TLRs and pro-inflammatory cytokines may become tolerised as a mechanism used in the oral mucosa to attempt to regulate local immune response31,37. In addition, IL-10, an anti-inflammatory cytokine that was highly expressed in Pg-LPS challenged animals, also appears to participate in the tolerance process by mediating immunosuppression and affecting production of pro-inflammatory cytokines38. In a previous study, cytokines showed dissimilar susceptibility to LPS challenge, consistent with our findings that TNF and IL-1 generated greater tolerance than other cytokines (e.g., IL-6) after LPS challenge31. TNF-α and IL-1 have been shown to be key molecules that contribute to several events essential for the initiation of an inflammatory response and, ultimately, tissue destruction39. They can also induce expression of other cytokines that amplify or sustain the inflammatory response and bone resorption as IL-6 and RANKL40. RANKL has been demonstrated to be increased during active periodontal disease in both soft and hard tissues.16, 53, 54 We believe that, in this study, TNF-α and IL-1, followed by IL-6, RANKL and other pro-inflammatory molecules, trigger a cascade of progressive bone destruction that persists during the experimental period, suggesting a similar mechanism of action to what is observed in natural disease. The observed up-regulation of IL-10 and OPG expression after Pg-LPS challenge is in parallel with previous studies that demonstrate a stimulatory effect of LPS on the expression of cytokines in gingival41,42 and periodontal ligament fibroblasts43. As mammalian cells are challenged by polymicrobial infection, they appear to display a defensive mechanism against tissue destruction elicited by bacterial challenge, once IL-10 and OPG stimulate anti-inflammatory and osteoclastogenesis inhibitory effects.
Pg-LPS challenged animals that were previously treated with AAV2/1-TNFR:Fc vector presented reduced inflammatory infiltrates in the palatal gingival tissue parallel to an inhibition of the expression of inflammatory cytokines and bone resorptive-related molecules (Figs 1; 3-5), which resulted in significantly less alveolar bone loss when compared with Pg-LPS treated animals (Fig. 2). Taken together, these results support the central role of TNF-α on the TLR-mediated immune-inflammatory cascade of events responsible for the tissue destruction in the chronic periodontal disease. More specifically, TNF-α stimulates the expression of chemoattractants and their receptors for inflammatory cells, OPG and RANKL by periodontal ligament fibroblasts, T and B lymphocytes, and osteoblastic/stromal cells10,16,43,45,46. TNF-α also has direct effects on osteoclast differentiation independent of the RANK-RANKL axis.11,12
To better investigate the AAV2/1-TNFR:Fc therapeutic effect on the alveolar bone resorption induced by Pg-LPS, we evaluated the presence of osteoclast-like cells at the bone crest surface and the expression of OPG and RANKL, which are key molecules in osteoclastic differentiation and activation47. In parallel with inhibition of cytokine expression, AAV2/1-TNFR:Fc-treated animals demonstrated lower levels of RANKL and OPG expression in the gingival tissue, confirming the association of TNFα and other pro-inflammatory cytokines on osteoclast differentiation/activation and subsequent bone loss47,48.
In summary, delivery of an AAV2/1-TNFR:Fc gene therapy vector suppresses experimental periodontal disease progression through key cytokine and bone-related molecule modulation, reminiscent to the effects observed in RA. AAV2/1-TNFR:Fc is a promising host modulation approach for use in periodontal disease co-management with anti-infective therapy.
MATERIALS AND METHODS
Plasmids/Vector production and purification
Recombinant AAV2 vectors were produced by a standard calcium phosphate transfection method in adherent human 293 cells, using the Ad helper, trans-packaging, and AAV vector plasmids as described previously49. Vector titers were determined by real-time PCR, using a Perkin-Elmer Applied Biosystems Prism 7900 sequence detector (Foster City, CA), and were between 5 and 20×1012 DRP/mL. Vector infectivity was assessed in a TCID50 assay using the HeLa-based B50 cell line50.
