

Abstract
Background
Cardiac myosin binding protein-C (cMyBP-C) phosphorylation modulates cardiac contractility. When expressed in cMyBP-C null (cMyBP-C(t/t)) hearts, a cMyBP-C phosphomimetic (cMyBP-CAllP+), rescued cardiac dysfunction and protected the hearts from ischemic-reperfusion (I/R) injury. However, cMyBP-C function may be dependent upon the myosin isoform type. Since these replacements were carried out in the mouse heart, which contains predominantly α-myosin heavy chain (α-MyHC), the applicability of the data to the human, whose cardiomyocytes contain predominantly β-MyHC, is unclear. We determined the effect(s) of cMyBP-C phosphorylation in a “humanized” mouse heart in which >80% of the α-MyHC was replaced by β-MyHC, which is the predominant myosin isoform in human cardiac muscle.
Methods and Results
To determine the effects of cMyBP-C phosphorylation in a β-MyHC background, transgenic mice expressing normal cMyBP-C (cMyBP-CWT), nonphosphorylatable cMyBP-C (cMyBP-CAllP-), or cMyBP-CAllP+ were bred into the β-MyHC background (β). These mice were then crossed into the cMyBP-C(t/t) background to ensure the absence of endogenous cMyBP-C. cMyBP-C(t/t)/β and cMyBP-CAllP-:(t/t)/β mice died prematurely due to heart failure, confirming that cMyBP-C phosphorylation is essential in the β-MyHC background. cMyBP-CAllP+:(t/t)/β and cMyBP-CWT:(t/t)/β hearts showed no morbidity and mortality and cMyBP-CAllP+:(t/t)/β hearts were significantly cardioprotected from I/R injury.
Conclusions
cMyBP-C phosphorylation is necessary for basal myocardial function in the β-MyHC background and can preserve function after I/R injury. Our studies justify exploration of cMyBP-C phosphorylation as a therapeutic target in the human heart.
Keywords: heart failure, myosin heavy chain, cardioprotection, myosin binding protein-C
Introduction
Cardiac myosin binding protein-C (cMyBP-C) phosphorylation is involved in the regulation of myocardial function and cardioprotection,1-4 but its precise functional roles remain obscure. cMyBP-C mutations cause heritable cardiomyopathies, accounting for ∼15-30% of all cardiomyopathic cases.5 A unique feature of cMyBP-C is its multiple phosphorylation sites.6 There are three phosphorylated serines in cMyBP-C (Ser-273, Ser-282 and Ser-302) that can serve as substrates for protein kinase A (PKA), protein kinase C and Ca2+-calmodulin-activated kinase II.3,4 Interestingly, the cardiomyopathic Gly278Glu, Gly279Ala and Arg282Trp mutations are located within the cMyBP-C phosphorylation motif.7
cMyBP-C phosphorylation is essential for normal cardiac function1 but decreases during the development of human heart failure (HF),8 and ischemia-reperfusion (I-R) injury.1 Previously we explored the role of cMyBP-C phosphorylation in cardiac function by mutating the three phosphorylation sites to either non-phosphorylatable alanines (cMyBP-CAllP-)1,9 or to aspartates (cMyBP-CAllP+), to create a phosphomimetic.2 Transgenic (TG) expression of cMyBP-CAllP- in cardiomyocytes resulted in depressed cardiac function and was unable to rescue the cMyBP-C null (cMyBP-C(t/t)) phenotype, in contrast with expression of wild-type cMyBP-C (cMyBP-CWT) in the cMyBP-C(t/t) background.1 Strikingly, expression of cMyBP-CAllP+ rescued the cMyBP-C(t/t) mice and, in addition, was cardioprotective during I-R injury.2
Rapid and reversible changes in thick filament structure and ordering of myosin heads can be produced in cardiac muscle by changes in cMyBP-C phosphorylation,10 and these changes in structure are accompanied by changes in force production.11,12 cMyBP-C’s interactions with myosin are modulated by cMyBP-C phosphorylation such that when cMyBP-C is dephosphorylated it interacts strongly with the S2 region of myosin, preventing its force-generating interaction with actin. In vitro studies show that when cMyBP-C sites are phosphorylated, myosin S2 interaction is weakened or ablated,2 promoting the interaction of the myosin head with actin, which activates cross-bridge cycling. Because of these interactions, cMyBP-C function is at least partially dependent upon the particular myosin isoform with which it interacts.10,13
How applicable are these mouse studies to the human heart? Two distinct myosin isoforms, termed V1 (two α-MyHC plus light chains) and V3 (two β-MyHC plus light chains), are in the mammalian heart, giving rise to differences in shortening velocity, force and actomyosin ATPase activity.14 The mouse heart consists of mostly α-MyHC while the human heart contains β-MyHC. Phosphorylation of Ser-273, Ser-282 and Ser-302 in cMyBP-C in vitro15 and in vivo2 causes the thick filaments to exhibit a relatively loose structure,2 with changes in myosin orientation, increased contractility and maximum Ca2+-activated force.15 Winegrad and coworkers examined thick filaments that contained either α-MyHC or β-MyHC and studied the effects of cMyBP-C phosphorylation on their structure and contractile parameters. 10 PKA-mediated phosphorylation of cMyBP-C and cMyBP-CAllP+ resulted in cross-bridge extension from the filament backbone, changes in overall orientation and a decrease in cross-bridge flexibility in the α-MyHC-containing filaments.2,10 In contrast, phosphorylation of cMyBP-C in β-MyHC-containing filaments had no effect on cross-bridge extension, the degree of order, or flexibility.10,13 These data call into question the relevance of altered cMyBP-C phosphorylation in modulating contractile parameters in the human heart.
We constructed mice in which the ventricular α-MyHC isoform was replaced with >80% β-MyHC (β-TG).14 Although mimicking the adult human myocardium’s MyHC isoform composition, it should be recognized that the Vmax ATPase activity of mouse β-MyHC is substantially larger than that of human β-MyHC in the “humanized” mouse heart. We then investigated the impact of cMyBP-C phosphorylation in these mice. The data demonstrate that cMyBP-C phosphorylation can improve myocardial function regardless of the myosin isoform with which it interacts and supports the hypothesis that altering cMyBP-C phosphorylation status in vivo may represent a novel therapeutic target in human HF.
Materials and Methods
An expanded Methods section is available in the Online-only Supplement.
