

Abstract
Macrophages undergo fusion to form multinucleated giant cells in several pathologic conditions, including the foreign body response (FBR). We detected high levels of matrix metalloproteinase (MMP)-9 during macrophage fusion in vitro and in foreign body giant cells (FBGCs) in vivo. Wild-type (WT) bone marrow-derived macrophages were induced to fuse with IL-4 in the presence of MMP-9 function-blocking antibodies and displayed reduced fusion. A similar defect, characterized by delayed shape change and abnormal morphology, was observed in MMP-9 null macrophages. Analysis of the FBR in MMP-9 null mice was then pursued to evaluate the significance of these findings. Specifically, mixed cellulose ester disks and polyvinyl alcohol sponges were implanted s.c. in MMP-9 null and WT mice and excised 2–4 weeks later. Histochemical and immunohistochemical analyses indicated equal macrophage recruitment between MMP-9 null and WT mice, but FBGC formation was compromised in the former. In addition, MMP-9 null mice displayed abnormalities in extracellular matrix assembly and angiogenesis. Consistent with a requirement for MMP-9 in fusion, we also observed reduced MMP-9 levels in MCP-1 null macrophages, previously shown to be defective in FBGC formation. Collectively, our studies show abnormalities in MMP-9 null mice during the FBR and suggest a role for MMP-9 in macrophage fusion.
Keywords: monocyte/macrophages, cell fusion, extracellular matrix, angiogenesis
INTRODUCTION
Cell fusion is a critical process in a number of physiological and pathological conditions, including fertilization, development, bone homeostasis, and the response to certain pathogens and foreign materials [1,2,3]. The latter involves the participation of macrophages that can undergo homotypic fusion to form multinucleated giant cells [4, 5]. Formation of these giant cells is believed to enhance the defensive capacities of macrophages. To date, a small number of surface receptors have been shown to participate in macrophage fusion, including CD44, CD47, CD200, signal regulatory protein 1-a, IL-4R, E-cadherin, and mannose receptor (reviewed in ref. [5]). In addition, blockade of molecules, such as DAP12, dendritic cell-specific transmembrane protein (DC-STAMP), Rac1, and STAT-6, have been shown to be effective in limiting macrophage fusion [6,7,8,9]. Despite the wealth of knowledge relating to macrophage activation and function, fusion as a terminal differentiation event remains poorly understood [10].
Foreign body giant cell (FBGC) formation from the fusion of macrophages is observed as a result of the response induced by biomaterials and other foreign bodies. As a result of their large size and surface characteristics, these substances elicit a prolonged series of reactions, a process known as the foreign body response (FBR) [5]. In many ways, the FBR reproduces the initial events associated with wound healing [11, 12]. Although inflammation and macrophage activation resolve in the later stages of wound healing to promote tissue remodeling, the FBR is characterized by the persistence of inflammatory cells, particularly macrophages, at the site of implantation. On the implant surface, macrophages fuse into large, multinucleated FBGCs. In comparison with single macrophages, FBGCs are capable of delivering a more effective and concentrated assault on the implant, degrading material and even causing failure of implanted devices [13]. Major efforts to engineer biocompatible devices that elude the FBR have been unsuccessful, perhaps because the complex biological effectors of the FBR are not fully understood [14].
Investigation of fusion in genetically modified mice or with cells derived from them has confirmed the requirement for several molecular modulators of FBGC formation, such as the receptors for IL-4, DAP12, DC-STAMP, STAT-6, plasma fibronectin, and CCL2/MCP-1 [6, 8, 9, 15,16,17]. Nevertheless, it is apparent that the FBR and FBGC formation are regulated by a complex network of molecular cues [5]. In fact, little is known about the potential overlap in the activities of the molecules mentioned above. Previously, we have shown that thrombospondin 2 null mice displayed an altered FBR, characterized by changes in extracellular matrix (ECM) deposition and increased angiogenesis [18]. Recent studies have suggested that these abnormalities are a result of increased extracellular levels of matrix metalloproteinase (MMP)-2 and MMP-9 [19,20,21,22]. In the process of examining the levels of MMPs during the FBR in wild-type (WT) and genetically modified mice, we detected high levels of MMP-9 in FBGC, an observation that served as impetus for the present study.
MMPs comprise a family of zinc-dependent endopeptidases with a large range of substrates, including ECM proteins, growth factors, chemokines, and cell-surface proteins [23, 24]. MMPs perform critical functions in many physiological and pathological processes, such as wound healing, angiogenesis, inflammation, and cancer. At the cellular level, these proteases have been shown to regulate cell invasion, migration, apoptosis, and proliferation [25, 26]. The pleiotropic effect of MMP-9 is mediated by its ability to process ECM components, such collagen I, IV, V, VII, X, and XI, elastin, fibronectin, and laminin [25, 27]. In addition, MMP-9 can cleave and activate a number of cytokines, chemokines, and growth factors, such as IL-1 and monocyte chemokine MCP-3/CCL7, and convert plasminogen to active antiangiogenic angiostatin [28, 29]. MMP-9 can also process the proinflammatory IL-8 to yield a more potent cleavage product and activates latent TGF-β [30, 31]. Furthermore, MMP-9-mediated cleavage can lead to deactivation of molecules such as stromal derived factor-1 [32]. These roles in modulating inflammatory signals implicate MMP-9 as a factor that might influence the FBR and FBGC formation.
Based on our observation of high levels of MMP-9 in FBGC in vivo and its functional complexity in inflammatory processes, we hypothesized that MMP-9 has the potential to modulate the FBR and FBGC formation. In the present study, we used an in vitro macrophage fusion assay and confirmed the participation and importance of MMP-9 in this process. Furthermore, we used two in vivo assays to induce FBGC formation and investigate the role of MMP-9 in the FBR. Specifically, we used s.c. implantation of Millipore filters composed of mixed cellulose esters. Previously, we had shown that this material elicits a FBR that is similar to that generated by biomaterials such as silicone, but it is preferable, as it is amenable to standard fixation and preparation protocols for histological and immunohistochemical analysis [33]. In addition, we used a polyvinyl alcohol (PVA) sponge granuloma model that elicits robust angiogenic and fibrogenic responses that can be quantitatively analyzed within a limited timeframe (1–3 weeks) [11]. Analyses of fusion and the FBR in these models indicated that in addition to compromised FBGC formation, MMP-9 null mice displayed abnormalities in ECM assembly and angiogenesis. Taken together, our observations suggest that MMP-9 plays a critical role in the development of the FBR. Specifically, we show that the fusion of macrophages requires MMP-9, making it the first extracellular enzyme to be linked to this process.