Porphyromonas gingivalis lipopolysaccharide (Pg-LPS)
Pg-LPS was isolated from P. gingivalis strain W83, following a previously described protocol51. Briefly, Pg strain W83 was cultured in an anaerobic chamber with modified Brucella-Broth medium. After growth, bacteria were centrifuged at 5,000 rpm for 30 min, resuspended in sterile water for washing and the final pellet was sequentially treated with lysozyme, DNAse, RNAse and proteases to extract and purify the lipopolysaccharide51.
Animal model of Pg-LPS-induced periodontal disease
A total of 45 adult male Sprague-Dawley rats (approximated weight 200 g, 8-10 weeks), were purchased from Charles River Laboratories (Wilmington, MA). All animal experimental procedures were approved by the University of Michigan Committee on the Use and Care of Animals. Animals were allowed to acclimate for seven days before experimentation and were given water and chow ad libitum. For all the procedures, animals were anesthetized with isoflurane (5% with O2 for induction and 3% maintenance) (Baxter Healthcare Corp., Deerfield, IL). Experimental periodontal disease induction was performed by administering 10 μL of Pg-LPS (1.0 mg/mL) into four palatal gingival tissue sites (total of 40 μL/animal) at the base of the interproximal gingival papillae between maxillary molars bilaterally as previously described52. The injections were performed three times weekly using custom-designed 0.375 in × 33 ga, 30° bevel needles attached to a 50 μL Hamilton syringe (Hamilton Company, Reno, NV). Disease induction was performed in two groups of animals (n=10/group): diseased group (Pg-LPS group) and treatment group (AAV2/1-TNFR:Fc + Pg-LPS group). Pg-LPS administrations were performed from baseline to sacrifice, for four or eight weeks after the first injection (n=5/group/timepoint).
Administration of AAV2/1-TNFR:Fc vector
After anesthesia, the skin overlying the quadriceps muscle areas was exposed by shaving and disinfection. A single dosage of rAAV2/1-TNFR:Fc viral vector (100 μl of 1×1011 DRP/animal) was delivered im, equally divided to both quadriceps (50 μl in each muscle), four weeks before Pg-LPS disease induction. AAV2/1-TNFR:Fc was administered via Hamilton syringes and the animals were placed in a recovery cage and observed for normal ambulation. Two groups of animals received vector therapy (n=5/group/timepoint): AAV2/1-TNFR:Fc only or AAV2/1-TNFR:Fc + Pg-LPS groups. The vehicle controls were treated as above with formulation buffer containing 20 mM Tris pH 8, 200 mM NaCl, 2 mM MgCl in 1% glycerol.
Short-term experiment
Based on the cytokine expression results for TNFα and IL1β found in this study, a short-term experiment subsequently was performed to evaluate the early cytokine response to a single injection of Pg-LPS in the palatal gingival tissue. A total of 52 rats were divided into three groups: Veh, Pg-LPS or AAV2/1-TNFR:Fc + Pg-LPS. The rats received the same treatment described for animals of the main experiment, except for Pg-LPS administration that consisted of a single injection. Subgroups of animals (n= 4/group/timepoint) were sacrificed at baseline, 12, 24, 48 and 72 h after Pg-LPS injection and TNFα and IL1β cytokine expression was evaluated by real-time PCR as described below.
ELISA TNFR serum level determination
Serum was obtained from blood samples collected by tail bleeding at -4 weeks (pre-baseline), 0 (baseline, disease induction), 4 and 8 weeks. Serum TNFR levels were determined by ELISA. Briefly, 96-well plates were coated with a goat anti-murine TNF receptor II antibody (R&D Systems, Minneapolis, MN) overnight at 4°C. Plates were washed 3x with phosphate-buffered saline supplemented with 0.05% Tween-20 (Fisher, Houston, TX), and blocked with 1% bovine serum albumin (Sigma, St Louis, MO) for 3 h at RT on an orbital shaker. After washing, the plates were incubated with serum samples for 1 h at RT on a shaker plate. Following sample incubation, the wells were washed, and incubated with a biotin-conjugated mouse anti-rat immunoglobulin G1 antibody (PharMingen/BD Biosciences, San Jose, CA) for 1 h, washed and then incubated with streptavidin-horseradish peroxidase (Zymed, San Francisco, CA) for 30 min at RT. The presence of rat TNFR:Fc protein was detected using the tetramethylbenzidine (TMB) colorimetric substrate (Pierce, Rockford, IL) and stopped with 1 M sulfuric acid (Sigma, St Louis, MO). Quantitation of rat TNFR:Fc protein was based on OD 450 values (Multiskan Ascent, Thermo Fisher Scientific Inc., Waltham, MA) compared with a standard curve of purified rat TNFR:Fc protein. The limit of detection of the assay was 0.219 ng/mL.