Transgenic and Targeted Mouse Models
To determine the efficacy of cMyBP-C phosphorylation in a β-MyHC background, the cMyBP-CWT (line 21)1, cMyBP-CAllP- (line 262)1 and cMyBP-CAllP+ (line 34)2 expressing mice were bred to the β-TG mice (line 137)14,16 to generate double TG animals which were subsequently crossed into the cMyBP-C(t/t) background17 to ensure the absence of endogenous cMyBP-C (cMyBP-CWT:(t/t)/β, cMyBP-CAllP-:(t/t)/β and cMyBP-CAllP+:(t/t)/β) as described.1 All mice (FVB/N) procedures were in accordance with the Guide for the Use of and Care of Laboratory Animals published by the National Institutes of Health.
Cardiac I-R Injury
To determine the cardioprotective effects of cMyBP-CAllP+ expression, cardiac I-R injury was performed at 8-10 weeks.2 Total area at risk (AAR), infarcted area (IA) and area not at risk. DNA laddering assay and the number of TUNEL positive nuclei were quantitated to assess cardiac apoptosis.2
Statistical Analysis
Results are presented as mean±SE. For comparisons of multiple groups, one-way ANOVA or ANOVA for repeated measurements followed by the Tukey-Kramer multiple comparisons test was used (SigmaStat V3.0). A P<0.05 was considered significant.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
The Phosphorylatable Domain of cMyBP-C Interacts with β-MyHC
Myosin is composed of two heavy chains and four light chains (Figure 1A). The globular head contains the actin binding and ATPase sites. Previously, we showed that cMyBP-C C1-C2 domains interact with α-MyHC at the S2 region.2 This interaction is dynamically regulated by phosphorylation/dephosphorylation of cMyBP-C at serines 273, 282 and 302 (Figure 1A). To confirm that phosphorylation of cMyBP-C also determines β-MyHC interaction, pull-down assays were performed using His-tagged C1-C2 peptides derived from cMyBP-CWT (C1-C2WT) or from C1-C2 peptides in which Ser-273, Ser-282 and Ser-302 were mutated to either alanines (C1-C2AllP–) or aspartates (C1-C2AllP+). The peptide fragments were tested against ventricular protein extracted from mouse predominantly expressing β-MyHC.14 C1-C2WT peptides were also phosphorylated with PKA to determine the relative myosin interaction. Results show that non-phosphorylated C1-C2WT and C1-C2AllP– proteins interact with β-MyHC, whereas PKA-treated C1-C2WT and untreated C1-C2AllP+ do not (Figure 1B). We conclude that dephosphorylated cMyBP-C C1-C2 interacts in vitro with β-MyHC and that phosphorylation ablates the interaction. We were not able to confirm these experiments in vivo, as there is a strong interaction of cMyBP-C through its COOH terminus domains with the light meromyosin region of either β- or α-MyHC, (Online Supplement Figure I), such that coimmunoprecipitation occurs irrespective of cMyBP-C phosphorylation when intact proteins are used.
Figure 1.
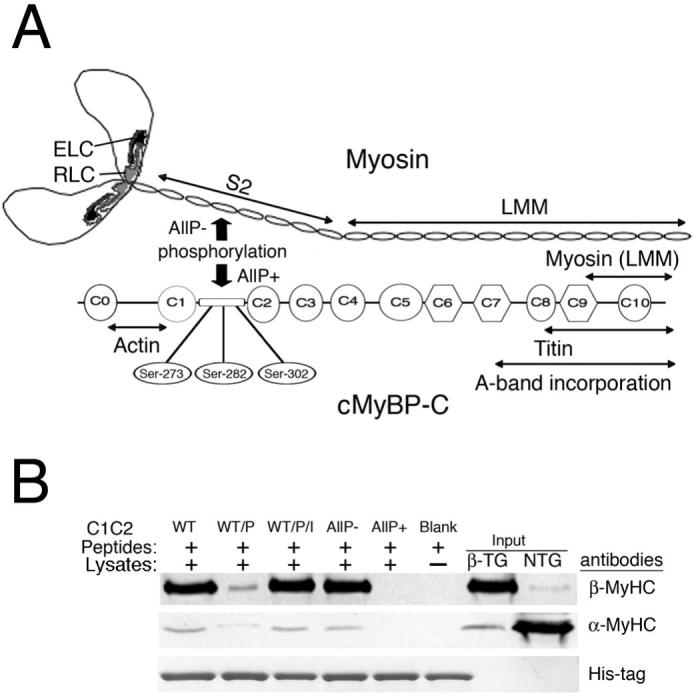
cMyBP-C interacts with both β-MyHC and α-MyHC in a phosphorylation-dependent fashion. A, The myosin and cMyBP-C domains are depicted with the region of interaction shown. The essential light chain (ELC), regulatory light chain (RLC), subfragment-2 (S2) and light meromyosin (LMM) regions are indicated. Phospho-ablation of cMyBP-C (AllP-) promotes the protein’s interaction with myosin S2, whereas the phosphomimetic (AllP+) abolishes the interaction. B, A pull-down assay demonstrates the phosphorylation-dependent interaction of C1-C2 with β-MyHC. Two hundred μg of total β-TG heart ventricular lysate was mixed with either 20 μg of His-tagged C1-C2WT peptide, PKA-treated WT peptides without (WT/P) and with PKA inhibitors (WT/P/I), C1-C2AllP- and C1C2AllP+ peptides and the interacting proteins collected with Ni-NTA resin as described.2 The proteins were separated in 4-15% SDS-PAGE and analyzed by western blots using anti-β-MyHC,14 anti-α-MyHC (BA-G5) and anti-His antibodies.
Phospho-ablation of cMyBP-C is Deleterious in a β-MyHC Background
To examine the effects of cMyBP-C phosphorylation in a β-MyHC background, cMyBP-CWT, cMyBP-CAllP- and cMyBP-CAllP+ mice were crossed with β-TG mice14 to generate double TG mice, which were then crossed into the cMyBP-C(t/t) background17 to ensure the absence of endogenous cMyBP-C.1 The gross cardiac anatomy of the seven groups was analyzed (Figure 2A). Both the nontransgenic (NTG), where α-MyHC is >95% of the total myosin content, and β-TG, where α-MyHC is replaced with >80% β-MyHC, hearts showed normal anatomy while cMyBP-C(t/t) hearts show markedly increased ventricular wall thickness, myocyte disarray and fibrosis.1,17
Figure 2.

Phospho-ablation (AllP-) of cMyBP-C is deleterious to the heart in a β-MyHC background. Representative hematoxylin-eosin stained longitudinal sections of mouse heart at 4X (A) and 20X (B). Masson’s trichrome-stained myocardial sections, 20X (C). D, Representative SYPRO Ruby stained glycerol gel shows MyHC isoform content of the samples shown in A. E, Survival curves show that cMyBP-C(t/t)/β and cMyBPCAllP-:(t/t)/β mice die within 7 weeks post-birth due to severe HF. NTG, cMyBP-C(t/t) β-TG and cMyBP-CWT:(t/t)/β mice showed normal survival. One mouse in the cMyBP-CAllP+:(t/t)/β group died from other causes (n=10 per group).