MATERIALS AND METHODS
Animal model
The generation of MMP-9 null mice has been described [34]. Three-month-old, sex-matched MMP-9 null and control mice on an FVB background were used. Control mice were generated by breeding MMP-9 null and WT mice and subsequent breeding of heterozygotes. The Institutional Animal Care and Use Committee at the University of Washington (Seattle, WA, USA) and Yale University (New Haven, CT, USA) approved all experiments.
Primary macrophage isolation
Bone marrow-derived macrophages were obtained from MMP-9 null, WT, and MCP-1 null mice as described previously [7]. Briefly, marrow flushed from the femurs of mice was collected in IMDM (Life Technologies, Inc., Gaithersburg, MD, USA), supplemented with 10% FBS, and depleted of MMP-9 using gelatin-coated beads, according to the manufacturer’s instructions (GE Healthcare, Piscataway, NJ, USA), and antibiotics (penicillin/streptomycin/fungizone). The cell suspension was layered over Lympholyte-M (Accurate Chemical, Westbury, NY, USA) and centrifuged according to the supplier’s instructions. The fraction containing mononuclear cells was collected, plated, and expanded in the presence of 1.5 ng/ml M-CSF (R&D Systems, Minneapolis, MN, USA) and 100 ng/ml fetal liver tyrosine kinase 3 ligand (R&D Systems). Cultures were fed on Day 5, and cells were collected by scraping on Day 10 and used for fusion experiments. Three mice per genotype were used in each isolation procedure.
Cell fusion and gelatin zymography
Fusion was induced in expanded macrophages as described previously [16]. Briefly, 1 × 106 cells/well (24-well plates) were induced to fuse with 10 ng/ml recombinant mouse (rm)GM-CSF (R&D Systems) and 10 ng/ml rmIL-4 (R&D Systems). Medium was changed once on Day 3, and all experiments were performed with MMP-9-depleted serum. This was necessary, as serum contained MMP-9, and fusion of MMP-9 null macrophages was not compromised in nondepleted serum (not shown). Selected wells of WT macrophages were treated with 1 or 5 μg anti-MMP-9 function-blocking antibody (Chemicon International, El Segundo, CA, USA; Catalog Number MAB13415) or 5 μg isotype control antibody. The function-blocking activity of the anti-MMP-9 antibody was demonstrated previously [35]. Conditioned media were collected daily from cultures of cells undergoing fusion over the 7-day treatment. SDS-PAGE gels supplemented with 0.1% gelatin (1 mg/mL) were used to detect gelatinases. rmMMP-9 (Chemicon International) was electrophoresed as a positive control. Gels were incubated in renaturing buffer (2.5% Triton X-100 v/v in distilled water), followed by developing buffer (50 mM Tris, 0.2 M NaCl, 5 mM CaCl2, 0.02% Brij 35 in distilled water) at 37°C overnight. Gels were stained with the Coomassie-based stain GelCode (Pierce, Rockford, IL, USA), and gelatinase activity was evident as clear bands, indicating gelatin degradation, against the Coomassie blue-stained background.
Cell staining
For morphological analysis and quantification of fusion, cultures were double-stained with May-Grunwald stain (Sigma Chemical Co., St. Louis, MO, USA) and Wright-Giemsa stain (Sigma Chemical Co.), according to standard protocols. All experiments were performed in triplicate, and a total of 25–50 high-power fields (hpf) were analyzed for each group. For visualization of the actin cytoskeleton and nuclei, cells were fixed in 4% paraformaldehyde (JT Baker, Phillipsburg, NJ, USA) for 20 min at room temperature and stained with rhodamine-phalloidin (Molecular Probes, Eugene, OR, USA) and 4′,6-diamidino-2-phenylindole (DAPI; Sigma Chemical Co.) as described previously [7]. All wells were mounted with Vectashield (Vector Labs) and examined with the aid of an Axiovert 200M Zeiss microscope equipped with fluorescent optics.
Implantation of biomaterials
s.c. implantations were performed as described previously [18, 19]. Five MMP-9 null and five WT mice, age 3–4 months, were used for each implantation model (10 mice of each genotype total). Each mouse received two implants, which were placed at least 2 cm apart, through a midline incision, allowing for 10 implants/group. Implants were excised en bloc at 2 weeks (PVA sponges) and 4 weeks (filters) following implantation, processed for histological analysis, and stained with Masson’s trichrome according to standard protocols. Dense invasion was quantified as the percentage of total sponge area occupied by mature collagen based on Masson’s trichrome stain as described previously [19]. In addition, sections were analyzed by immunohistochemistry with antibodies against MMP-9 (Chemicon International), macrophage antigen-3 (Mac-3), PECAM-1 (BD PharMingen, San Diego, CA, USA), and fibronectin (Sigma Chemical Co.) as described previously [36].
RESULTS
MMP-9 participates in macrophage fusion in vitro
Zymography of conditioned media samples from fusing macrophages following IL-4/GM-CSF treatment revealed MMP-9 induction as early as Day 2 and peaking by Day 3, concomitantly with fusion (Fig. 1A). Based on the fusion assay, which required media change on Day 3, we collected serial samples on Days 1–3 and 4–7. Sustained high expression of MMP-9 at Day 4 represents continued production between Days 3 and 4, and Days 1–3 represent cumulative production over 72 h. Consistent with the observation in receptor activator of NF-κB ligand (RANK-L)-induced osteoclasts, we detected predominantly pro-MMP-9 [37]. The temporal expression of MMP-9 suggested that it might play a role in fusion. To examine this possibility, WT macrophages were induced to fuse in the presence of a function-blocking MMP-9 antibody (Fig. 1C) or isotype IgG control (Fig. 1B). Analysis of fusion at Day 7 revealed that addition of 5 μg antibody caused a reduction in overall fusion, characterized by the presence of smaller FBGCs (Fig. 1C). Enumeration of FBGC/hpf and the number of nuclei/FBGC revealed that they were increased and decreased by the function-blocking antibody, respectively (Fig. 1, D and F). In addition, overall fusion was reduced to below 50% in this group. Taken together, these observations suggest that inhibition of MMP-9 resulted in the formation of smaller and more numerous FBGC, leading to a reduction in fusion.
Fig. 1.

