

Abstract
We studied the survival of cone photoreceptors following the degeneration of rods in the rd mouse. Cones were visualized by selective expression of green fluorescent protein (GFP) following transduction with an adeno-associated virus (AAV) vector. As previously reported, many cones survive after the initial degeneration of the rods. Soon after the initial degeneration, they lose their outer segments and all but a vestigial inner segment; and they partially retract or lose their axon and synaptic pedicle. However, they retain many fundamental features of the cone phenotype, and for many weeks show a polarized morphology indicative of substantial regrowth of processes. The cells retain their laminar position, forming a cell row just distal to a much thinned outer plexiform layer. The somata subsequently enlarge. Most of the cells extend bipolar processes, recreating the original bipolar morphology of a photoreceptor cell -- though now turned on its side relative to the native position. The cells express short- or middle– wavelength opsins, recoverin and connexin 36. One or more of the polarized processes could often be shown to contain synaptic ribbons, as visualized by antibodies against RIBEYE. The cones do not express protein kinase C alpha, Go alpha, ChX10 or calbindin, markers of bipolar or horizontal cells. The partially differentiated cone morphology persists for at least several months, after which the processes begin to retract and there is slow loss of the cells. Thus, during the time following the loss of their rod-dominated microenvironment, the cones achieve a semi-stable state in which much of their normal phenotype is preserved. Cone photoreceptors in retinas of human RP donors appear from their morphology to undergo a similar progression. The therapeutic window for rescue of cone photoreceptors may be longer than would have been thought.
Introduction
In many forms of retinal degeneration, the primary pathological event is death of the rod photoreceptors. This can occur for a wide variety of reasons, including many different mutations of the rhodopsin gene or other components of the phototransduction cascade, as well as transcription factors etc. (Blanks et al., 1974; Farber and Lolley, 1974; Bowes et al., 1990; Rosenfeld et al., 1992; Marc et al., 2003). In most cases, the cones follow: for reasons that remain unclear the cones cannot survive indefinitely in the absence of the normal microenvironment dominated by rods. Even when rods contain the mutated protein and are the initial cells to degenerate, it is actually the secondary loss of cones that is the most handicapping to the afflicted individual, because acute vision in the modern environment takes place primarily under photopic conditions.
Here, we report a study of the secondary degeneration of cones in a model of human retinitis pigmentosa (RP), the rd1 mouse. In this well known strain, a mutation of the beta subunit of the rod-specific phosphodiesterase gene leads to the massive degeneration of the rod photoreceptors. The degeneration of rods begins at about postnatal day 8-10 and is virtually complete by day 30. This leads eventually to degeneration of the cones and to remodeling of the horizontal and bipolar cells (Strettoi and Pignatelli, 2000; Strettoi et al., 2002; Jones et al., 2003). It has long been known that some cones survive after degeneration of the rods (Carter-Dawson et al., 1978; García-Fernández et al., 1995; Jiménez et al., 1996; LaVail et al., 1997). A morphological study described sprouting of neurites from the remaining cone cells at postnatal day P8 and the formation of ectopic connections with rod bipolar cells around P20 (Peng et al., 2000). However, the surviving cones have not been studied in much detail or at longer times, except to show convincing evidence that the final outcome is an almost complete degeneration (Blanks et al., 1974; Berson, 1993; Chang et al., 1993; Farber et al., 1994; Milam et al., 1998).
We began with the serendipitous finding that an adeno-associated virus (AAV) vector, injected subretinally, selectively labels the surviving cones (Lin et al., 2008). As has been speculated for lentiviral vectors (Pang et al., 2006), it is possible that AAV preferentially invades degenerating photoreceptors (see discussion). At low viral titers, a subset of these cones express the green fluorescent protein (GFP). This meant that individual cones were readily visualized, allowing a longitudinal study of changes in their overall morphology. Immunohistochemical staining for cone-specific proteins confirmed the identity of the transduced cells and allowed quantification of their density, chromatic type, and transretinal distribution. We wanted particularly to learn the detailed character of their neuritic processes; how much of the original molecular phenotype of a cone is retained at various stages; and the duration and progression of changes during their survival. Availability of retinal tissue from eyes of human donors suffering from RP (of unknown genetic origin, donation from the Foundation Fighting Blindness bank) allowed comparison with cones surviving in a similar human disease.
Methods
Plasmid Preparation and AAV Vector Packaging
The pGFP plasmid was used to create AAV-GFP-WPRE. Flanked by AAV terminal repeats, the expression cassette of pGFP contained the hybrid cytomegalovirus (CMV) immediate early enhancer/chicken β-actin (CBA) promoter, GFP cDNA downstream of the CBA promoter. The construct was packaged into AAV2 serotype virus at the Harvard Vector Core. The packaged viruses were concentrated and purified in PBS at titer 7.8 × 1012 genome copies/ml.