Importantly, in the β-MyHC background, the cMyBP-C(t/t) mouse phenotype was significantly more severe, with cMyBP-C(t/t)/β mouse hearts exhibiting gross pathology and concentric hypertrophy (Figure 2A-C) at 6 weeks of age. Expression of normal cMyBP-C or the phosphomimetic in the β-MyHC and cMyBP-C null background (cMyBP-CWT:(t/t)/β and cMyBP-CAllP+:(t/t)/β, respectively) resulted in a rescue of the aberrant cardiac gross anatomy (Figure 2A-D). In marked contrast, the phospho-ablated cMyBP-CAllP-:(t/t)/β experimental group died by seven-weeks post-birth (Figure 2E). We hypothesize that the inability of cMyBP-CAllP- to rescue the null lies in its tendency to strongly bind myosin S2, mimicking chronic, complete dephosphorylation, whereas, cMyBP-CWT continues to be dynamically and differentially phosphorylated on some of its phosphorylatable residues. Similarly to NTG, cMyBP-C(t/t) and β-TG mice, the cMyBP-CWT:(t/t)/β and cMyBP-CAllP+:(t/t)/β mice show essentially no signs of increased morbidity and mortality throughout their life spans, confirming the necessity of cMyBP-C phosphorylation in a ®-MyHC background for normal cardiac function. Levels of cMyBP-C phosphorylation were similar among NTG, β-TG and cMyBP-CWT:(t/t)/β hearts as quantified by phospho-site-specific antibodies and one-dimensional isoelectric focusing (Online Supplement Figures II and III).
cMyBP-CAllP+ Effects a complete rescue of cMyBP-C(t/t) in the β-MyHC Background
To determine the effects of cMyBP-C phosphorylation on whole organ function in a β-MyHC background, four groups; NTG, β-TG, cMyBP-CWT:(t/t)/β and cMyBP-CAllP+:(t/t)/β, were chosen for further studies at 10-12 weeks. SDS-PAGE (Figure 3A) and Western blots using anti-cMyBP-C (Figure 3B) show normal myofilament stoichiometry in the cMyBP-CAllP+:(t/t)/β hearts compared to control hearts. RNA analysis revealed (Figure 3C) that the overexpression of β-MyHC is consistent with protein levels (Figure 2D) in the β-TG, cMyBP-CWT:(t/t)/β and cMyBP-CAllP+:(t/t)/β mice. Atrial natriuretic peptide expression, a sensitive molecular marker for cardiac stress/hypertrophy, was unchanged among the four groups as were the heart/body weight ratios (Figure 3D). Furthermore, M-mode echocardiography showed that NTG, β-TG, cMyBP-CWT:(t/t)/β and cMyBP-CAllP+:(t/t)/β mice had normal cardiac function (Figure 3E and Table 1). cMyBP-CAllP+:(t/t)/β hearts had normal levels of cardiac troponin I, myosin essential light chain and phospholamban phosphorylation (Online Supplement Figure IV). Altogether, these data suggest that cMyBP-CAllP+:(t/t) in the β-MyHC background does not cause a discernible pathology at either the whole organ, molecular or cellular level.
Figure 3.
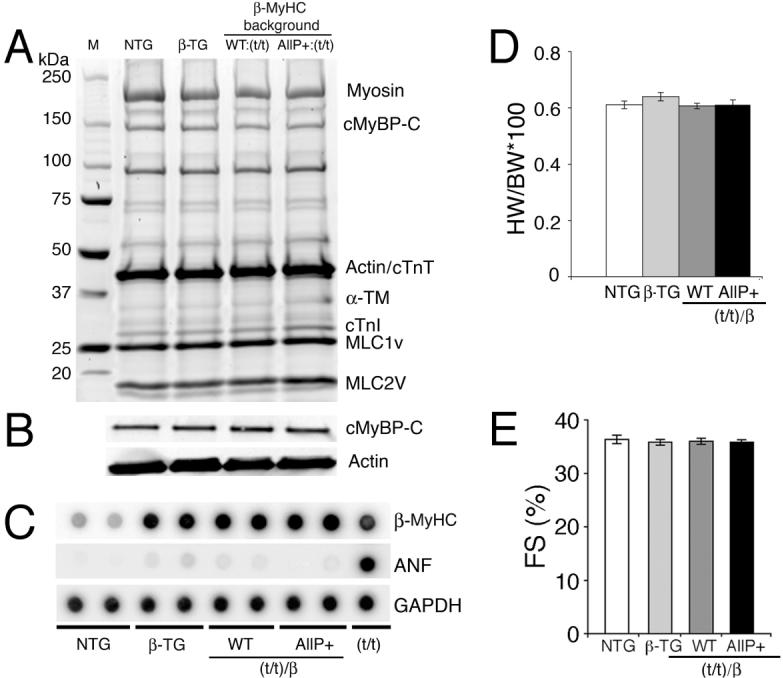
cMyBP-CAllP+ in a β-MyHC background. A, SDS-PAGE analyses of myofibrillar proteins from NTG, β-TG, cMyBP-CWT:(t/t)/β and cMyBP-CAllP+:(t/t)/β hearts. B, Representative western blot analyses using anti-cMyBP-C antibodies show that total cMyBP-C content is unchanged. α-sarcomeric actin was used as a loading control. C, RNA dot-blot analyses. As expected, β-MyHC is up-regulated in the β-MyHC background. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control (n=3). D, Heart weight/body weight (HW/BW) ratios and E, percentages of fractional shortening (%FS) are unchanged among the four groups (n=6) at 12 weeks.
Table 1.