Histological analysis of the inflammatory cell infiltrates
Rats were sacrificed in a CO2 chamber at designated timepoints. Hemi-maxillae were harvested and fixed for 48 h in 10% neutral-buffered formalin and transferred to 70% ethanol for μCT scanning. Samples were subsequently decalcified with 10% ethylenediaminetetraacetic acid disodium salt (EDTA) for 3 weeks, embedded in paraffin, and cut into 4- to 5 μm-thick serial sections. Transverse sections of the maxillary 2nd molar teeth parallel to the long axis were stained with H&E for stereometric analysis by a masked, calibrated examiner. The presence of inflammatory cell infiltrate and tissue destruction was evaluated in the periodontal region. A point-counting technique was used to evaluate the proportion of the following structures: fibroblastic cells, collagen, vascular structures and inflammatory cells on the H/E-stained sections. Two areas were assessed individually in comparison to the experimental groups: a submarginal area, representing the connective tissue subjacent to the gingival sulcus (apical border of junctional epithelium and tooth structure as the coronal and medial limits, respectively); and a bone crest region, representing the connective tissue in adjacent to the bone crest (coronal portion of the bone crest and tooth structure as apical and medial limits, respectively). A 500 μm2 rectangular-lattice grid with 50 intersection points was constructed using image software and the type of structure found on the intersection of the grid lines was counted on an optical microscope (Diastar-Cambridge Instruments) under 200X magnification.
Microcomputed Tomography scanning and bone loss analysis
Microcomputed tomography provides high qualification and accurate quantification of the mineralized tissues such as alveolar bone and teeth52. Each maxillary specimen was scanned and reconstructed at 18μm3 voxels using a μCT system (GE Healthcare, London, ON, Canada). A 3-D volume viewer and analyzer software (Microview Analysis+ v.2.1.2 software, GE Healthcare) was used as the tool for both 3-D and 2-D visualization and quantification as previously described52. In brief, linear bone loss was measured as the distance from the cementoenamel junction (CEJ) to the alveolar bone crest or to the base of the alveolar intrabony defect at the palatal surface of the mesio- and disto-palatal roots of the maxillary second molar teeth. For the volumetric analysis, the roof of the furcation and the root apex of the 2nd maxillary molars, the mesial roots of the 3rd molars and the distal roots of the 1st molars were used as reproducible landmarks for estimation of alveolar bone loss. 2-D ROIs were drawn at regular intervals, (average 8 data slices) on a coronal view and reconstructed as a 3-D structure in order to quantify volumetric parameters, bone volume fraction (BVF), and bone mineral density (BMD) (mg/cc). The measurements of coded specimens were made by two independent, calibrated examiners.