MMP-9 expression and participation in macrophage fusion. Macrophages were stimulated to fuse with IL-4/GM-CSF, and the levels of MMP-9 were monitored by zymography. (A) Serum used in the assay was depleted of MMP-9 as shown (Lane M), and macrophages did not express MMP-9 prior to fusion (Day 1). MMP-9 was strongly induced at Day 3, and the levels remained high throughout the fusion assay. rmMMP-9 was used as positive control (Lane MMP9). (B and C). Antibody-mediated inhibition of MMP-9 reduces fusion. Representative phase-contrast images of WT macrophages induced to fuse in the presence of a function-blocking antibody (C) or isotype control IgG (B) for 7 days. Cultures treated with control IgG displayed robust FBGC formation (arrows denote FBGCs). Cultures treated with 5 μg-blocking antibody displayed formation of small FBGCs (arrows in C). Original bars = 50 μm. (D–F) Analysis of fusion at Day 7 by enumeration of various parameters in May-Grunwald-stained cultures revealed a fusion defect in antibody-treated macrophages that involved the formation of smaller FBGCs. *, P ≤ 0.05; n = 9 (D–F).
To address the role of MMP-9 specifically in FBGC formation, we compared the ability of MMP-9 null and WT macrophages to fuse. May-Grunwald stained cultures (Fig. 2, A and B) were analyzed for fusion, as measured by number of giant cells, percent fusion, and number of nuclei per FBGC. MMP-9 null macrophages displayed a decrease in overall fusion (Fig. 2D), as a result of the formation of FBGC that contained fewer nuclei (Fig. 2E). Analysis of the distribution of nuclei/FBGC confirmed the trend for a reduced number of nuclei in MMP-9 null macrophages (Fig. 2F). A trend toward a higher number of FBGC in MMP-9 null cultures was observed, but the difference was not significant (Fig. 2C). MMP-9 null macrophages that remained under fusogenic conditions for an extra 3 days failed to undergo additional fusion (not shown). Based on these observations, we conclude that there is an inherent defect in fusion in the absence of MMP-9, suggesting that MMP-9 is an important modulator of FBGC formation.
Fig. 2.

MMP-9 null macrophages display reduced fusion. Representative phase-contrast images of WT (A) and MMP-9 null macrophages (B) at Day 3 showing reduced elongation and FBGC formation in the latter. (A and B) Arrows and arrowheads denote elongated cells and FBGC, respectively. Original bars = 25 μm. (C–E) Analysis of fusion at Day 7 by enumeration of various parameters in May-Grunwald-stained cultures revealed a fusion defect in MMP-9 null macrophages that involved the formation of smaller FBGCs. *, P ≤ 0.05; n = 9. (F) Histogram demonstrating the distribution of nuclei per FBGC.
MMP-9 null macrophages fail to undergo changes in cell shape
Previously, it was shown that macrophages undergo dynamic cytoskeletal changes prior to fusion in response to fusogenic stimuli [7]. We therefore analyzed the ability of IL-4-treated MMP-9 null macrophages to undergo morphological changes by visualizing the actin cytoskeleton with rhodamine phalloidin. A significant delay in the response was observed at 24 h, at which time, MMP-9 null macrophages retained peripheral actin (Fig. 3B). In contrast, WT macrophages displayed elongation, lamellipodia formation, and accumulation of punctate actin at the cell edges (Fig. 3A). Such formations could be observed in MMP-9 null macrophages at Day 3, but multinucleated cells, which could be observed in WT cultures, were still absent (Fig. 3D). Taken together, these observations indicate that MMP-9 null macrophages are defective in their early response to a fusogenic stimulus, and although cytoskeletal changes prior to fusion are observed, these alterations are inconsistent with typical fusion and do not precede fusion.
Fig. 3.

MMP-9 null macrophages display delayed and abnormal fusion-induced cytoskeletal reorganization. Representative images of WT (A and C) and MMP-9 null (B and D) IL-4-treated macrophages from Day 1 (A and B) and Day 3 (C and D) culture stained with phalloidin and DAPI are shown. At Day 1, WT macrophages exhibited normal elongation, accumulation of punctate actin (white arrows), and lamellipodia formation (yellow arrows), which were absent in MMP-9 null macrophages. Arrows in B identify the presence of peripheral actin. At Day 3, WT macrophages displayed FBGC formation and extensive lamellipodia formation (yellow arrows in C). (D) At this time-point, MMP-9 null macrophages displayed changes consistent with preparation for fusion, such as lamellipodia formation (yellow arrow) and cytoskeletal reorganization (white arrow), but FBGC formation was not evident. Original bars = 25 μm (A–D).
MMP-9 expression in FBGC in vivo
To investigate the role of MMP-9 in FBGC formation in vivo, we chose to use two different implantation models to induce a FBR and study FBGC formation in the context of processes that have been shown to be influenced by MMP-9, such as inflammation, ECM remodeling, and angiogenesis. Specifically, Millipore filters and PVA sponges were implanted s.c. in MMP-9 null and WT mice for 4 and 2 weeks, respectively. Detection of MMP-9 by immunohistochemistry revealed high levels of MMP-9 in FBGC that formed in response to both implants (Figs. 4A, and see 6A). MMP-9 was also present in single cells surrounding implants but at comparatively much lower levels (Figs. 4A, and see 6A).
Fig. 4.

MMP-9 participates in FBGC formation and the FBR in vivo. (A) A section of the Millipore filter (*) implanted s.c. in WT mice for 4 weeks was stained with anti-MMP-9 antibody and visualized with the peroxidase reaction (brown color). Arrows indicate immunoreactive FBGC. (B and C) Sections of filters (*) from WT (B) and MMP-9 null (C) mice stained with Masson’s trichrome. Arrows in B denote FBGCs, which are absent in the image from MMP-9 null mice (C). (C, inset) An abnormal MMP-9 null FBGC. Examination of collagen deposition (blue color in B and C) reveals irregular assembly in MMP-9 null mice. (D) Immunohistochemical detection of macrophages with anti-Mac-3 revealed normal levels of individual macrophages (arrowheads) in MMP-9 null mice. Sections were counterstained with methyl green (A and D). Original bars = 25 μm (A–D).
Fig. 6.