Evaluation of cardiac function
| Groups | LVED (mm) | LVES (mm) | AW (mm) | PW (mm) | HR (bpm) | E-TIME (ms) | FS (%) |
|---|---|---|---|---|---|---|---|
| NTG (n=8) | 3.69±0.1 | 2.35±0.1 | 0.75±0.05 | 0.78±0.04 | 551±13 | 45±1.8 | 36.3±2 |
| β-TG (n=7) | 3.62±0.1 | 2.34±0.1 | 0.79±0.04 | 0.80±0.03 | 594±16 | 43±2.5 | 35.7±2 |
| cMyBP-CWT:(t/t)/β (n=6) | 3.75±0.1 | 2.39±0.1 | 0.79±0.04 | 0.82±0.03 | 579±19 | 45±2.0 | 36.3±1 |
| cMyBP-CAllP+:(t/t)/β (n=7) | 3.52±0.1 | 2.26±0.1 | 0.78±0.04 | 0.77±0.02 | 585±19 | 44±1,0 | 35.8±1 |
Hearts were visualized with a Hewlett-Packard (Palo Alto, CA, USA) Sonos 5500 instrument and a 15 MHz transducer as described.1 Measurements were averaged from ≥ three separate cardiac cycles: left ventricular (LV) end diastolic thickness (LVED), LV end systolic thickness (LVES), anterior wall thickness (AW), posterior wall thickness (PW), aortic ejection time (E-TIME in milliseconds), fractional shortening (FS) and heart rate (HR). Data are expressed as mean±SE. No significant differences were observed. mm; millimeter, bpm; beats per minute, ms; milliseconds.
cMyBP-CAllP+ Improves Myocardial Contractility in a β-MyHC Background
Myocardial β-adrenergic receptor (β-AR) function is impaired in human HF.8 In order to determine if cMyBP-C phosphorylation plays a role in mediating the positive inotropic effect of β-AR stimulation in the intact animal, cardiac hemodynamic rates were measured at baseline and during β-AR agonist infusion in the intact closed chest model.1 The β-MyHC TG mouse hearts exhibited decreased systolic and diastolic rates compared to the control NTG mice (Figure 4A, 4B).14 Heart rate and LV pressure were significantly improved in the cMyBP-CAllP+:(t/t)/β mouse hearts compared to β-TG and cMyBP-CWT:(t/t)/β mouse hearts (Table 2). In contrast with our previous data obtained in the normal, α-MyHC background,2 cMyBP-CAllP+:(t/t)/β has both significantly improved contraction (Figure 4A) and relaxation (Figure 4B) compared to β-TG and cMyBP-CWT:(t/t)/β cohorts although these parameters did not reach NTG values, emphasizing the critical role that myosin isoform content plays in determining cardiac hemodynamics.
Figure 4.
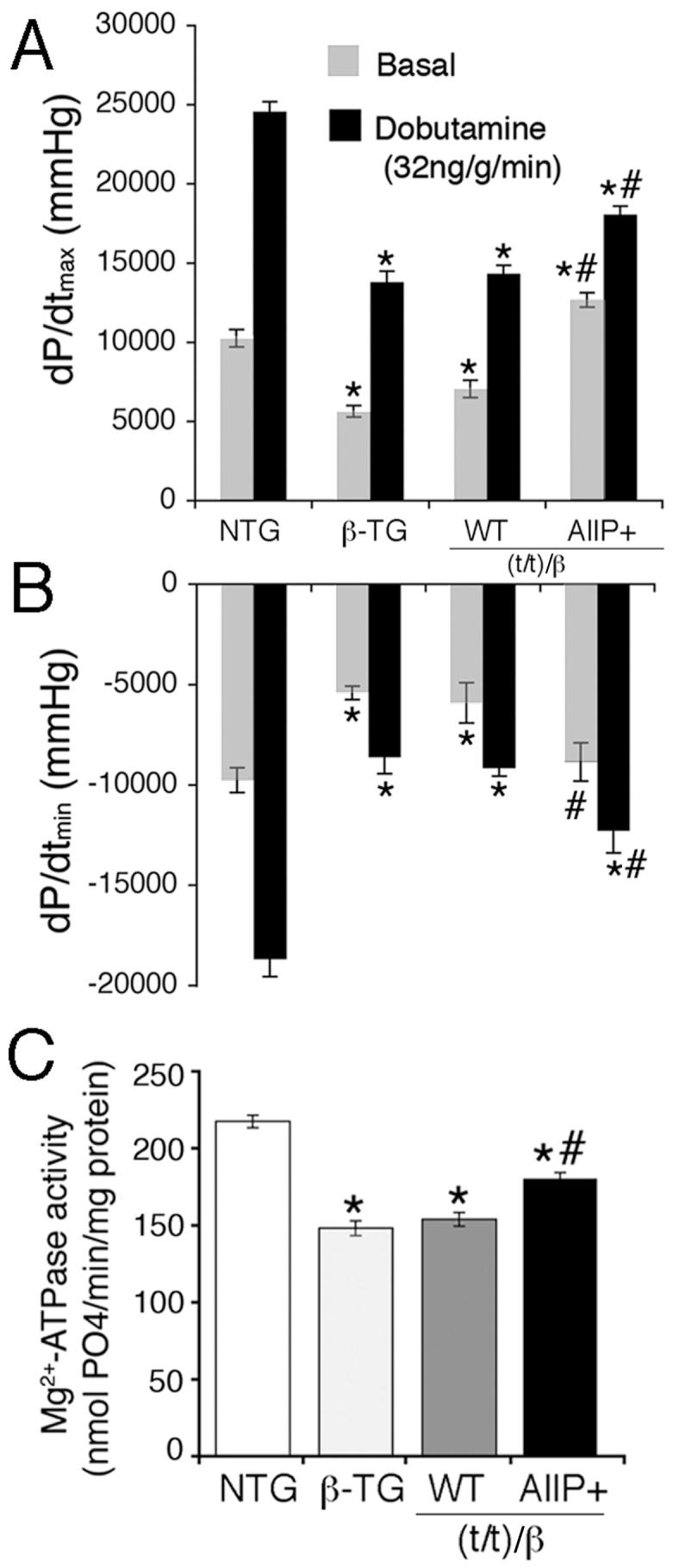
cMyBP-CAllP+ in a β-MyHC background improves myocardial function in vivo. cMyBP-CAllP+:(t/t)/β mouse hearts show enhanced in vivo contraction (dP/dtmax, A) and relaxation (dP/dtmin, B), compared to β-MyHC TG and cMyBP-CWT:(t/t)/β mouse hearts, which show decreased function compared to the NTG controls (n=6, Table 2). Measurements were made at basal levels and during β-agonist stimulation. C, Effects on maximal Ca2+-activated Mg2+-ATPase activities of the cMyBP-C phosphomimetic (AllP+) in a β-MyHC background (n=5) at pCa 4.0 (Table 3). *P<0.05 versus NTG and #P<0.05 versus β-TG.
Table 2.