Subretinal injections
Experiments were carried out on 65 C3Hpderd1 mice, homozygous for rd1 mutation (rd1/rd1) (The Jackson Laboratory) at the ages of postnatal day 14 to 12 months, which were maintained at the Massachusetts General Hospital. All experimental procedures were performed in accordance with institutional guidelines. Animals were anesthetized with a mixture of ketamine hydrochloride (30-40 mg/kg) and xylazine (3-6 mg/kg). Pupillary dilation was achieved with 1% tropicamide and 2.5% phenylephrine eye drops. Guided by an operating microscope, a small incision was made in sclera, and 0.5–1 μl of vector suspension was slowly injected through the incision into the subretinal space in approximately 30 s using a 5 μl syringe (Hamilton) equipped with a 33-gauge blunt needle. Following subretinal delivery, the needle was gently withdrawn, to prevent the vector from leakage.
Immunocytochemistry and Imaging
After 1 - 4 weeks post injection, the animals were anesthetized with the mixture of ketamine hydrochloride and xylazine as above. Eyes were quickly enucleated after a reference point was taken to label the superior pole and the retinas were dissected free of the vitreous and sclera in carboxygenated Ames' Medium (Sigma, St. Louis, MO), and then fixed in 4% paraformaldehyde (PFA) for 0.5 - 1 hour. Some of the retinas were sectioned serially at a thickness of 15 – 50 μm on either a cryostat or a vibratome. Whole retinas and sections were blocked in a solution containing 3% normal goat serum (NGS), 1% bovine serum albumin (BSA), and 0.3% Triton X-100 in phosphate-buffered saline (PBS; pH 7.4) for 1 hour to reduce nonspecific labeling. The following primary antibodies were applied: rabbit anti-GFP (1:1000 or 2 μg/ml, Molecular Probes, Eugene, OR); mouse anti-PKCα clone MC5 (1:500 or 0.2 μg/ml, Amersham, Arlington Heights, IL); mouse anti-CtBP2 (1:500 or 0.05 μg/ml, BD Biosciences, San Jose, CA); rabbit anti-connexin36 (1:1000 or 0.005 μg/ml, Zymed Laboratories, San Francisco, CA); rabbit anti-recoverin; rabbit anti-blue opsin, rabbit anti-red/green opsin, mouse anti-rhodopsin and mouse anti-Goα were bought from Chemicon (Temecula, CA) and diluted at 1:500 or 2 μg/ml; rabbit anti-CHX10 (1:500 or 0.5 μg/ml,) was kindly provided by Dr. Constance Cepko (Harvard Medical School). The primary antibodies were diluted with a blocking solution (1% NGS, 1% BSA, 0.1% Triton X-100 in PBS) and applied overnight at 4°C. After rinsing and blocking, a secondary antibody conjugated to Alexa TM 488 (1:500; Molecular Probes, Eugene, OR) or Alexa TM 594 (1:500; Molecular Probes, Eugene, OR) was applied for 2 hours at room temperature. In double-labeling experiments using primary antibodies from different hosts, the primary antibodies were applied simultaneously and then visualized by application of appropriate secondary antibodies. Labeled sections and whole retinas were rinsed, coverslipped in a Vectashield mounting medium (Vector Laboratories, Burlingame, CA).
Confocal micrographs of fluorescent specimens from retinal flat-mounted preparations and vertical sections were captured using a Bio-Rad Radiance confocal microscope (Hercules, CA, USA) equipped with a krypton-argon laser at a resolution of 1024 -1024 pixels. Zeiss Plan Apochromat 25-/0.8 and C-Apochromat 63-/1.2 W Korr lenses were used. Images scale was calibrated, and if necessary, brightness and contrast were adjusted using Photoshop 8.0 software (Adobe Systems, San Jose, CA, USA).
To qualify the number of surviving S cones and M cones, we counted all the surviving cones, identified by immunostaining, in six 240 μm by 240 μm regions across either the dorsal-ventral axis or nasal-temporal axis of retinal wholemounts.
Immunocytochemistry on human RP retinas
Retinal fragments were dissected from the eye cup of one human donor, a 72 years old female, diagnosed at age 36 as a typical RP patient. The eye sample had the identification code RP 93-45/46, was isolated at the University of Washington, RP Histopathology Center, and obtained through the courtesy of the Foundation Fighting Blindness eye bank. Primary fixation was achieved 12 hrs postmortem in 4% PAF and 0.5% glutaraldheyde and lasted for 1 year. Afterwards, the specimen was transferred to 2% paraformaldehyde alone. After rinsing in buffer, retinal fragments from the central and peripheral retina were infiltrated in 30% sucrose, embedded in OCT, frozen at -20°C and sectioned vertically at a cryostat. Immunocytochemistry with retinal-specific antibodies was performed as for the mouse retina. Primary antibodies which produced satisfactory reactivity were calbindin, PKC alpha, Go Alpha, blue-cone opsin and recoverin. Retinal sections were examined and imaged using a Leica TCS-Sp confocal microscope.
