

Abstract
Learning and memory are two of the fundamental cognitive functions that confer us the ability to accumulate knowledge from our experiences. Although we use these two mental skills continuously, understanding the molecular basis of learning and memory is very challenging. Methylation modification of DNA is an epigenetic mechanism that plays important roles in regulating gene expression, which is one of the key processes underlying the functions of cells including neurons. Interestingly, a genome-wide decline in DNA methylation occurs in the brain during normal aging, which coincides with a functional decline in learning and memory with age. It has been speculated that DNA methylation in neurons might be involved in memory coding. However, direct evidence supporting the role of DNA methylation in memory formation is still under investigation. This particular function of DNA methylation has not drawn wide attention despite several important studies that have provided supportive evidence for the epigenetic control of memory formation. To facilitate further exploration of the epigenetic basis of memory function, we will review existing studies on DNA methylation that are related to the development and function of the nervous system. We will focus on studies illustrating how DNA methylation regulates neural activities and memory formation via the control of gene expression in neurons, and relate these studies to various age-related neurological disorders that affect cognitive functions.
Keywords: DNA methylation, Epigenetics, Cognition, Nutrition, Neurodegenerative disease, Aging
1. Introduction
Learning and memory are two cognitive functions through which we acquire new knowledge and store the learned information over time. There are both gains and losses in cognitive functions with increased age. Cognitive gains mainly refer to the accumulation of knowledge and information, whereas cognitive losses are usually exemplified by the reduced efficiency or impairment of cognitive activities in the elderly. Considering the prevalence and socioeconomic burden of several major age-related neurological disorders such as Alzheimer’s disease, the age-dependent loss in cognitive functions has received an increasing amount of attention for investigations into the underlying causes. Among the fundamental questions to be addressed, understanding the molecular basis of learning and memory formation remains to be a challenging area of study, although such knowledge will be imperative for the ultimate understanding of how the aging process influences cognitive functions.
Long-term potentiation (LTP) was first described as a form of synaptic transmission in the hippocampus (Bliss and Lomo, 1973). It is now widely accepted that LTP is an enduring form of synaptic plasticity underlying learning and memory. The N-methyl-d-aspartate (NMDA) receptor-activated LTP is one of the best-understood forms of LTP. An antagonist of the NMDA receptor, (6)-2-amino-5-phosphonovaleric acid, can block amygdaloid LTP induction and thus impair the expression of conditional fear (Kim et al., 1993; Lee and Kim, 1998), which further supports the view that LTP mediates learning and memory. LTP can be divided into two phases: an early, protein synthesis-independent phase (E-LTP) that lasts for a few hours; and a late, protein synthesis-dependent phase (L-LTP) that lasts from days to months (Huang et al., 2004; Nguyen and Kandel, 1997). L-LTP is believed to result from pronounced strengthening of the postsynaptic response largely through the synthesis of new proteins. These proteins include glutamate receptors, transcription factors, and structural proteins that enhance existing synapses and form new connections. However, more experimental evidence is needed to support this view. Based on the length of retention in the brain, memory can be categorized into short-term memory (STM) and long-term memory (LTM). STM is a limited-capacity memory system that holds information in awareness for a brief period of time. STM is often conceptualized as ‘working memory’ that processes information for transfer to LTM. Formation of LTM often requires a repetitive or intensified process of neural activity that is referred to as the ‘consolidation’ process. Existing evidence suggests that STM formation does not require protein synthesis but the modifications of existing proteins, whereas LTM formation depends on new RNA or protein synthesis (Huang et al., 1994; Nguyen et al., 1994). The medial temporal lobes of the brain are consistently identified to be involved in the creation of LTM (Yonelinas et al., 2002).