Real time PCR for detection of pro-inflammatory cytokines and bone resorption related molecules
Total RNA was extracted from gingival tissue biopsies harvested from a standardized region of the palatal region of the maxillary molar teeth, comprising an ∼5 × 2 mm rectangular area from the medial of the first molar to the distal of the third molar extending from the gingival margin to the palatine suture. RNeasy Mini Kit (Qiagen, Valencia, CA) complemented with RNase-Free DNase Set (Qiagen) for RNA purification were used following the protocols recommended by the manufacturer. After determining RNA concentration and purity by spectrophotometry, complementary DNA (cDNA) was synthesized using 1μg of RNA by a reverse transcription reaction (TaqMan Reverse Transcription Reagents, Applied Biosystems, Foster City, CA). Real time quantitative PCR was performed in an ABI Prism 7500 Real Time PCR System using TaqMan Gene Expression Assays (Applied Biosystems). For each reaction (30 μL), 1 μL of cDNA was added to 1.5 μL of specific TaqMan Gene Expression Assay, 15 μL of TaqMan 2x PCR MasterMix (Applied Biosystems) and 12.5 μL of RNase/DNase free water. The PCR conditions were 50°C (2 min), 95°C (10 min), followed by 40 cycles of 95°C (15 sec) and 6 0°C (1 min) and by the standard denaturation curve. The target gene, ABI ID # and reporter probe sequence of each specific TaqMan Gene Expression Assay were: glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ID Rn99999916_s1, CGGGAAACCCATCACCATCTTCCAG; interleukin-1 beta, ID Rn00580432_m1, CATAAGCCAACAAGTGGTATTCTCC; tumor necrosis factor alpha, ID Rn99999017_m1, CACACTCAGATCATCTTCTCAAAAC; interleukin-6, ID Rn00561420_m1, GAGAAAAGAGTTGTGCAATGGCAAT; interleumin-10, ID Rn00563409_m1, osteoprotegerin, ID Rn00563499_m1, GCTGTGCACTCCTGGTGTTCTTGGA; Receptor Activator of Nuclear Factor-Kappa B Ligand (RANKL), IDRn00589289_m1, TGCCGACATCCCATCGGGTTCCCAT.
Immunohistochemistry of TRAP following gene delivery
Serial sections of the maxillary second molar teeth were prepared as previously described and immunostained for TRAP detection. Deparaffinized tissue sections were pretreated in antigen retrieval buffer (Dako; Glostrup, Denmark) in a pressure chamber (Biocare Medical; Concord, CA) for 20 min. and cooled at RT. ABC Staining System (sc-2023, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used following the manufacturer’s instructions. Primary antibody was raised in goat and consisted of: TRAP (K17): sc-30833 (4 μg/mL, Santa Cruz Biotechnology, Inc). Control sections were incubated with normal IgG (R&D Systems) pre-immunoserum in the same concentrations to assess background staining. All sections were counterstained with Hematoxylin Gill’s Formulation #1 (Fisher Scientific).
Osteoclast enumeration was based on the presence of TRAP-positive multinucleated cells, adjacent to the alveolar bone surface in a pit area. Photomicrographs of the most coronal 1 mm of the palatal bone crest were analyzed by a masked, calibrated examiner and the number of osteoclast-like cells on the palatal bone plate surface were enumerated and in response to Pg-LPS administration.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. Data were pooled by experimental group and the mean ± SD were calculated. One-way ANOVA followed by Tukey’s post hoc test were performed to determine the presence of any significant difference between groups for serum TNFR:Fc levels, linear bone loss and cytokine expression. P-values less than 0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
The authors appreciate the assistance of Charles E. Shelburne (Department of Biologic and Material Sciences, University of Michigan, Ann Arbor, MI), Heather H. Huffer, Timothy J. Daws and Nancy I. Chen. This study was supported by NIDCR DE 016619 to WVG, NIH P-30-AR 46024 to Steven A. Goldstein and CAPES -BEX0495/05-0 and FAPESP 2006/01970-0 to JAC.