Results
Retinal cone photoreceptors in the rd mouse are disorganized and disoriented
Adeno-associated virus (AAV) vectors provided a useful way for gene transfer to retinal cone photoreceptor cells. After 1 to 4 weeks post-subretinal injection of AAV-eGFP, highly efficient transduction of retinal cone photoreceptor cells was achieved in young and adult retinas (Fig. 1). The transduced cone photoreceptors were initially restricted to injection sites. With the passage of time, however, AAV-eGFP particles spread out from the injection site to cover, in many cases, the whole retina. Uniform transduction of retinal cone photoreceptor cells across the whole retina could usually be achieved by four week post-injection. Following subretinal injection, AAV-eGFP vectors appeared to target cone photoreceptors more or less selectively (Fig. 1), although a small number of horizontal cells, bipolar cells or amacrine cells were weakly labeled (data not shown). The entire remaining processes of individual surviving cone photoreceptors were clearly visualized by GFP labeling (Fig. 1), which allowed us to investigate when and how cone photoreceptors alter their morphology during the course of photoreceptor degeneration.
Fig. 1.

A - C, GFP expression in retinal whole mounts (focus on the photoreceptor layer) from rd1 mice following subretinal injection of AAV-eGFP. Highly efficient transduction of the retinal cone photoreceptors with GFP was evident about 4 weeks after the subretinal injection and remained for many months. Strong GFP expression occured across the whole retina. D - I, Abnormal morphology of GFP-expressing retinal cone photoreceptors in rd1 mice shown at higher magnification. The outer and inner segments and cone pedicles of surviving cone photoreceptors were clearly labeled by GFP. Degenerating photoreceptors show abnormal monopolar to multipolar morphologies across various ages. Shortening or loss of outer segments or both outer and inner segments were evident in some GFP positive cones (arrows), while others showed a novel process of abnormal neurite sprouting (arrowheads). Scale bars, 20 μm in A, B and C; 10 μm in D-I.
Remodeling of cone photoreceptor cells started shortly after postnatal day 8 (P8), the time when rod photoreceptors begin degenerating in the rd1 mice due to a rod-specific gene mutation. Cone outer segments initially develop more or less normally (Nir et al., 1989; Gouras and Tanabe, 2003). However, the outer segments of S cones began to degenerate as early as postnatal days 8, followed by the inner segments and cell bodies. By the age of P25, most of the cone photoreceptors had lost outer segments, as well as part of their axon and pedicles (Figs. 1A and 1D, arrows), and some appeared to lack all processes except cell bodies.
Interestingly, after they lost outer segments and other processes, cone photoreceptors began substantial outgrowth of new processes (Fei, 2002) and generated a new polarized morphology. Most of the degenerating cone photoreceptors restored a bipolar morphology (Fig. 1F-I). Others extended long, multipolar processes (Fig. 1G, 1H). However, these new processes, which emerged either from a cell body (Fig. 1G) or from a remaining axon process (Fig. 1H), were disorganized and wildly oriented toward different directions.
With increasing age, the atrophy of processes in cone photoreceptor cells was more extensive. After 120 days of age, the majority of cone photoreceptors were already gone. A few were observed in the process of another round of retraction of their processes, in the end-stage leaving most cone photoreceptors with shortened processes or no processes at all (Fig. 1C).
Preservation of a molecular phenotype in degenerating cone photoreceptor cells
Counterstaining of the retinal sections and whole mounts with cone cell-specific antibodies identified the majority of GFP expressing cells as cone photoreceptor cells. Based on the expression of opsins, cone photoreceptors of the mouse retina can be classified into short wavelength opsin-expressing cones and middle wavelength opsin-expressing cones (Jacobs et al., 1991; Szél et al., 1992; Applebury et al., 2000). S opsins were found to be expressed in a fraction of the GFP expressing cells (Fig. 2 A-C, arrows), while M opsins were present in other GFP expressing cells (Fig. 2 D-F, arrows). In the wild type mouse retina, the M and S opsins are generally confined to the photoreceptor outer segments (Figs. 6A-D, 7A-B). In the rd1 retina, the cell membrane of somata, axons and cone pedicles of degenerating cones were all positive for M or S opsins (Fig. 2), suggesting the redistributions of the opsins in degenerating cones after they lost outer segments and/or inner segments. This redistribution of opsins was best appreciated in high-resolution images of the cone photoreceptors in retinal whole mounts (Fig. 3 B and E). Recoverin, another cone photoreceptor marker, was also present in degenerating cones (Fig. 2 G-I, arrows). Therefore, cone photoreceptors preserve their molecular phenotype and were still capable of synthesizing their native proteins, though cone photoreceptors had undergone dramatic remodeling in morphology.