Abnormal sponge granuloma formation in MMP-9 null mice. Representative images of PVA sponges (*) implanted s.c. for 2 weeks in WT (A, C, and E) and MMP-9 null (B, D, and F) mice are shown. (A) Sections stained anti-MMP-9 antibody and visualized with the peroxidase reaction (brown color) revealed high levels of expression in FBGC (arrows). (B) Background immunoreactivity was observed in MMP-9 null sections (negative control). Sections of sponges (*) from WT (C) and MMP-9 null (D) mice stained with Masson’s trichrome are shown. Examination of collagen deposition (blue color in C and D) revealed reduced deposition in MMP-9 null mice. (E and F) Immunohistochemical detection of fibronectin deposition in WT (E) and MMP-9 null (F) mice revealed abnormal deposition in the latter, characterized by thinner fibers. Sections were counterstained with methyl green (A, B, E, and F). Original bars = 25 μm (A–F).
Abnormal FBR in MMP-9 null mice
Histological examination of the FBR in MMP-9 null mice revealed a striking paucity of FBGCs and irregular deposition of collagen fibers within the capsule (Fig. 4C). Specifically, unlike the FBR in WT mice (Fig. 4B), MMP-9 null mice formed capsules that contained mature collagen fibers that lacked organization, were disrupted, and did not form dense layers. In addition, FBGC formation in MMP-9 null mice involved the formation of columnar FBGC that displayed nuclear orientation toward their apical surface and contained dense cytoplasmic material (see inset in Fig. 4C). Immunohistochemical detection of macrophages with the Mac-3 antibody revealed the presence of single macrophages at the implant site with some cells in close proximity to the implant (Fig. 4D). Thus, a defect in macrophage recruitment cannot account for the reduced number of FBGC. In fact, enumeration of single macrophages revealed similar levels between WT and MMP-9 null implants (37.9±11.1 cells/300 μm2 for WT and 41.3±15.2 cells/300 μm2 for MMP-9 null).
Evaluation of FBGC formation, angiogenesis, and capsule thickness indicated that only the number of FBGC was decreased significantly in MMP-9 null mice (Fig. 5A). A difference in vascular density was not observed (Fig. 5B), and despite the irregular collagen deposition in MMP-9 null mice, capsule thicknesses were similar between the two groups (Fig. 5C).
Fig. 5.
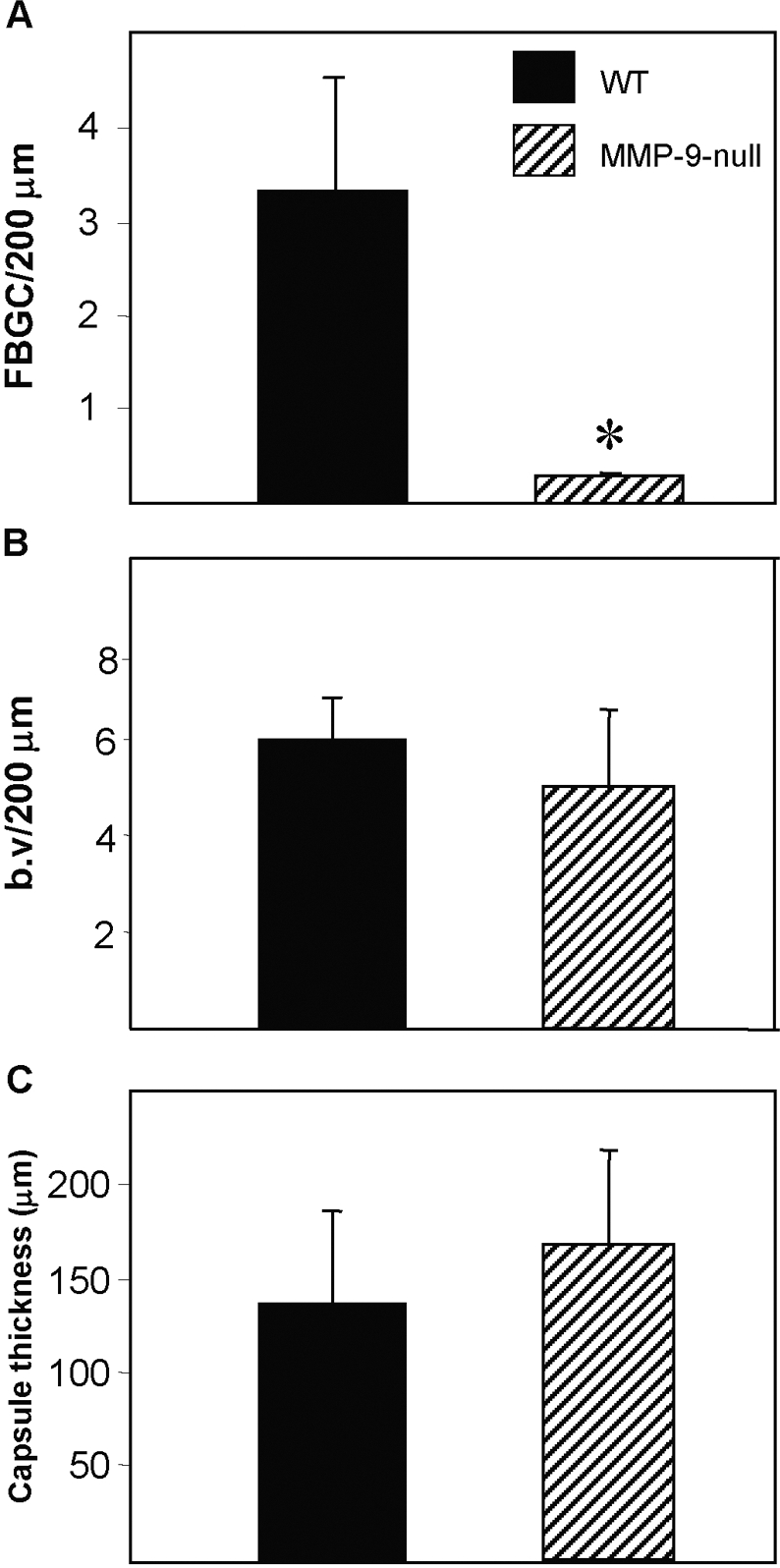
MMP-9 null mice display reduced FBGC formation. Filters were implanted s.c. for 4 weeks, explanted, and analyzed by histochemistry and immunohistochemistry. (A) The number of FBGC/unit length (surface of filter) was determined and found to be significantly lower in MMP-9 null mice. *, P ≤ 0.05; n = 5. Enumeration of blood vessels (b.v; B) and capsule thickness (C) indicated normal levels in MMP-9 null mice. A total of 50 sections from five mice/group was analyzed.