REFERENCES
- 1.Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 2.Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DR, Jr., Sacco RL, et al. Periodontal microbiota and carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology Study (INVEST) Circulation. 2005;111:576–582. doi: 10.1161/01.CIR.0000154582.37101.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 4.Giannobile WV. Host-response therapeutics for periodontal diseases. J Periodontol. 2008;79:1592–1600. doi: 10.1902/jop.2008.080174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy MS, Geurs NC, Gunsolley JC. Periodontal host modulation with antiproteinase, anti-inflammatory, and bone-sparing agents. A systematic review. Ann Periodontol. 2003;8:12–37. doi: 10.1902/annals.2003.8.1.12. [DOI] [PubMed] [Google Scholar]
- 6.Graves DT, Cochran D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J Periodontol. 2003;74:391–401. doi: 10.1902/jop.2003.74.3.391. [DOI] [PubMed] [Google Scholar]
- 7.Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. 2000;275:4858–4864. doi: 10.1074/jbc.275.7.4858. [DOI] [PubMed] [Google Scholar]
- 8.Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol. 1999;11:255–260. doi: 10.1016/s0955-0674(99)80034-9. [DOI] [PubMed] [Google Scholar]
- 9.Okada H, Murakami S. Cytokine expression in periodontal health and disease. Crit Rev Oral Biol Med. 1998;9:248–266. doi: 10.1177/10454411980090030101. [DOI] [PubMed] [Google Scholar]
- 10.Graves DT, Oskoui M, Volejnikova S, Naguib G, Cai S, Desta T, et al. Tumor necrosis factor modulates fibroblast apoptosis, PMN recruitment, and osteoclast formation in response to P. gingivalis infection. J Dent Res. 2001;80:1875–1879. doi: 10.1177/00220345010800100301. [DOI] [PubMed] [Google Scholar]
- 11.Kim N, Kadono Y, Takami M, Lee J, Lee SH, Okada F, et al. Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J Exp Med. 2005;202:589–595. doi: 10.1084/jem.20050978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191:275–286. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 14.Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- 15.Abu-Amer Y, Ross FP, Edwards J, Teitelbaum SL. Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its P55 receptor. J Clin Invest. 1997;100:1557–1565. doi: 10.1172/JCI119679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garlet GP, Cardoso CR, Campanelli AP, Ferreira BR, Avila-Campos MJ, Cunha FQ, et al. The dual role of p55 tumour necrosis factor-alpha receptor in Actinobacillus actinomycetemcomitans-induced experimental periodontitis: host protection and tissue destruction. Clin Exp Immunol. 2007;147:128–138. doi: 10.1111/j.1365-2249.2006.03260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klareskog L, van der Heijde D, de Jager JP, Gough A, Kalden J, Malaise M, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363:675–681. doi: 10.1016/S0140-6736(04)15640-7. [DOI] [PubMed] [Google Scholar]
- 18.van de Putte LB, Atkins C, Malaise M, Sany J, Russell AS, van Riel PL, et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63:508–516. doi: 10.1136/ard.2003.013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keystone EC, Schiff MH, Kremer JM, Kafka S, Lovy M, DeVries T, et al. Once-weekly administration of 50 mg etanercept in patients with active rheumatoid arthritis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50:353–363. doi: 10.1002/art.20019. [DOI] [PubMed] [Google Scholar]
- 20.Scott DL, Kingsley GH. Tumor necrosis factor inhibitors for rheumatoid arthritis. N Engl J Med. 2006;355:704–712. doi: 10.1056/NEJMct055183. [DOI] [PubMed] [Google Scholar]
- 21.van der Heijde DM. Overview of radiologic efficacy of new treatments. Rheum Dis Clin North Am. 2004;30:285–293. doi: 10.1016/j.rdc.2004.01.002. vi. [DOI] [PubMed] [Google Scholar]