Fig. 2.

Preservation of native cone cell proteins in degenerating cone photoreceptors. These images also illustrate the near-total absenceof transduced (GFP – expressing) neurons other than the degenerating cones: In these fields there are no GFP –expressing neurons in the INL or GCL. Counterstaining of the GFP- expressing cone photoreceptors in retinal sections with cone cell-specific antibodies showed that GFP positive cones still expressed S- opsins (A, B and C), M- opsins (D, E and F) and recoverin (G, H and I) (arrows), suggesting the continuing syntheses of these proteins in degenerating cone photoreceptors. INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars, 10 μm.
Fig. 6.

The distribution and number of surviving S cones at different ages. Images were taken from four different regions across ventral-dorsal axis. A-D, S opsin expressing cones in the retina of a wild type with the focus on the cone outer segments. The large majority of S cones were in the ventral retina (A-C), only a few S cones in the dorsal retina (D). S cones in the rd1 mouse retina start losing their outer segments as early as P8. By P12, most of S cones have lost their outer segments; some appear to have died and left open free spaces (E-H). I-N, over the following months, S cones disappeared from the dorsal and central retina (L), and the number of cells rapidly declined. Scale bars, 10 μm.
Fig. 7.

Temporal patterning of surviving M cones at different ages. Representative images were taken from two dorsal regions of retinal whole mounts (H). A-B, M opsin expressing cones in the retina of wild type, with the focus on the cone outer segments. C-D, Most of M cones lost their outer segments by P12, some have lost the inner segments. E-F, The retinas of P47 showed an advanced level of degeneration. M cones disappeared from the central and ventral retina, the remaining M cones lost their outer and/or inner segments. There was hypertrophy of the surviving somas. G, At P95, only scattered M cones were observed in the far peripheral retina. No outer segments could be seen in the vast majority of surviving M cones. Scale bars, 10 μm.
Fig. 3.

Confirmation of GFP positive cells as cone photoreceptors in whole mounts. Some of GFP positive cells express S – opsins, which were ectopically redistributed to the membranes of remaining cell bodes, axons and cone pedicles (arrows in A - C), while others synthesize M- opsins (arrows in D - F). Scale bars, 10 μm.
The degenerating cone photoreceptors did not express a series of molecular markers known normally not to be present in cones. Immunolabeling of the GFP- expressing cone photoreceptors in retinal sections with calbindin, PKCα and CHX10, which specifically label horizontal cells, rod bipolar cells and all population of bipolar cells, respectively, revealed the expression of none of them in the GFP- expressing cone photoreceptors (Fig. 4, arrowheads).
Fig. 4.
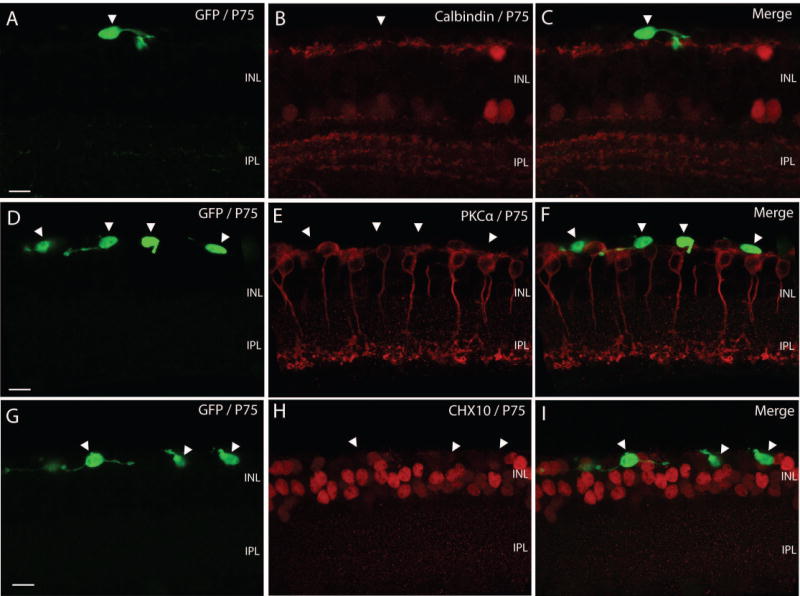
Absence of labeling for proteins known not to be present in cones. Immunolabeling of the GFP- expressing cone photoreceptors in retinal sections with calbindin, PKCα and Ch×10. A – C, GFP - expressing cone photoreceptors that were calbindin negative retain their laminar position in the markedly thinning ONL. They are situated on top of the band formed by horizontal cells, which were revealed by calbindin (arrowhead). D – F, GFP – expressing cone photoreceptors of PKCα negative formed one no continuous monolayer of cells (arrows), which was adjacent to rod bipolar cells (red). G – I, GFP positive cones did not express bipolar cell marker (arrowheads). Some of GFP positive cones migrated into the INL and extended neurite sproutings horizontally. INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars, 10 μm.