Abnormal sponge granuloma formation in MMP-9 null mice
Because of the established functions of MMPs in neovascularization and the striking abnormality in ECM assembly in MMP-9 null Millipore filter capsules, it was surprising not to detect changes in vascular density. We presume that this might be a result of the low angiogenic response elicited in this model. We therefore performed implantation of PVA sponges, which is a more robust model for the study of biomaterial-induced angiogenic responses [11]. Examination of PVA sponges implanted s.c. in WT and MMP-9 null mice for 2 weeks revealed differences in FBGC formation and in the overall fibrovascular invasion. Specifically, FBGCs were mostly absent from sponges implanted in MMP-9 null mice (Fig. 6, B, D, and F). Furthermore, examination of Masson’s trichrome-stained sections revealed reduced matrix formation and invasion in MMP-9 null mice (Fig. 6D). To investigate the abnormality in ECM assembly further, we stained sections with an antifibronectin antibody and observed diffuse deposition of thin, immature fibers in MMP-9 null sponges in comparison with WT sponges (Fig. 6F).
Analysis of digital images from Mac-3-stained sections revealed that the total number of macrophages in the PVA sponges was similar between MMP-9 null and WT mice (Fig. 7B), suggesting that the deficiency in MMP-9 did not affect their recruitment. However, MMP-9 null mice had significantly fewer FBGCs within the PVA sponges (Fig. 7A), suggesting a defect in fusion but not in inflammatory signaling. Fewer blood vessels were also detected within the PVA sponges implanted into MMP-9 null mice, as determined by PECAM-1 immunodetection (Fig. 7C). Dense invasion, an index of mature ECM formation within the area of the implanted sponge, was also reduced in MMP-9 null mice, indicating a defect in the assembly and/or maturation of collagen fibers (Fig. 7D).
Fig. 7.

MMP-9 null mice display reduced FBGC formation and angiogenesis in sponge granulomas. PVA sponges were implanted s.c. for 2 weeks, explanted, and analyzed by histochemistry and immunohistochemistry. (A and B) The number of FBGCs (A) and macrophages (B) per hpf was measured in Mac-3-stained sections, and the former was found to be lower in MMP-9 null mice. (C) Enumeration of blood vessels from PECAM-1-stained sections indicated a decrease in MMP-9 null mice. (D) Less-dense invasion within sponges, determined from Masson’s trichrome-stained sections, was observed in MMP-9 null mice. A total of 50 sections from five mice/group was analyzed. *, P ≤ 0.05; n = 5.
Reduced MMP-9 levels in MCP-1 macrophages
We have shown previously that MCP-1 null macrophages are deficient in FBGC formation [7]. Specifically, these cells displayed reduced overall fusion and formation of smaller FBGC in vitro. Here, we asked whether these cells express normal levels of MMP-9 and undergo normal shape changes in response to IL-4. Examination of cells at 2 days following addition of IL-4 revealed predominantly rounded cells with peripheral actin deposition (Fig. 8A). Some cells displayed cytoplasmic actin deposition but were not elongated. Examination of cultures at Day 7 revealed few small FBGCs, minimal elongation, and numerous single-nucleated cells (Fig. 8B). The appearance of these cultures is consistent with our previous quantitative analysis of FBGC formation by these cells [7]. Conditioned media from IL-4-treated MCP-1 null cells were analyzed by zymography, which revealed weak expression of MMP-9 at Day 2 (Fig. 8C). MMP-9 levels peaked by Day 3, dropped by Day 4, and were below detection at Days 5 and 6. Media were changed at Day 3, indicating some expression between Days 3 and 4. Overall, the expression of MMP-9 in MCP-1 null cells was low, suggesting that the reduced FBGC formation is a result, in part, of low MMP-9 levels.
Fig. 8.

Reduced MMP-9 levels in fusion-defective MCP-1 null macrophages. Representative images of MCP-1 null macrophages induced to fuse with IL-4 for 2 (A) and 7 days (B) and visualized with rhodamine-phalloidin and DAPI. (A) Arrows and arrowheads denote single cells with peripheral and cytoplasmic actin distribution, respectively. (B) Formation of FBGC (arrowhead) and single cells with peripheral actin (arrows) can be observed in Day 7 cultures. Original bars = 50 μm. (C) The levels of MMP-9 were monitored by zymography and revealed that MCP-1 null macrophages express low levels of MMP-9 during fusion. rmMMP-9 (lane MMP9) was used as positive control. Lane Mr, molecular weight standards.
DISCUSSION
In the present study, we provide evidence for the participation of MMP-9 in the process of macrophage fusion and FBGC formation. Experiments in vitro and in vivo show high expression of MMP-9 in FBGC and defective fusion of MMP-9 null macrophages. Furthermore, the expression of MMP-9 was reduced in fusion-defective MCP-1 null macrophages. Consistent with these findings, a recent study has shown that macrophages cultured on biomaterials expressed MMP-9 and that its expression was regulated by the nature of the biomaterial [38]. In the same study, the use of specific MMP inhibitors suggested that MMP-9 was not implicated in the fusion process. However, recent examination of the specificity of these MMP inhibitors revealed a wider range than reported originally [39]. Other studies have shown that MMP-9 is critical for various aspects of macrophage function, including migration [40, 41]. As the number of macrophages in the FBR in WT and MMP-9 null mice is equal, we conclude that MMP-9 is not required for their recruitment in vivo. This finding is not unprecedented in the context of the FBR, as we previously showed reduced fusion but normal recruitment of macrophages in mice that lack the potent monocyte chemokine CCL-2/MCP-1 [16].