- 22.Catrina AI, af Klint E, Ernestam S, Catrina SB, Makrygiannakis D, Botusan IR, et al. Anti-tumor necrosis factor therapy increases synovial osteoprotegerin expression in rheumatoid arthritis. Arthritis Rheum. 2006;54:76–81. doi: 10.1002/art.21528. [DOI] [PubMed] [Google Scholar]
- 23.Assuma R, Oates T, Cochran D, Amar S, Graves DT. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol. 1998;160:403–409. [PubMed] [Google Scholar]
- 24.Delima AJ, Oates T, Assuma R, Schwartz Z, Cochran D, Amar S, et al. Soluble antagonists to interleukin-1 (IL-1) and tumor necrosis factor (TNF) inhibits loss of tissue attachment in experimental periodontitis. J Clin Periodontol. 2001;28:233–240. doi: 10.1034/j.1600-051x.2001.028003233.x. [DOI] [PubMed] [Google Scholar]
- 25.Graves DT, Delima AJ, Assuma R, Amar S, Oates T, Cochran D. Interleukin-1 and tumor necrosis factor antagonists inhibit the progression of inflammatory cell infiltration toward alveolar bone in experimental periodontitis. J Periodontol. 1998;69:1419–1425. doi: 10.1902/jop.1998.69.12.1419. [DOI] [PubMed] [Google Scholar]
- 26.Oates TW, Graves DT, Cochran DL. Clinical, radiographic and biochemical assessment of IL-1/TNF-alpha antagonist inhibition of bone loss in experimental periodontitis. J Clin Periodontol. 2002;29:137–143. doi: 10.1034/j.1600-051x.2002.290208.x. [DOI] [PubMed] [Google Scholar]
- 27.Lima V, Vidal FD, Rocha FA, Brito GA, Ribeiro RA. Effects of tumor necrosis factor-alpha inhibitors pentoxifylline and thalidomide on alveolar bone loss in short-term experimental periodontal disease in rats. J Periodontol. 2004;75:162–168. doi: 10.1902/jop.2004.75.1.162. [DOI] [PubMed] [Google Scholar]
- 28.Taylor PC. Anti-tumor necrosis factor therapies. Curr Opin Rheumatol. 2001;13:164–169. doi: 10.1097/00002281-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Chan JM, Villarreal G, Jin WW, Stepan T, Burstein H, Wahl SM. Intraarticular gene transfer of TNFR:Fc suppresses experimental arthritis with reduced systemic distribution of the gene product. Mol Ther. 2002;6:727–736. doi: 10.1006/mthe.2002.0808. [DOI] [PubMed] [Google Scholar]
- 30.Sandalon Z, Bruckheimer EM, Lustig KH, Burstein H. Long-term suppression of experimental arthritis following intramuscular administration of a pseudotyped AAV2/1-TNFR:Fc Vector. Mol Ther. 2007;15:264–269. doi: 10.1038/sj.mt.6300043. [DOI] [PubMed] [Google Scholar]
- 31.Muthukuru M, Jotwani R, Cutler CW. Oral mucosal endotoxin tolerance induction in chronic periodontitis. Infect Immun. 2005;73:687–694. doi: 10.1128/IAI.73.2.687-694.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.WHO . Evidence for action. World Health Organization; 2003. Adherence to long-term therapies. Available from: http://www.who.int/mediacentre/releases/2003/pr2054/en/. [Google Scholar]
- 33.Warrington KH, Jr., Herzog RW. Treatment of human disease by adeno-associated viral gene transfer. Hum Genet. 2006;119:571–603. doi: 10.1007/s00439-006-0165-6. [DOI] [PubMed] [Google Scholar]
- 34.Bartold PM, Marshall RI, Haynes DR. Periodontitis and rheumatoid arthritis: a review. J Periodontol. 2005;76:2066–2074. doi: 10.1902/jop.2005.76.11-S.2066. [DOI] [PubMed] [Google Scholar]
- 35.Socransky SS, Haffajee AD. The bacterial etiology of destructive periodontal disease: current concepts. J Periodontol. 1992;63:322–331. doi: 10.1902/jop.1992.63.4s.322. [DOI] [PubMed] [Google Scholar]
- 36.Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72:5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broad A, Jones DE, Kirby JA. Toll-like receptor (TLR) response tolerance: a key physiological "damage limitation" effect and an important potential opportunity for therapy. Curr Med Chem. 2006;13:2487–2502. doi: 10.2174/092986706778201675. [DOI] [PubMed] [Google Scholar]
- 38.Grutz G. New insights into the molecular mechanism of interleukin-10-mediated immunosuppression. J Leukoc Biol. 2005;77:3–15. doi: 10.1189/jlb.0904484. [DOI] [PubMed] [Google Scholar]