We asked whether the long, newly synthesized processes of degenerating cones contained synaptic ribbons. C-terminal binding protein 2 (CtBP2), a RIBEYE homolog, which labels presynaptic ribbons in photoreceptors, was used to visualize ribbon profiles in the cone pedicles. Some of the long processes indeed possessed several synaptic ribbons, which had approximately the normal shape and the normal location within a swelling at the end of a long process (Fig. 5A-F). We surveyed 40 cones with one or more long processes from different regions at different ages. In these cells, 29 out of 91 long processes had more than one synaptic ribbon.
Fig. 5.

Expression of synaptic ribbons but not gap junction molecules by degenerating cone pedicles. Counterstaining of the retinal sections (A-C) and whole mounts (D-F) with antibodies against Ribeye showed synaptic ribbons (red) located within cone pedicles (arrowheads), and had a normal shape. INL, inner nuclear layer; IPL, inner plexiform layer. Scale bars, 10 μm.
The pedicles of neighboring cones in a normal retina form gap junctions via connexin36. In the adult rd retina, however, counterstaining the GFP expressing cones with an antibody against connexin36 showed the long processes of the degenerating cones no longer to express connexin36 in their cone pedicles (data not shown).
Distribution pattern and number of the surviving cones
Antibodies specific for middle wavelength sensitive cones (M cones) and short wavelength sensitive cones (S cones), were used to reveal the temporal and topographic distributions of the surviving M and S cones. A low power view of the retina demonstrated that the remodeling of S cone photoreceptor cells was widespread, i.e. it was not confined to a few cones or a specific region of the retina (Fig. 6E-N). In a normal mouse retina, S cones form a distinct gradient across the ventral-dorsal retina: the majority of S cones reside in the ventral half and scattered S cones in the dorsal half (Fig. 6A-D). This distribution pattern remained in the rd1 mouse retina at an early stage of degeneration, with the exceptions that S cones in the rd1 retina no longer had a normal morphology and the absolute number of S cones was decreased due to the cone degeneration (Figs. 6-8). The outer segments of S cones began to degenerate as early as postnatal days 8, followed by the inner segments and cell bodies. By postnatal day 12, a substantial population of S cones had lost outer segments; as for the overall population of cones, the degeneration took place initially from the central retina and subsequently proceeded towards the periphery (Carter-Dawson et al., 1978; García-Fernández et al., 1995; Jiménez et al., 1996; LaVail et al., 1997) (Fig. 6E-H). The gradient was surprisingly steep: S cones had disappeared from the very central retina at this age but S cones residing in the most peripheral ventral retina looked almost normal in morphology. By P47, the cone free zone in the central retina was much larger and most S cones lacked outer segments and/or inner segments; S cones with portions of inner segments were only observed in the far periphery of the ventral retina (Fig. 6I). At P95, surviving S cones were restricted to the very far periphery of the ventral retina. Almost all lacked outer and inner segments; only a few cones close to the extreme edge of the retina had inner segments, and those were severely atrophic (Fig. 6M).
Fig. 8.

Spatial topographies of surviving M and S cones. The maps shown in the top panels are a a schematic representation of the temporal pattern for S cones (A, top panel) and M cones (B, middle panel) degenerative events. The degeneration and death of both S and M cones advanced from the central to the peripheral retina. By P90, remaining S cones were restricted to the far periphery of the ventral retina, while remaining M cones were located in the far periphery of the dorsal retina. The size of solid dots represents the density of remaining cones, with larger dots representing higher densities. C and D, show decreasing densities, as measured for a series of retinas at different ages, of surviving S cones (C) and M cones (D). Counting was done along a ventral-dorsal axis, with the optic nerve head taken as the zero point. n, the number of retinas. Scale bars, 1 mm.
In contrast to S cones, M cones in the ventral retina of rd mice were more vulnerable to the insult than those in the dorsal retina, and degenerated and disappeared at a faster rate than those in the ventral retina (Figs. 7-8). Like S cones, however, the pattern of degeneration for M cones in the rd1 mouse also advanced from the central to peripheral retina (Figs. 7-8). M cones in the central retina began degenerating considerably earlier than those in the rest of the retina and M cones resisted terminal degeneration longer in the periphery than in the central retina. With progression, more M cones across the retina are affected and the region devoid of M cones in the center retina increase. By P90, a small number of surviving M cones were found to be located in the far periphery of the dorsal retina only. These surviving cones lacked outer segments and inner segments.