Our studies in vitro show that MMP-9 is induced in macrophages under fusogenic conditions. Previously, FBGC formation in vitro was shown to occur after 48 h of exposure to IL-4 [7]. Thus, the temporal expression of MMP-9, which is present at high levels at 48 h, suggests its active participation in fusion. A potential role for MMP-9 in pre-fusion events might include modulation of cell adhesion, which has been shown to be critical for fusion in the context of integrin-ECM-mediated interactions [42]. However, we did not observe an adhesion defect in MMP-9 null macrophages (not shown). Furthermore, the present study revealed reduced fusion in WT macrophages induced to fuse in the presence of a function-blocking MMP-9 antibody. Collectively, the effects of MMP-9 blockade on fusion, the irregular remodeling of the cytoskeleton in MMP-9 null FBGC, and the abnormal morphology of FBGC in MMP-9 null mice suggest a role for MMP-9 in fusion per se. Specifically, the delayed cytoskeletal rearrangement and irregular actin assembly during fusion indicate that MMP-9 macrophages can receive a fusogenic stimulus but are unable to carry out proper fusion. We have reported recently on the importance of cytoskeletal rearrangements as a prerequisite to macrophage fusion [7]. Consistent with this report, we detected reduced MMP-9 expression and delayed shape change in MCP-1 null macrophages. We have also detected low MMP-9 levels in MCP-1 null FBGC in vivo, but these cells are smaller and infrequent, which makes it difficult to compare with WT cells (not shown). It is tempting to assume that the fusion defect in MCP-1 null macrophages is solely a result of reduced MMP-9 levels. However, differences in the in vitro and in vivo fusion phenotypes between MCP-1 and MMP-9 null cells suggest that the defects are not identical. Specifically, MCP-1 FBGCs do not display irregular morphology in vivo. Furthermore, despite not being completely void of MMP-9, MCP-1 null macrophages display a greater reduction in percent fusion and number of nuclei per FBGC [16]. Thus, we speculate that MCP-1 null cells have additional defects. To our knowledge, there are no studies providing evidence that MCP-1 is involved in the regulation of MMP-9 expression in macrophages. However, a study involving bleomycin-induced pulmonary fibrosis in mice that lack the receptor for MCP-1, CCR2, showed reduced MMP-2 and MMP-9 expression in macrophages. These findings prompted the authors to speculate that the MCP-1/CCR2 pathway might enhance the expression of MMPs [43].
MMP-9 null FBGCs display abnormal morphology in vivo, consistent with our in vitro findings. Because of the functional complexity of MMP-9, which includes its ability to cleave ECM components, activate or deactivate growth factors, and act as a sheddase by releasing molecules from the cell surface, it is difficult to associate a specific function to the fusion defect. Interestingly, in an in vitro model of granuloma-associated macrophage fusion, it was shown that the process requires a disintegrin and metalloprotease domain 9 (ADAM9) [44]. Specifically, it was shown that the expression of this transmembrane protein with metalloproteinase activity was induced during mycobacterial lipomannan-induced fusion, and an ADAM9 function-blocking antibody inhibited fusion. Consistent with this observation, ADAM9 expression was induced during RANK-L and anti-fusion regulatory protein 1/CD98-induced macrophage fusion [45]. As the role of ADAM9 in fusion has not been elucidated, it is intriguing to speculate that it might involve its sheddase activity. Similarly, MMP-9 could perform a similar function in FBGC formation. It should be noted that proteins such as plasma fibronectin, vitronectin, and osteopontin have been linked to FBGC formation, and these observations implicate macrophage-ECM interactions as possible regulators of fusion [17, 46, 47]. Thus, multiple, nonexclusive mechanisms that can be affected by MMP-9 may act in parallel.
MMP-9 levels remained high in macrophage culture medium following fusion, as well as in FBGC in vivo, suggesting that MMP-9 has a role post-fusion. Similarly, MMP-9 is expressed highly in osteoclasts, which were formed by RANK-L-treated macrophages and are related to FBGC [37]. Consistent with the present findings, only pro-MMP-9 was stimulated by RANK-L-induced fusion, possibly as a result of activity of a small cell surface-localized pool of MMPs not detectable from the whole media sample. Further, a recent study revealed that the formation of FBGC and osteoclasts depends on the activation of DC-STAMP, suggesting a significant overlap between the two cell types [6]. MMP-9 has been shown to contribute to the ability of osteoclasts to degrade bone, and its overexpression has been linked to various bone pathologies [48, 49]. Thus, the expression of MMP-9 in FBGC might be part of their enzymatic repertoire in their effort to combat pathogens and biomaterials. Interestingly, the fusion process of macrophages has been shown to be similar to that of myoblasts [50]. It is unknown at this time if MMP-9 is playing a role in the fusion of the latter.
Our studies revealed additional defects in the FBR of MMP-9 null mice, including irregular encapsulation of Millipore filters and decreased fibrovascular invasion in PVA sponges. To our knowledge, MMP-9 is the first molecule that can affect each of the three major aspects of the FBR, specifically encapsulation, angiogenesis, and FBGC. Furthermore, MMP-9 is the first secreted extracellular enzyme shown to participate in macrophage fusion. Alteration of the FBR in MMP-9 null mice is not surprising, as MMP-9 can modulate ECM components, growth factors, cytokines, and chemokines. For example, alterations in the processing of the ECM might change the extracellular milieu and cause the observed alterations in encapsulation and angiogenesis. Consistent with this hypothesis, a study involving the use of GM6001 in wound healing and PVA sponge granuloma formation has shown changes in collagen expression and assembly [51].
MMP-9 has also been shown to play a modulating role in angiogenesis by its ability to release soluble vascular endothelial growth factor sequestered in the ECM [52, 53]. Here, we report decreased blood vessel density in MMP-9 null mice in a PVA sponge model but not in a Millipore filter implant model. We believe that this disparity is a result of the different angiogenic responses elicited in the two models and the duration of the experiment [11]. Specifically, the sponge granuloma model involves a robust, angiogenic response that can be observed in the first 2–3 weeks following implantation, followed by a more classic FBR. In contrast, Millipore filters are a solid implant model and elicit a classic FBR implantation model, typically characterized by a modest angiogenic response that usually regresses by 3–4 weeks. As MMP-9 can be considered a proangiogenic molecule, it is not surprising that we can detect a decrease in blood vessel density in MMP-9 null mice only in the PVA model. In fact, by using this model, we have shown previously an association between high levels of MMP-9 expression and increased angiogenesis [19].
Collectively, our observations identify MMP-9 as a key modulator of several processes in the FBR, including a newly discovered role in macrophage fusion. Our in vitro and in vivo studies suggest that MMP-9 is required for the fusion process and plays a role in FBGC function. Consistent with roles of MMP-9 in ECM remodeling and angiogenesis established previously, this study demonstrates abnormalities in collagenous encapsulation, ECM deposition, and blood vessel formation in MMP-9 null mice. Ongoing studies are focused on the development of strategies for MMP-9 blockade and on the identification of intracellular pathways that are modulated by the activity of MMP-9.
Acknowledgments
This work was supported by National Institutes of Health grants GM 072194-01 (to T. R. K.), AR45418 (to P. B.), and HL47328 (to R. M. S.) and the Alan A. and Edith L. Wolff Charitable Trust/Barnes-Jewish Hospital Foundation (to R. M. S.). We thank Dr. Joseph Madri for helpful discussions and Jianmin Zeng, Michael Michaud, and Farah Laiwalla for technical assistance.