- 39.Ridderstad A, Abedi-Valugerdi M, Moller E. Cytokines in rheumatoid arthritis. Ann Med. 1991;23:219–223. doi: 10.3109/07853899109148051. [DOI] [PubMed] [Google Scholar]
- 40.Nanes MS. Tumor necrosis factor-alpha: molecular and cellular mechanisms in skeletal pathology. Gene. 2003;321:1–15. doi: 10.1016/s0378-1119(03)00841-2. [DOI] [PubMed] [Google Scholar]
- 41.Nagasawa T, Kobayashi H, Kiji M, Aramaki M, Mahanonda R, Kojima T, et al. LPS-stimulated human gingival fibroblasts inhibit the differentiation of monocytes into osteoclasts through the production of osteoprotegerin. Clin Exp Immunol. 2002;130:338–344. doi: 10.1046/j.1365-2249.2002.01990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almasri A, Wisithphrom K, Windsor LJ, Olson B. Nicotine and lipopolysaccharide affect cytokine expression from gingival fibroblasts. J Periodontol. 2007;78:533–541. doi: 10.1902/jop.2007.060296. [DOI] [PubMed] [Google Scholar]
- 43.Wada N, Maeda H, Yoshimine Y, Akamine A. Lipopolysaccharide stimulates expression of osteoprotegerin and receptor activator of NF-kappa B ligand in periodontal ligament fibroblasts through the induction of interleukin-1 beta and tumor necrosis factor-alpha. Bone. 2004;35:629–635. doi: 10.1016/j.bone.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 44.Wang PL, Azuma Y, Shinohara M, Ohura K. Toll-like receptor 4-mediated signal pathway induced by Porphyromonas gingivalis lipopolysaccharide in human gingival fibroblasts. Biochem Biophys Res Commun. 2000;273:1161–1167. doi: 10.1006/bbrc.2000.3060. [DOI] [PubMed] [Google Scholar]
- 45.Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, et al. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275:768–775. doi: 10.1006/bbrc.2000.3379. [DOI] [PubMed] [Google Scholar]
- 46.Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006;169:987–998. doi: 10.2353/ajpath.2006.060180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 48.Teng YT. Protective and destructive immunity in the periodontium: Part 2--T-cell-mediated immunity in the periodontium. J Dent Res. 2006;85:209–219. doi: 10.1177/154405910608500302. [DOI] [PubMed] [Google Scholar]
- 49.Sandalon Z, Bruckheimer EM, Lustig KH, Rogers LC, Peluso RW, Burstein H. Secretion of a TNFR:Fc fusion protein following pulmonary administration of pseudotyped adeno-associated virus vectors. J Virol. 2004;78:12355–12365. doi: 10.1128/JVI.78.22.12355-12365.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao GP, Qu G, Faust LZ, Engdahl RK, Xiao W, Hughes JV, et al. High-titer adeno-associated viral vectors from a Rep/Cap cell line and hybrid shuttle virus. Hum Gene Ther. 1998;9:2353–2362. doi: 10.1089/hum.1998.9.16-2353. [DOI] [PubMed] [Google Scholar]
- 51.Darveau RP, Hancock RE. Procedure for isolation of bacterial lipopolysaccharides from both smooth and rough Pseudomonas aeruginosa and Salmonella typhimurium strains. J Bacteriol. 1983;155:831–838. doi: 10.1128/jb.155.2.831-838.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park CH, Abramson ZR, Taba M, Jr., Jin Q, Chang J, Kreider JM, et al. Three-dimensional micro-computed tomographic imaging of alveolar bone in experimental bone loss or repair. J Periodontol. 2007;78:273–281. doi: 10.1902/jop.2007.060252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu D, Xu JK, Fibliomeni L, et al. Expression of RANKL and OPG mRNA in periodontal disease: Possible involvement in bone destruction. Int J Mol Med. 2003;11:17–21. doi: 10.3892/ijmm.11.1.17. [DOI] [PubMed] [Google Scholar]
- 54.Wara-aswapati N, Surarit R, Chayasadom A, Boch JA, Pitiphat W. RANKL upregulation associated with periodontitis and Porphyromonas gingivalis. J Periodontol. 2007;78:1062–1069. doi: 10.1902/jop.2007.060398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.