Stereotyped remodeling of cones in mouse and human retinas
Despite the limitations imposed by postmortem time and long exposure to fixative, we succeeded in revealing cone morphology from specimens of human RP donors labeled with recoverin and other photoreceptor-specific markers (Fig. 9). Remarkably, remodeling of surviving cones appeared morphologically identical to that observed for the degenerating mouse retina. Different stages of remodeling could be observed from retinal areas in which a higher or lower number of photoreceptors survived. In the first instance, cones appeared still mostly bipolar in shape, although they seemed to be confluent in a single layer. Note that cone photoreceptor cells of the extrafoveal retina normally form a regularly spaced mosaic, each cone separated from its neighbors by a defined interval. A continuous row of adjacent cones thus suggests that the degenerating cones migrate from their original positions into clumps. This was also observed in the mouse. In retinal areas of lower photoreceptor densities, cones had lost their original shape: the outer and inner segments were atrophic and the major cellular axis was oriented parallel to the outer limiting membrane. The mutation responsible for RP in the human donor examined here was not known. However, given the low incidence of phosphodiesterase mutations in humans, it is unlikely that the present phenotype could be assigned to an “rd-like” genetic origin. This suggests that, independent of the underlying genetic defect, remodeling of cones in RP occurs through stereotyped stages, comprising initial retention of cell polarity, progressive atrophy of outer and inner segments, ovoidal morphology and lateral migration.
Fig. 9.

Abnormal morphology of cone photoreceptors from the retina of a human RP donor. Recoverin (green) staining and nuclear (red) counterstaining. A: numerous cones persist in this retinal area. They exhibit anomalous morphologies with bulbous-shaped outer segments (arrows). B: in this area of more severe disorganization, photoreceptor morphology is more affected and the typical bipolar shape of cones has been partially lost. ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars, 10 μm.
Discussion
By expressing GFP in the surviving cones and by immunostaining against cone-specific marker proteins, we obtained an overview of the changes in morphology and the synthesis of certain key proteins during the months following the degeneration of the rods. That some cones survive for some time following the rod degeneration is of course well known (Carter-Dawson et al., 1978; García-Fernández et al., 1995; Jiménez et al., 1996; LaVail et al., 1997). We found that the cones achieve a more or less stable state that persists for many months without signs of a progressive degeneration. This state appears to be shared in human RP and mouse models of the same disease. Our goal here was to more completely characterize the biological state of the cones during this window of time.
Continued expression of elements of the cone phenotype
It was immediately evident that cones not only survive, but achieve a complex morphology. Furthermore, this morphology is reproducible from cell to cell. In other words, the cells do not simply de-differentiate, they achieve a different phenotype than the one maintained when the surrounding complement of rods is present.
The most striking deformation of the surviving cones is the loss of the outer segment and of all but a vestige of an inner segment. Although the cells continue to synthesize many of the proteins of a normal cone, they never recover these structures and it seems likely, as is widely supposed, that the failure of these structures is due to the loss of mechanical or tropic support from the now-absent neighboring rods.
Nonetheless, the cones retain a substantial fraction of their normal phenotype. They remain in the retinal layer normally occupied by cones. They continue to synthesize opsins, and like normal cones they synthesize predominantly one opsin or the other (Applebury et al., 2000). They extend long processes. Various proteins characteristic ribbons can be labeled within these processes of the dying cones. We used only one of them, RIBEYE, to visualize the ribbons, but this protein decorates the whole ribbon surface and it was clear that the ribbons had at least approximately the normal shape and the normal location within a swelling at the end of a long process. These quasi-normal features are suggestive that the cells would have a certain regenerative potential. Not only are they synthesizing many of the normal proteins of a cone, they remain polarized cells with a clear tendency to the extension of pedicles, and they form endings reminiscent of their normal synaptic terminals. Whether or not they retain an ability to assemble inner and outer segments is unclear; this capability is strikingly lacking in most cells transplanted to the subretinal space from other sources (reviewed in Lamba et al., 2008).
The distribution of surviving cones across the retina
The temporal patterns of photoreceptor degeneration in the rd1 mouse retina were revealed by antibodies specific to green sensitive cones (M cones) and blue sensitive cones (S cones). A general consequence for both classes of cones in the rd1 retina is that the vast majority of them eventually will die. A small number of cones in the far peripheral regions of the retina, however, do survive for a long time, perhaps even for the life of the animal, even though they lack outer segments, inner segments, and connections with other retinal neurons and are clearly nonfunctional.