In vivo hemodynamics
| NTG | β-TG | cMyBP-CWT:(t/t)/β | cMyBP-CAllP+:(t/t)/β | |||||
|---|---|---|---|---|---|---|---|---|
| Basal | Dobut. | Basal | Dobut. | Basal | Dobut. | Basal | Dobut. | |
| Heart rate (bpm) | 382±10 | 597±17 | 295±21* | 399±35* | 322±11* | 425±15* | 346±16 | 561±20# |
| LVP (mm Hg/s) | 108±5 | 121±7 | 77±4* | 86±2* | 79±5* | 94±3* | 103±4# | 113±4# |
| dP/dtmax (mm Hg/s) | 10253±561 | 24575±627 | 5631±363* | 13789±689* | 6555±396* | 14322±529* | 12680±455*# | 18395±479*# |
| dP/dtmin (mm Hg/s) | -9767±-617 | -18687±-867 | -5410±-339* | -8630±-806* | -5916±-1006* | -9164±-404* | -8860±-951# | -12291±-1084*# |
Cardiac catheterization was used to evaluate LV function.1 Heart rate, peak LV pressure (LVP) maximum, LV dP/dtmax (an index of myocardial contractility) and LV dP/dtmin (an index of myocardial relaxation), were determined at baseline (basal) and during infusion of dobutamine (Dobut; 32 ng/g/min). All data are presented as mean±SE.
P<0.001 versus NTG
P<0.05 versus cMyBP-CWT:(t/t)/β (n=6).
Data suggest that cMyBP-C phosphorylation influences actomyosin Mg2+-ATPase activity, the kinetics of cross-bridge cycling and the rate of relaxation.4 In addition, β-AR stimulation has differential effects on the actomyosin Mg2+-ATPase activity depending upon whether α- or β-MyHC is present.18 Therefore, to assess the role of cMyBP-CAllP+ in a β-MyHC background, actomyosin Mg2+-ATPase activity was measured in the four groups. As expected, actomyosin Mg2+-ATPase activity was depressed in the β-TG hearts compared to NTG hearts (Figure 4C). However, the cardiac myofibrils of the cMyBP-CAllP+:(t/t)/β showed increased maximal actomyosin Mg2+-ATPase activity compared to cMyBP-CWT:(t/t)/β without changes in the Hill coefficients and Ca2+ sensitivity (Table 3). Our data suggest that increased actomyosin Mg2+-ATPase contributes to improved cardiac function in the cMyBP-CAllP+:(t/t)/β heart.
Table 3.
Ca2+-activated actomyosin Mg2+-ATPase activity
| Genotype | Vmax (nmol Pi/min/mg) | EC50 (pCa) | Hill Coefficient |
|---|---|---|---|
| NTG | 217±4 | 5.79±0.025 | 2.32±0.08 |
| β-TG | 148±2* | 5.84±0.016 | 2.50±0.28 |
| cMyBP-CWT:(t/t)/β | 152±2* | 5.84±0.018 | 2.30±0.12 |
| cMyBP- CAllP+:(t/t)/β |
180±3*# | 5.83±0.024 | 2.34±0.10 |
Myofibril Mg2+-ATPase activity and Ca2+-sensitivity were measured at pCa of 8.0-4.0 at 18°C. Maximum actomyosin Mg2+-ATPase activity (Vmax) was measured at pCa 4.0. All data are presented as mean±SE (n=5).
P<0.001 compared to NTG
P<0.001 compared WT:(t/t)/β and β-TG. pCa unit is referred as -log[Calcium2+].
cMyBP-CAllP+ Enhances Myosin Kinetics at the Isolated Fiber Level
cMyBP-CAllP+ mimics the effects of PKA-mediated phosphorylation of cMyBP-C. The β-TG mice permit testing the hypothesis that cMyBP-CAllP+ has consequences on cross-bridge cycling in the presence of the slow myosin isoform. Fiber kinetic studies were carried out in which cMyBP-CAllP+:(t/t)/β skinned fibers were compared to control fibers in the absence or presence of PKA treatment. As previously determined, NTG fibers show increased shortening velocity (Figure 5A), power output (Figure 5B) and unloaded shortening velocity (Figure 5C) compared to β-TG fibers.14 PKA treatment of NTG fibers (NTG/PKA) led to increased shortening velocity (Figure 5A), power output (Figure 5B) and unloaded shortening velocity (Figure 5C) as well as decreased Ca2+ sensitivity (Figure 5D and Table 4). In contrast, β-TG fibers exhibited reduced shortening velocity (Figure 5A), power output (Figure 5B) and unloaded shortening velocity (Figure 5C) at baseline relative to NTG fibers. Upon PKA treatment (β-TG/PKA), the β-TG/PKA fibers show similar responses (Figure 5D and Table 4). cMyBP-CAllP+:(t/t)/β fibers showed increased shortening velocity (Figure 5E), power output (Figure 5F) and unloaded shortening velocity (Figure 5G) compared to cMyBP-CWT:(t/t)/β and β-TG fibers in the absence of altered myofibrillar Ca2+ sensitivity. Treatment of the cMyBP-CAllP+:(t/t)/β fibers with PKA resulted in decreased maximal Ca2+ sensitivity (Figure 5H) compared to cMyBP-CWT:(t/t)/β. Maximum Ca2+-activated isometric forces, calcium sensitivity (pCa 5.0) and Hill coefficients were no different in all three cases (Figure 5I-L and Table 4). Taken together, the data show that cMyBP-CAllP+ contributes to faster myosin kinetics in a β-MyHC background and that cMyBP-C phosphorylation plays a significant role in regulating the kinetics in human cardiac muscle.
Figure 5.

β-MyHC kinetics. A, E, Isotonic quick release data were utilized to determine the force-velocity relationships and (I) maximum shortening velocities at pCa 5.0 and sarcomere length of 2.1 μm.33 m.l./s; muscle lengths per second. B, F, Normalized power output-force relationship and (J) maximum power output at pCa 5.0. C, G, Rate of force redevelopment and (K) maximum unloaded shortening velocities were determined using the slack test. The changes in sarcomere length (Δlength) amplitude versus duration (Δtime) of unloaded shortening are shown (C and G) to determine cross-bridge turnover. The isometric force at different calcium (pCa) concentrations (D and H) and maximum force relationships (L) are shown before and after treatment with PKA at 22°C. PKA treatment accelerates the kinetics of cross-bridge rate and Ca2+ sensitivity. The values of fiber force, shortening velocity and power output are summarized in Table 4. Data are mean±SE (n=5). *P<0.05 versus NTG and #P<0.05 versus β-TG and cMyBP-CWT:(t/t)/β.
Table 4.