The function and behavior of neurons, like any other types of cells, are ultimately determined by the genes expressed in them. Synaptic connectivity among neurons serves as the physical basis for memory formation, which often entails gene products (mRNA or protein) from a vast number of neural activity-related genes. The molecular basis of synapse-dependent LTM formation can thus be understood by studying the regulatory mechanisms of gene expression in the neural network. Indeed, both transcription and translation have been implicated as important mechanisms underlying the formation of LTM that involves many signaling path-ways and the regulation of numerous genes (Roberson and Sweatt, 1999; Squire et al., 1975). A recent bioinformatics study shows that consolidation of long-term contextual memory in the hippocampus triggers altered expression of numerous genes encompassing many aspects of neuronal function (Levenson et al., 2004a). Classical studies of gene regulation focus on identifying regulatory DNA elements and their interacting transcription factors. In the past two decades, epigenetic modifications of genes and their chromatin environment due to DNA methylation have been recognized as a major player in regulating gene activities. This also highlights the importance of the epigenetic hypothesis that proposes the pattern of DNA methylation in neurons being an integral part in memory coding (Holliday, 1999, 2004).
Epigenetic mechanisms play important roles in normal development. DNA methylation and histone modifications are two of the most extensively studied epigenetic mechanisms. They regulate gene expression via reversible and dynamic chromatin remodeling processes. The importance of epigenetics, however, was not recognized until the recent two decades following the observation that abnormal DNA methylation events are associated with cancer (Feinberg and Vogelstein, 1983). Since then, cancer epigenetics has drawn extensive attention with a focus on DNA methylation studies (Jones and Laird, 1999), which subsequently leads to the study of another major epigenetic regulatory mechanism via posttranslational modifications of histone proteins (Landsberger and Wolffe, 1997). Initial studies of chromatin remodeling were focused on histone acetylation (Brehm et al., 1998; Magnaghi-Jaulin et al., 1998), a reversible biochemical process that confers either open or condensed chromatin conformations to alter gene expressions. Several studies have shown that DNA methylation and histone acetylation can regulate gene expression synergistically through protein mediators such as the methyl-CpG binding protein MeCP2 (Jones et al., 1998; Nan et al., 1997). Recent advances in the field of epigenetics have uncovered other histone-based epigenetic codes such as methylation, sumoylation, ubiquitination, ADP-ribosylation, and biotinylation (Hassan and Zempleni, 2006; Nicholson et al., 2004; Peterson and Laniel, 2004). Although DNA methylation modification of the genome occurs primarily on cytosines located in CpG dinucleotides, posttranslational modifications of histone proteins are much more complex and affect multiple residues (Arg, Lys, Pro, Ser) at over 30 sites within the N-terminal tails of histones (Iizuka and Smith, 2003; Jenuwein and Allis, 2001). Histone methylation alone can appear in the form of mono-, di-, and tri-methylation (Cheung and Lau, 2005). Even more complex, these different forms of methylation can occur on different amino acid residues that are located at different positions (e.g., H3 Lys 4, 9, 27, 36, 79; H3 Arg 2, 17, 16) (Nicholson et al., 2004).
Epigenetic regulation of memory function through histone acetylation is well-supported by studies on Rubinstein-Taybi syndrome (RTS) patients who suffer from mental retardation and defects in learning and memory. RTS patients are associated with heterozygous mutations in the gene encoding the cAMP-responsive element binding protein [CRBP]-binding protein (CBP). CBP is a transcriptional co-activator containing an endogenous histone acetyltransferase activity that catalyses histone acetylation (Chan and La Thangue, 2001). CBP-mutant mice display defects in long-term memory formation, which indicates that decreased levels of histone acetylation can impair memory function (Alarcon et al., 2004). Consistent with these observations, elevated levels of histone acetylation have been shown to enhance long-term potentiation in the hippocampus in vitro and improve LTM formation in rats as measured by contextual fear conditioning studies (Levenson et al., 2004b). Other studies on the control of LTM formation via histone modification have been discussed in detail in a previous review (Levenson and Sweatt, 2005). Although such complex modes of histone modifications confer enormous functional potentials to the genome, they also present a huge challenge to crack the so-called “histone codes” underlying memory function. Given the relative simplicity of the DNA methylation system, we will review existing evidence that supports the function of DNA methylation in LTM formation and explore what other studies can be done to further advance this area of research.