On the global level, we have shown here that the degeneration of two classes of cones in the rd1 retina occurs in a sequence. Confirming previous observations, the cone degeneration starts initially from the central retina (Carter-Dawson et al., 1978; García-Fernández et al., 1995; Jiménez et al., 1996; LaVail et al., 1997). Subsequently, the degeneration spreads radially towards the retinal periphery at differential rates, also consistent with previous findings (García-Fernández et al., 1995; Jiménez et al., 1996; LaVail et al., 1997). Two types of cones, however, were found to exhibit different regional specific degeneration in the present study. The degeneration for M cones proceeds more rapidly in the ventral retina and more gradually in the dorsal retina. By postnatal day 60, a large number of M cones have been lost in the ventral retina, while many M cones survive in the dorsal retina. By postnatal day 90, there are still a number of M cones in the far peripheral retina of dorsal half and but almost nothing elsewhere. Conversely, S cones are affected more severely in the dorsal retina and degenerate faster there than elsewhere, so that at 90 days of age, there are still a small number of S cones in the far periphery of the ventral retina (Fig. 8). Thus, the surviving cones from two classes of cone photoreceptors show a marketed topographic separation. They form two independent groups separated by millimeters across the ventral and dorsal axis.
On both a local and a global level, we have observed variation in number of surviving cones from animal to animal. This confirms a previous report by LaVail and colleagues (1997). The variability is very great. For instance, animals from the same age group can show quite different densities of surviving cones even at the same retinal region (Fig. 8). In one extreme case, the number of surviving S cones in the ventral retina at P60 was greater than that at P30 (Fig. 8). This variability in the degeneration may be to be expected, since the cone photoreceptors in the rd1 retina die secondarily to a genetic lesion not in themselves but in the rod photoreceptors. This global variation, together with the tendency of the degenerating cones to aggregate locally into clumps, may explain apparently conflicting results in some earlier studies, especially those in which sections instead of whole mounts were studied.
Selective transduction of cone photoreceptors by AAV-2
It has been known for some time that AAV-2, when injected subretinally, preferentially transduces photoreceptor cells. This seems to have been the case in these experiments, although here the only available photoreceptor cells were the surviving cones. When injected intravitreally, in either wild type or rd mice, AAV-2 primarily transduces ganglion cells, with the addition of a very few bipolar or amacrine cells (Martin et al., 2002). It has been tacitly assumed that this was partly a matter of geometry – that the viral particles are most likely to transduce the cells to which they have the most direct access. The present results suggest that there may be an additional selectivity. Although the surviving cones indeed lay on the surface of the retina closest to the site of injection of viral particles, the spatial separation between those cones and the horizontal cells or bipolar cells was very slight. Furthermore, there were substantial gaps between individual cones; they did not form a sheet of cells that would screen the bipolar and horizontal cells from exposure to the virus. This suggests that there is a viral tropism that favors transduction of cones and works against transduction of horizontal and bipolar cells. Alternatively, it is conceivable (though seemingly less likely) that a barrier of some sort exists between the cones and the outer margin of the inner nuclear layer; this could perhaps lie within the outer limiting membrane or the vestigial outer plexiform layer.
Whatever the mechanism, the result suggests that AAV-2 might be useful as a vehicle for delivering to the residual cones genes that could promote their survival or enhance their function. This would apply to cases, such as the one studied here, where the cone degeneration is secondary, but also to cone-based diseases. There would be many barriers to such a therapy for human photoreceptor degenerations, among them that the AAV2 vector may not transduce all of the surviving cones (see Figs. 2 and 3). Nonetheless, such a strategy does seem worth evaluating in animal models, if only as an experimental tool.