References
- Sapir A, Avinoam O, Podbilewicz B, Chernomordik L V. Viral and developmental cell fusion mechanisms: conservation and divergence. Dev Cell. 2008;14:11–21. doi: 10.1016/j.devcel.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren-Suissa M, Podbilewicz B. Cell fusion during development. Trends Cell Biol. 2007;17:537–546. doi: 10.1016/j.tcb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Chen E H, Olson E N. Unveiling the mechanisms of cell-cell fusion. Science. 2005;308:369–373. doi: 10.1126/science.1104799. [DOI] [PubMed] [Google Scholar]
- Vignery A. Macrophage fusion: the making of osteoclasts and giant cells. J Exp Med. 2005;202:337–340. doi: 10.1084/jem.20051123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J M, Rodriguez A, Chang D T. Foreign body reaction to biomaterials. Semin Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, Morita K, Ninomiya K, Suzuki T, Miyamoto K, Oike Y, Takeya M, Toyama Y, Suda T. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345–351. doi: 10.1084/jem.20050645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay S M, Skokos E, Laiwalla F, Krady M M, Kyriakides T R. Foreign body giant cell formation is preceded by lamellipodia formation and can be attenuated by inhibition of Rac1 activation. Am J Pathol. 2007;171:632–640. doi: 10.2353/ajpath.2007.061213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno J L, Mikhailenko I, Tondravi M M, Keegan A D. IL-4 promotes the formation of multinucleated giant cells from macrophage precursors by a STAT6-dependent, homotypic mechanism: contribution of E-cadherin. J Leukoc Biol. 2007;82:1542–1553. doi: 10.1189/jlb.0107058. [DOI] [PubMed] [Google Scholar]
- Helming L, Tomasello E, Kyriakides T R, Martinez F O, Takai T, Gordon S, Vivier E. Essential role of DAP12 signaling in macrophage programming into a fusion-competent state. Sci Signal. 2008;1:ra11. doi: 10.1126/scisignal.1159665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming L, Gordon S. The molecular basis of macrophage fusion. Immunobiology. 2007;212:785–793. doi: 10.1016/j.imbio.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Kyriakides T R, Bornstein P. Matricellular proteins as modulators of wound healing and the foreign body response. Thromb Haemost. 2003;90:986–992. doi: 10.1160/TH03-06-0399. [DOI] [PubMed] [Google Scholar]
- Luttikhuizen D T, Harmsen M C, Van Luyn M J. Cellular and molecular dynamics in the foreign body reaction. Tissue Eng. 2006;12:1955–1970. doi: 10.1089/ten.2006.12.1955. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Topham N, Anderson J M, Hiltner A, Lodoen G, Payet C R. Foreign-body giant cells and polyurethane biostability: in vivo correlation of cell-adhesion and surface cracking. J Biomed Mater Res. 1991;25:177–183. doi: 10.1002/jbm.820250205. [DOI] [PubMed] [Google Scholar]
- Ratner B D. Reducing capsular thickness and enhancing angiogenesis around implant drug release systems. J Control Release. 2002;78:211–218. doi: 10.1016/s0168-3659(01)00502-8. [DOI] [PubMed] [Google Scholar]
- Helming L, Gordon S. Macrophage fusion induced by IL-4 alternative activation is a multistage process involving multiple target molecules. Eur J Immunol. 2007;37:33–42. doi: 10.1002/eji.200636788. [DOI] [PubMed] [Google Scholar]
- Kyriakides T R, Foster M J, Keeney G E, Tsai A, Giachelli C M, Clark-Lewis I, Rollins B J, Bornstein P. The CC chemokine ligand, CCL2/MCP1, participates in macrophage fusion and foreign body giant cell formation. Am J Pathol. 2004;165:2157–2166. doi: 10.1016/S0002-9440(10)63265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keselowsky B G, Bridges A W, Burns K L, Tate C C, Babensee J E, LaPlaca M C, Garcia A J. Role of plasma fibronectin in the foreign body response to biomaterials. Biomaterials. 2007;28:3626–3631. doi: 10.1016/j.biomaterials.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakides T R, Leach K J, Hoffman A S, Ratner B D, Bornstein P. Mice that lack the angiogenesis inhibitor, thrombospondin 2, mount an altered foreign body reaction characterized by increased vascularity. Proc Natl Acad Sci USA. 1999;96:4449–4454. doi: 10.1073/pnas.96.8.4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakides T R, Zhu Y H, Yang Z, Huynh G, Bornstein P. Altered extracellular matrix remodeling and angiogenesis in sponge granulomas of thrombospondin 2-null mice. Am J Pathol. 2001;159:1255–1262. doi: 10.1016/S0002-9440(10)62512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Kyriakides T R, Bornstein P. Matricellular proteins as modulators of cell-matrix interactions: adhesive defect in thrombospondin 2-null fibroblasts is a consequence of increased levels of matrix metalloproteinase-2. Mol Biol Cell. 2000;11:3353–3364. doi: 10.1091/mbc.11.10.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroen B, Heymans S, Sharma U, Blankesteijn W M, Pokharel S, Cleutjens J P, Porter J G, Evelo C T, Duisters R, van Leeuwen R E, Janssen B J, Debets J J, Smits J F, Daemen M J, Crijns H J, Bornstein P, Pinto Y M. Thrombospondin-2 is essential for myocardial matrix integrity: increased expression identifies failure-prone cardiac hypertrophy. Circ Res. 2004;95:515–522. doi: 10.1161/01.RES.0000141019.20332.3e. [DOI] [PubMed] [Google Scholar]
- Hahn-Dantona E, Ruiz J F, Bornstein P, Strickland D K. The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9) by mediating its cellular catabolism. J Biol Chem. 2001;276:15498–15503. doi: 10.1074/jbc.M100121200. [DOI] [PubMed] [Google Scholar]
- Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- Ra H J, Parks W C. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26:587–596. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott J D, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–564. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks W C, Wilson C L, Lopez-Boado Y S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- Niyibizi C, Chan R, Wu J J, Eyre D. A 92 kDa gelatinase (MMP-9) cleavage site in native type V collagen. Biochem Biophys Res Commun. 1994;202:328–333. doi: 10.1006/bbrc.1994.1931. [DOI] [PubMed] [Google Scholar]