Measurements in skinned fibers
| NTG | NTG/PKA | β-TG | β-TG/PKA | WT:(t/t)/β | WT:(t/t)/β/PKA | AllP+:(t/t)/β | AllP+:(t/t)/β/PKA | |
|---|---|---|---|---|---|---|---|---|
| pCa50 | 5.87±0.01 | 5.70±0.01a | 5.87±0.01 | 5.70±0.01a | 5.85±0.02 | 5.69±0.01a | 5.88±0.01 | 5.69±0.01a |
| Hill coefficient | 4.48±0.16 | 4.68±0.61 | 4.35±0.22 | 5.31±0.57 | 6.31±0.55 | 5.30±0.58 | 4.92±0.26 | 5.78±0.44 |
| Maximum absolute force (μN/length) | 10.16±0.23 | 9.59±0.29 | 9.46±0.32 | 9.04±0.36 | 9.42±0.24 | 9.01±0.11 | 9.71±0.19 | 9.33±0.12 |
| Unloaded-shortening velocity (m.l./s) | 4.24±0.09 | 5.67±0.27a | 3.05±0.06b | 4.15±0.15a,d | 3.07±0.14b | 4.11±0.18a,d | 4.08±0.16c | 4.47±0.12d |
| Maximum shortening velocity (m.l./s) | 3.15± 0.08 | 4.33±0.10a | 2.48±0.12b | 3.4±0.23a d | 2.26±0.12b | 3.45±0.13a,d | 3.13±0.16c | 3.6±0.24d |
| Maximum power output (m.l./s) | 0.59±0.02 | 0.85±0.04a | 0.45±0.03b | 0.67±0.02a,d | 0.41±0.04b | 0.64±0.02a,d | 0.61±0.05c | 0.67±0.02d |
Contractile properties of fibers isolated from the papillary muscles of 12 week old mice.14 The unloaded shortening and maximum shortening velocities and relative power that the fibers developed were measured. To determine the effects of PKA phosphorylation on the pCa-force relationship in vitro, skinned fibers were treated with PKA after measurements of the pCa-force relationship under standard conditions. All values are expressed as mean±SE (n=6). Statistical significance was determined using the paired t-test (n=5).
P<0.05 versus respective untreated PKA controls
P<0.05 versus NTG
P<0.05 versus WT:(t/t)/β
P<0.05 versus NTG/PKA
cMyBP-CAllP+ Protects the Heart from I-R Injury in a β-MyHC Background
In the mouse, cMyBP-CAllP+ is cardioprotective during I-R injury, but is associated with a reduction in β-MyHC levels.2 To determine if this phenomenon might be relevant in the human heart, NTG, β-TG, cMyBP-CAllP+:(t/t)/β and cMyBP-CWT:(t/t)/β hearts were subjected to LV cardiac ischemia for 1 hour followed by 24 hours of reperfusion.2 β-TG mice were treated with isoproterenol as a positive control to protect the heart against ischemic injury. Results show that cMyBP-CAllP+:(t/t)/β hearts had a significantly reduced (22±1%) infarcted area (IA), when normalized to the area at risk (AAR) from I-R injured NTG (32±2%), β-TG (36±3%) and cMyBP-CWT:(t/t)/β (40±3%) controls (Figure 6A, 6B). The AAR was not significantly different among the four groups (Figure 6B). The degree of cardioprotection is essentially equivalent to that observed in the normal mouse α-MyHC background.2 As expected, the preconditioned β-TG hearts showed the largest reduction in IA (9±1%). Cardiac injury was apparent in the cMyBP-CWT:(t/t)/β and β-TG hearts, whereas the cMyBP-CAllP+:(t/t)/β hearts were relatively less affected. NTG hearts with I-R injuries show a modest β-MyHC induction but this did not occur in the β-MyHC background (Figure 6C). Cardioprotection in the cMyBP-CAllP+:(t/t)/β hearts was accompanied by decreased TUNEL positive nuclei (Figure 6D-E), DNA fragmentation (Figure 6F) and cMyBP-C degradation (Figure 6G) compared to β-TG and cMyBPCWT:(t/t)/β hearts. Fractional shortening (Figure 6H) and fractional area change (Figure 6I) were completely preserved in the cMyBP-CAllP+:(t/t)/β mice, whereas these parameters were decreased in β-TG and cMyBP-CWT:(t/t)/β mice at four weeks. These results demonstrate that cMyBP-C phosphorylation protects the myocardium from cell injury and death irrespective of myosin background.
Figure 6.

I-R injury. A, Representative I-R injured mouse hearts stained with Evans blue and triphenyltetrazolium chloride. The white areas represent the infarcted region and the red shows the area at risk region. B, Area at risk (AAR) and infarcted area (IA) normalized to total LV area and AAR, respectively (n=6; *P<0.05). C, Myosin isoform shift in sham (S) and I-R injured hearts. D, Immunohistochemistry shows TUNEL-positive nuclei (red), cardiac TnI (green) and nuclei (blue). E, Quantitation of TUNEL-positive cardiomyocytes expressed as a percentage of total cardiomyocytes in hearts after sham and I-R injury. Values are expressed as mean±SE (*P<0.01 versus NTG, β-TG and cMyBP-CWT:(t/t)/β, n=3 hearts/group). F, DNA fragmentation assays by ligation-mediated PCR as an indication of apoptosis after I-R. G, Western blot of cMyBP-C shows decreased cMyBP-C degradation after I-R in cMyBP-CAllP+:(t/t)/β hearts compared to NTG, β-TG and cMyBP-CWT:(t/t)/β hearts (small arrow= cMyBP-C degradation products). H, Percentage of fractional shortening (%FS) and I, fractional area change (%FAC) assessed by echocardiography (*P<0.001, **P<0.05 versus sham; #P<0.05 versus NTG, β-TG and cMyBP-CWT:(t/t)/β after I-R (n=6)).
Discussion
cMyBP-C phosphorylation has consequences on cross-bridge cycling that differs depending upon its phosphorylation state and the myosin isoform present.18,19 The present study was directed at determining the structural and functional aspects of cMyBP-C phosphorylation in a mouse heart whose myosin isoform complement mimics that found in the human heart, establishing a rationale for cMyBP-C phosphorylation modulation after I-R as a cardioprotective strategy. Our data clearly show that expression of a cMyBP-C phosphomimetic in a β-MyHC background has different consequences in vivo. Unlike the case for when cMyBP-CAllP+ was expressed in the normal mouse background of cardiac α-MyHC, expression in the β-MyHC background led to improved contraction and relaxation (Figures 4 and 5) compared to β-TG and cMyBP-CWT:(t/t)/β cohorts. At the same time, placing the cMyBP-C(t/t) genotype in the β-MyHC background exacerbated morbidity and resulted in a dramatically decreased life expectancy with expression of a nonphosphorylatable cMyBP-C being completely ineffective at rescuing the mouse from death. These results underscore the differences expression of either normal or mutant cMyBP-C can have on cardiac function in a β-MyHC background and emphasizes the importance of cMyBP-C in hearts that contain predominantly β-MyHC.