Acknowledgments
This work was supported by NIH grant EY 017169. RHM is a Senior Investigator of Research to Prevent Blindness. ES was supported by the Italian CNR and NIH grant RO1 EY12654.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Applebury ML, Antoch MP, Baxter LC, Chun LL, Falk JD, Farhangfar F, Kage K, Krzystolik MG, Lyass LA, Robbins JT. The murine cone photoreceptor: a single cone type expresses both S and M opsins with retinal spatial patterning. Neuron. 2000;27:513–523. doi: 10.1016/s0896-6273(00)00062-3. [DOI] [PubMed] [Google Scholar]
- Berson EL. Retinitis pigmentosa (The Friedenwald Lecture) Invest Ophthalmol Vis Sci. 1993;34:1659–1676. [PubMed] [Google Scholar]
- Blanks JC, Adinolfi AM, Lolley RN. Photoreceptor degeneration and synaptgenesis in retinal-degenerative (rd) mice. J Comp Neurol. 1974;156:81–94. doi: 10.1002/cne.901560108. [DOI] [PubMed] [Google Scholar]
- Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM, Sidman RL. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest Ophthalmol Vis Sci. 1978;17:489–498. [PubMed] [Google Scholar]
- Chang GQ, Hao Y, Wong F. Apoptosis: Final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- Farber DB, Lolley RN. Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science. 1974;186:449–451. doi: 10.1126/science.186.4162.449. [DOI] [PubMed] [Google Scholar]
- Farber DB, Flannery JG, Bowes-Rickman C. The rd mouse story: seventy years of research on an animal model of inherited retinal degeneration. Prog Retin Eye Res. 1994;13:31–64. [Google Scholar]
- Fei Y. Cone neurite sprouting: an early onset abnormality of the cone photoreceptors in the retinal degeneration mouse. Mol Vis. 2002;27:306–314. [PubMed] [Google Scholar]
- García-Fernández JM, Jimenez AJ, Foster RG. The persistence of cone photoreceptors within the dorsal retina of aged retinally degenerate mice (rd/rd): implications for circadian organization. Neurosci Letter. 1995;187:33–36. doi: 10.1016/0304-3940(95)11330-y. [DOI] [PubMed] [Google Scholar]
- Gouras P, Tanabe T. Ultrastructure of adult rd mouse retina. Graefes Arch Clin Exp Ophthalmol. 2003;241:410–417. doi: 10.1007/s00417-003-0649-1. [DOI] [PubMed] [Google Scholar]
- Jacobs GH, Neitz J, Deegan JF. Retinal receptors in rodents maximally sensitive to ultraviolet light. Nature. 1991;353:655–656. doi: 10.1038/353655a0. [DOI] [PubMed] [Google Scholar]
- Jiménez AJ, García-Fernández JM, González B, Foster RG. The spatio-temporal pattern of photoreceptor degeneration in the aged rd/rd mouse retina. Cell Tissue Res. 1996;284:193–202. doi: 10.1007/s004410050579. [DOI] [PubMed] [Google Scholar]
- Jones BW, Watt CB, Frederick JM, Baehr W, Chen CK, Levine EM, Milam AH, Lavail MM, Marc RE. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- Lamba D, Kar IM, Reh TA. Neural regeneration and cell replacement: a view from the eye. Cell Stem Cell. 2008;2:538–549. doi: 10.1016/j.stem.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Matthes MT, Yasumura D, Steinberg RH. Variability in rate of cone degeneration in the retinal degeneration (rd/rd) mouse. Exp Eye Res. 1997;65:45–50. doi: 10.1006/exer.1997.0308. [DOI] [PubMed] [Google Scholar]
- Lin B, Koizumi A, Tanaka N, Panda S, Masland RH. Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc Natl Acad Sci U S A. 2008;105:16009–16014. doi: 10.1073/pnas.0806114105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc RE, Jones BW, Strettoi E. Neural remodeling in retinal degeneration. Prog Retin Eye Res. 2003;22:607–655. doi: 10.1016/s1350-9462(03)00039-9. [DOI] [PubMed] [Google Scholar]
- Martin KR, Klein RL, Quigley HA. Gene delivery to the eye using adeno-associated viral vectors. Methods. 2002;28:267–275. doi: 10.1016/s1046-2023(02)00232-3. [DOI] [PubMed] [Google Scholar]
- Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res. 1998;17:175–205. doi: 10.1016/s1350-9462(97)00012-8. [DOI] [PubMed] [Google Scholar]
- Nir I, Agarwal N, Sagie G, Papermaster DS. Opsin distribution and synthesis in degenerating photoreceptors of rd mutant mice. Exp Eye Res. 1989;49:403–421. doi: 10.1016/0014-4835(89)90050-x. [DOI] [PubMed] [Google Scholar]
- Pang J, Cheng M, Haire SE, Barker E, Planelles V, Blanks JC. Efficiency of lentiviral transduction during development in normal and rd mice. Mol Vis. 2006;12:756–767. [PubMed] [Google Scholar]
- Peng YW, Hao Y, Petters RM, Wong F. Ectopic synaptogenesis in the mammalian retina caused by rod photoreceptor-specific mutations. Nature Neurosci. 2000;3:1121–1127. doi: 10.1038/80639. [DOI] [PubMed] [Google Scholar]
- Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992;1:209–213. doi: 10.1038/ng0692-209. [DOI] [PubMed] [Google Scholar]
- Strettoi E, Pignatelli V. Modifications of retinal neurons in a mouse model of retinitis pigmentosa. Proc Natl Acad Sci U S A. 2000;97:11020–11025. doi: 10.1073/pnas.190291097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strettoi E, Porciatti V, Falsini B, Pignatelli V, Rossi C. Morphological and functional abnormalities in the inner retina of the rd/rd mouse. J Neurosci. 2002;22:5492–5504. doi: 10.1523/JNEUROSCI.22-13-05492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szél A, Röhlich P, Caffé AR, Juliusson B, Aguirre G, Van Veen T. Unique topographic separation of two spectral classes of cones in the mouse retina. J Comp Neurol. 1992;325:327–342. doi: 10.1002/cne.903250302. [DOI] [PubMed] [Google Scholar]