- Schonbeck U, Mach F, Libby P. Generation of biologically active IL-1 β by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 β processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- Patterson B C, Sang Q A. Angiostatin-converting enzyme activities of human matrilysin (MMP-7) and gelatinase B/type IV collagenase (MMP-9) J Biol Chem. 1997;272:28823–28825. doi: 10.1074/jbc.272.46.28823. [DOI] [PubMed] [Google Scholar]
- Van den Steen P E, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-α and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–2681. [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- McQuibban G A, Butler G S, Gong J H, Bendall L, Power C, Clark-Lewis I, Overall C M. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276:43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- Kyriakides T R, Hartzel T, Huynh G, Bornstein P. Regulation of angiogenesis and matrix remodeling by localized, matrix-mediated antisense gene delivery. Mol Ther. 2001;3:842–849. doi: 10.1006/mthe.2001.0336. [DOI] [PubMed] [Google Scholar]
- Vu T H, Shipley J M, Bergers G, Berger J E, Helms J A, Hanahan D, Shapiro S D, Senior R M, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher D W, Hoyhtya M, Ottosen S K, Liang C M, Sarkar F H, Crissman J D, Fridman R. Enhanced expression of tissue inhibitor of metalloproteinase-2 (TIMP-2) in the stroma of breast carcinomas correlates with tumor recurrence. Int J Cancer. 1994;59:339–344. doi: 10.1002/ijc.2910590308. [DOI] [PubMed] [Google Scholar]
- Kyriakides T R, Tam J W, Bornstein P. Accelerated wound healing in mice with a disruption of the thrombospondin 2 gene. J Invest Dermatol. 1999;113:782–787. doi: 10.1046/j.1523-1747.1999.00755.x. [DOI] [PubMed] [Google Scholar]
- Sundaram K, Nishimura R, Senn J, Youssef R F, London S D, Reddy S V. RANK ligand signaling modulates the matrix metalloproteinase-9 gene expression during osteoclast differentiation. Exp Cell Res. 2007;313:168–178. doi: 10.1016/j.yexcr.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Jones J A, McNally A K, Chang D T, Qin L A, Meyerson H, Colton E, Kwon I L, Matsuda T, Anderson J M. Matrix metalloproteinases and their inhibitors in the foreign body reaction on biomaterials. J Biomed Mater Res A. 2008;84:158–166. doi: 10.1002/jbm.a.31220. [DOI] [PubMed] [Google Scholar]
- Antczak C, Radu C, Djaballah H. A profiling platform for the identification of selective metalloprotease inhibitors. J Biomol Screen. 2008;13:285–294. doi: 10.1177/1087057108315877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabot V, Reverdiau P, Iochmann S, Rico A, Senecal D, Goupille C, Sizaret P Y, Sensebe L. CCL5-enhanced human immature dendritic cell migration through the basement membrane in vitro depends on matrix metalloproteinase-9. J Leukoc Biol. 2006;79:767–778. doi: 10.1189/jlb.0804464. [DOI] [PubMed] [Google Scholar]
- Rahat M A, Marom B, Bitterman H, Weiss-Cerem L, Kinarty A, Lahat N. Hypoxia reduces the output of matrix metalloproteinase-9 (MMP-9) in monocytes by inhibiting its secretion and elevating membranal association. J Leukoc Biol. 2006;79:706–718. doi: 10.1189/jlb.0605302. [DOI] [PubMed] [Google Scholar]
- McNally A K, Anderson J M. β1 and β2 integrins mediate adhesion during macrophage fusion and multinucleated foreign body giant cell formation. Am J Pathol. 2002;160:621–630. doi: 10.1016/s0002-9440(10)64882-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuma T, Terasaki Y, Kaikita K, Kobayashi H, Kuziel W A, Kawasuji M, Takeya M. C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J Pathol. 2004;204:594–604. doi: 10.1002/path.1667. [DOI] [PubMed] [Google Scholar]
- Puissegur M P, Lay G, Gilleron M, Botella L, Nigou J, Marrakchi H, Mari B, Duteyrat J L, Guerardel Y, Kremer L, Barbry P, Puzo G, Altare F. Mycobacterial lipomannan induces granuloma macrophage fusion via a TLR2-dependent, ADAM9- and β1 integrin-mediated pathway. J Immunol. 2007;178:3161–3169. doi: 10.4049/jimmunol.178.5.3161. [DOI] [PubMed] [Google Scholar]
- Namba K, Nishio M, Mori K, Miyamoto N, Tsurudome M, Ito M, Kawano M, Uchida A, Ito Y. Involvement of ADAM9 in multinucleated giant cell formation of blood monocytes. Cell Immunol. 2001;213:104–113. doi: 10.1006/cimm.2001.1873. [DOI] [PubMed] [Google Scholar]
- McNally A K, Jones J A, Macewan S R, Colton E, Anderson J M. Vitronectin is a critical protein adhesion substrate for IL-4-induced foreign body giant cell formation. J Biomed Mater Res A. 2008;86:535–543. doi: 10.1002/jbm.a.31658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A T, Rice J, Scatena M, Liaw L, Ratner B D, Giachelli C M. The role of osteopontin in foreign body giant cell formation. Biomaterials. 2005;26:5835–5843. doi: 10.1016/j.biomaterials.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Wucherpfennig A L, Li Y P, Stetler-Stevenson W G, Rosenberg A E, Stashenko P. Expression of 92 kD type IV collagenase/gelatinase B in human osteoclasts. J Bone Miner Res. 1994;9:549–556. doi: 10.1002/jbmr.5650090415. [DOI] [PubMed] [Google Scholar]
- Kusano K, Miyaura C, Inada M, Tamura T, Ito A, Nagase H, Kamoi K, Suda T. Regulation of matrix metalloproteinases (MMP-2, -3, -9, and -13) by interleukin-1 and interleukin-6 in mouse calvaria: association of MMP induction with bone resorption. Endocrinology. 1998;139:1338–1345. doi: 10.1210/endo.139.3.5818. [DOI] [PubMed] [Google Scholar]
- Pajcini K V, Pomerantz J H, Alkan O, Doyonnas R, Blau H M. Myoblasts and macrophages share molecular components that contribute to cell-cell fusion. J Cell Biol. 2008;180:1005–1019. doi: 10.1083/jcb.200707191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte M B, Thornton F J, Kiyama T, Efron D T, Schulz G S, Moldawer L L, Barbul A. Metalloproteinase inhibitors and wound healing: a novel enhancer of wound strength. Surgery. 1998;124:464–470. [PubMed] [Google Scholar]
- Belotti D, Paganoni P, Manenti L, Garofalo A, Marchini S, Taraboletti G, Giavazzi R. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: implications for ascites formation. Cancer Res. 2003;63:5224–5229. [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu T H, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]