Interestingly, the positions of the cross-bridges are different in filaments containing α- and β-MyHC,14 even though the region to which the C1-C2 domain of cMyBP-C binds is highly homologous between the two myosin isoforms.10,20,21 Using recombinant C1-C2 peptides we confirmed that cMyBP-C binds to both myosins in the same region in a phosphorylation dependent manner. In contrast to when it is placed into the α-MyHC background,2 cMyBP-CAllP+ improves myocardial contractility at baseline in the β-TG hearts, demonstrating that the influence of cMyBP-C phosphorylation on cardiac contractility is indeed myosin isoform dependent in the whole organ context and that these experiments are relevant to human cardiac function. In contrast to cMyBP-CWT and cMyBP-CAllP+ in vivo, the constant state of cMyBP-CAllP- binding with the myosin S2 region may associate with increased inhibition of myosin extension during cross-bridge cycles and dysregulate myosin organization in the β-MyHC background than the α-MyHC background for the observed detrimental effect. In conclusion, presence of either cMyBP-CAllP- or absence of cMyBP-C (cMyBP-C(t/t)) causes decreased cardiac function and cardiac hypertrophy,1,17 suggesting the necessity of cMyBP-C and its phosphorylation for normal cardiac function.
Does phosphorylation of cMyBP-C play a direct role in regulating contraction? The data are convincing that cMyBP-C influences actomyosin Mg2+-ATPase activity and the kinetics of cross-bridge cycling and that cMyBP-C phosphorylation modifies actomyosin Mg2+-ATPase activity and the rate of relaxation.4,22,23 However, failure of cMyBP-C phosphorylation to alter the Ca2+ sensitivity of actomyosin Mg2+-ATPase activity in reconstituted contractile protein systems has been cited by some as strong evidence against a role for phosphorylation of cMyBP-C in regulating contraction. 22,24 Reconstituted thick filament proteins do not, however, reproduce the normal thick-filament structure or the steric arrangement of contractile proteins in a filament lattice. Indeed it is necessary to study the overall effect of total cMyBP-C phosphorylation in an intact mouse model with different myosin backgrounds and PKA-mediated phosphorylation has differential effects on actomyosin Mg2+-ATPase activity depending on whether α-MyHC or β-MyHC is present.25 Our data show that under normal conditions, cMyBP-C phosphorylation increases actomyosin Mg2+-ATPase activity without altering thin filament Ca2+ sensitivity in a β-MyHC background, suggesting that cMyBP-C phosphorylation may not directly modulate myofilament Ca2+-sensitivity.22 However, changes in Ca2+-sensitivity due to length-dependent activation have not been excluded26 and cMyBP-C phosphorylation may play a direct role in the cooperative activation of the filament system, force generation, regulating cross-bridge rates of the myosin-actin motors, as well as functioning as a tether.12,27 Our in vitro data suggest that cMyBP-C phosphorylation releases myosin from a myosin interaction constraint, which ultimately allows myosin to increase cross-bridge formation resulting in faster cycle kinetics.12 These results are consistent with previous studies using fibers in which cMyBP-C has been completely ablated. Those data documented an increase in the unloaded shortening velocity.28 The data, in conjunction with the Mg2+-ATPase activity, suggest that cMyBP-C phosphorylation can accelerate β-MyHC kinetics and myocardial contractility without affecting Ca2+ sensitivity of the thin filaments in a β-MyHC background.
Myofilament protein phosphorylation represents a point of convergence for complex signaling events that ultimately result in cardioprotection and changes in contractile function. Direct manipulation of the contractile apparatus and endpoints, bypassing receptor-ligand signaling pathways, could have significant advantages in more precisely targeting a therapeutic action, and is now a focus of drug development.29 Widespread myocardial ischemia can cause contractile dysfunction, which often persists even after blood flow has been restored. A major finding of the present work is that cMyBP-CAllP+ is associated with significant protection from myocardial injury with better cardiac function and less cellular damage after I-R injury in the humanized mouse heart. One possible explanation is that cMyBP-CAllP+ is relatively resistant to calpain cleavage,30 which can be activated during I-R. Our recent data show that cMyBP-C is a substrate of calpain and phosphorylation of cMyBP-C renders the protein resistant to calpain-mediated proteolysis.31 This is consistent with the observation that phosphorylation of cardiac troponin I by PKA significantly reduced its cleavage by calpain.32 Therefore, the presence of an intact active cMyBP-C is required to regulate the formation of the weakly bound state between actin and myosin that leads to entry into force-generation by driving the main molecular components-the thick and thin filaments. Furthermore, improved cardiac function after I-R injury could be mediated by cMyBPCAllP+ resulting from direct enhancement of the myosin-actin interaction. The absence of cMyBP-CAllP+ interaction with the myosin S2 region leading to increased thick filament spacing2 and resistance to calpain-mediated proteolysis, may associate with cardioprotection, independent of myosin background. These data suggest that improved cardiac function after I-R injury could be mediated by directed phosphorylation of cMyBP-C and that contractile protein-based cardioprotection may represent a therapeutic avenue to improve myocardial contractility in the ischemic and failing heart.
Acknowledgments
Sources of Funding This research was supported by NIH grants P01HL69799, P50HL074728, P50HL077101, P01HL059408, R01HL087862 and the International Collaboration Research Project with the Japanese Health Science Foundation (J.R.) and by the American Heart Association, Ohio Valley Affiliate (S.S.)
Footnotes
Disclosures
None
Author Disclosures
Sakthivel Sadayappan: Research Grant: AHA beginning investigator, Amount: >= $10,000
James Gulick: No disclosures
Raisa Klevitsky: No disclosures
John N Lorenz: Research Grant: NIH PPG, Amount: >= $10,000
Michelle Sargent: No disclosures
Jeffery D Molkentin: Research Grant: Multiple NIH R01’s and a PPG component, Amount: >= $10,000
Jeffrey Robbins: Research Grant: Multiple R01’s, a Program Project Grant and Japanese Pioneer Grant, Amount: >= $10,000
References
- 1.Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, Klevitsky R, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein-C phosphorylation and cardiac function. Circ Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein-C phosphorylation is cardioprotective. Proc Natl Acad Sci USA. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohamed AS, Dignam JD, Schlender KK. Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch Biochem Biophys. 1998;358:313–319. doi: 10.1006/abbi.1998.0857. [DOI] [PubMed] [Google Scholar]
- 4.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? Embo J. 1995;14:1952–1960. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775–785. doi: 10.1056/NEJM199703133361107. [DOI] [PubMed] [Google Scholar]
- 6.Yuan C, Guo Y, Ravi R, Przyklenk K, Shilkofski N, Diez R, Cole RN, Murphy AM. Myosin binding protein C is differentially phosphorylated upon myocardial stunning in canine and rat hearts-- evidence for novel phosphorylation sites. Proteomics. 2006;6:4176–4186. doi: 10.1002/pmic.200500894. [DOI] [PubMed] [Google Scholar]
- 7.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 8.El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh YH, Nattel S, Dobrev D, Eschenhagen T, Carrier L. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol. 2007;43:223–229. doi: 10.1016/j.yjmcc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Nagayama T, Takimoto E, Sadayappan S, Mudd JO, Seidman JG, Robbins J, Kass DA. Control of in vivo left ventricular contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation. Circulation. 2007;116:2399–2408. doi: 10.1161/CIRCULATIONAHA.107.706523. [DOI] [PubMed] [Google Scholar]
- 10.Weisberg A, Winegrad S. Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc Natl Acad Sci USA. 1996;93:8999–9003. doi: 10.1073/pnas.93.17.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofmann PA, Greaser ML, Moss RL. C-protein limits shortening velocity of rabbit skeletal muscle fibres at low levels of Ca2+ activation. J Physiol. 1991;439:701–715. doi: 10.1113/jphysiol.1991.sp018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lecarpentier Y, Vignier N, Oliviero P, Guellich A, Carrier L, Coirault C. Cardiac Myosin-binding protein C modulates the tuning of the molecular motor in the heart. Biophys J. 2008;95:720–728. doi: 10.1529/biophysj.107.127787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winegrad S. Myosin binding protein C, a potential regulator of cardiac contractility. Circ Res. 2000;86:6–7. doi: 10.1161/01.res.86.1.6. [DOI] [PubMed] [Google Scholar]
- 14.Krenz M, Sanbe A, Bouyer-Dalloz F, Gulick J, Klevitsky R, Hewett TE, Osinska HE, Lorenz JN, Brosseau C, Federico A, Alpert NR, Warshaw DM, Perryman MB, Helmke SM, Robbins J. Analysis of myosin heavy chain functionality in the heart. J Biol Chem. 2003;278:17466–17474. doi: 10.1074/jbc.M210804200. [DOI] [PubMed] [Google Scholar]
- 15.Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RH. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res. 2000;86:51–58. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- 16.Krenz M, Robbins J. Impact of beta-myosin heavy chain expression on cardiac function during stress. J Am Coll Cardiol. 2004;44:2390–2397. doi: 10.1016/j.jacc.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 17.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–1244. doi: 10.1172/JCI7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weisberg A, Winegrad S. Relation between crossbridge structure and actomyosin ATPase activity in rat heart. Circ Res. 1998;83:60–72. doi: 10.1161/01.res.83.1.60. [DOI] [PubMed] [Google Scholar]
- 19.Stelzer JE, Brickson SL, Locher MR, Moss RL. Role of myosin heavy chain composition in the stretch activation response of rat myocardium. J Physiol. 2007;579:161–173. doi: 10.1113/jphysiol.2006.119719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gruen M, Gautel M. Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol. 1999;286:933–949. doi: 10.1006/jmbi.1998.2522. [DOI] [PubMed] [Google Scholar]
- 21.Gruen M, Prinz H, Gautel M. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 1999;453:254–259. doi: 10.1016/s0014-5793(99)00727-9. [DOI] [PubMed] [Google Scholar]
- 22.Stelzer JE, Patel JR, Walker JW, Moss RL. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ Res. 2007;101:503–511. doi: 10.1161/CIRCRESAHA.107.153650. [DOI] [PubMed] [Google Scholar]
- 23.McClellan G, Weisberg A, Winegrad S. cAMP can raise or lower cardiac actomyosin ATPase activity depending on alpha-adrenergic activity. Am J Physiol. 1994;267:H431–442. doi: 10.1152/ajpheart.1994.267.2.H431. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann PA, Lange JH., 3rd Effects of phosphorylation of troponin I and C protein on isometric tension and velocity of unloaded shortening in skinned single cardiac myocytes from rats. Circ Res. 1994;74:718–726. doi: 10.1161/01.res.74.4.718. [DOI] [PubMed] [Google Scholar]
- 25.Winegrad S, Weisberg A. Isozyme specific modification of myosin ATPase by cAMP in rat heart. Circ Res. 1987;60:384–392. doi: 10.1161/01.res.60.3.384. [DOI] [PubMed] [Google Scholar]
- 26.Cazorla O, Szilagyi S, Vignier N, Salazar G, Kramer E, Vassort G, Carrier L, Lacampagne A. Length and protein kinase A modulations of myocytes in cardiac myosin binding protein C-deficient mice. Cardiovasc Res. 2006;69:370–380. doi: 10.1016/j.cardiores.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 27.Stelzer JE, Moss RL. Contributions of stretch activation to length-dependent contraction in murine myocardium. J Gen Physiol. 2006;128:461–471. doi: 10.1085/jgp.200609634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korte FS, McDonald KS, Harris SP, Moss RL. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ Res. 2003;93:752–758. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 29.Cleland JG, Coletta AP, Clark AL. Clinical trials update from the Heart Failure Society of America meeting: FIX-CHF-4, selective cardiac myosin activator and OPT-CHF. Eur J Heart Fail. 2006;8:764–766. doi: 10.1016/j.ejheart.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Decker RS, Decker ML, Kulikovskaya I, Nakamura S, Lee DC, Harris K, Klocke FJ, Winegrad S. Myosin-binding protein C phosphorylation, myofibril structure, and contractile function during low-flow ischemia. Circulation. 2005;111:906–912. doi: 10.1161/01.CIR.0000155609.95618.75. [DOI] [PubMed] [Google Scholar]
- 31.Sadayappan S, Greis KD, Robbins J. Phosphorylation-dependent proteolysis and pathogenesis of cardiac myosin binding protein–C. J Mol Cell Cardiol. 2008;44:S44. [Google Scholar]
- 32.Di Lisa F, De Tullio R, Salamino F, Barbato R, Melloni E, Siliprandi N, Schiaffino S, Pontremoli S. Specific degradation of troponin T and I by mucalpain and its modulation by substrate phosphorylation. Biochem J. 1995;308(Pt 1):57–61. doi: 10.1042/bj3080057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krenz M, Sadayappan S, Osinska HE, Henry JA, Beck S, Warshaw DM, Robbins J. Distribution and structure-function relationship of myosin heavy chain isoforms in the adult mouse heart. J Biol Chem. 2007;282:24057–24064. doi: 10.1074/jbc.M704574200. [DOI] [PubMed] [Google Scholar]