2. DNA methylation in aging and CNS development
DNA methylation is catalyzed by DNA methyltransferases (Dnmts), which add a methyl moiety to the cytosine located within CpG dinucleotides. The establishment of normal DNA methylation is essential for development, and DNA methylation abnormalities are frequently associated with tumorigenesis and cell aging (Jones and Laird, 1999; Liu et al., 2003). Several Dnmts have been identified in vertebrates, including Dnmt1, 3a and 3b, which are encoded by independent genes (Hendrich and Bird, 2000). Dnmt1 is the most abundant enzyme that preferentially methylates hemimethylated DNA and is responsible for stabilizing methylation patterns established early in development. Homozygous mutation of Dnmt1 in mice is embryonic lethal, which strongly supports that DNA methylation is crucial to normal development (Li et al., 1992). Dnmt3a and 3b appear to be the main players for creating methylation patterns de novo during development. Although Dnmt3b seems to be expressed in a limited time window during early CNS development, Dnmt3a expression is detected both in neuronal precursor cells during embryogenesis and in postmitotic neurons during postnatal development (Feng et al., 2005). It has been proposed that memory function may be tightly linked with the methylation of DNA in neurons (Holliday, 1999, 2004). However, the exact function of DNA methylation in learning and memory remains elusive despite that multiple studies on neurological disorders such as Rett syndrome suggest that abnormal methylation events are linked with neurological disorders and defective memory function.
As in all other parts of the body, the DNA in brain cells is subject to dynamic methylation modifications. Maintenance of genetic and epigenetic integrity is expected to be critically important for cells like neurons that have to function for the lifetime. DNA methylation levels in the brain undergo dynamic changes perinatally (Tawa et al., 1990). The level of DNA methylation is higher in adult brain than in other tissues (Tawa et al., 1990; Wilson et al., 1987). CNS neurons appear to express an extremely high level of Dnmt1, which may be necessary to keep their DNA precisely methylated to fulfill the many specific and precise functions of the brain (Brooks et al., 1996; Inano et al., 2000; Veldic et al., 2004). Our preliminary studies indicate that Dnmt1 levels vary significantly among different regions of the brain (Liu et al., unpublished observations). Because the maintenance methylation activity of Dnmt1 is usually associated with DNA replications in dividing cells, the biological function of a high level of Dnmt1 activity in postmitotic neurons remains unclear. Dnmt1 is proposed to function to remethylate cytosine residues that have undergone DNA mismatch repair in neurons following spontaneous deamination of methyl cytosine to thymine residues (Brooks et al., 1996). However, experimental support to this interesting idea still lacks. Conditional deletion of Dnmt1 in neuronal precursors leads to highly demethylated neurons and glial cells that are rapidly eliminated postnatally, but this deletion does not affect the survival of postmitotic neurons (Fan et al., 2001), suggesting that DNA methylation is crucial in regulating neurogenesis. A recent study has linked the underlying mechanisms with the epigenetic control of the JAK-STAT signaling pathway during brain development (Fan et al., 2005).
A long-standing dogma pertaining to mammalian adult neurogenesis has now been resolved with conclusive experimental support showing that adult neurogenesis exists in both rodents and humans (Cameron and McKay, 1999; Eriksson et al., 1998). Although lower vertebrates such as lizards can regenerate the entire brain, mammalian adult neurogenesis seems to be restricted mainly in two brain regions: the sub-granular layer of the dentate gyrus of the hippocampus and the subventricular zone (SVZ) of the lateral ventricles (Gage, 2000; Lois and Alvarez-Buylla, 1994; Luskin, 1993). It is estimated that up to 50 percent of neurons die after they form synaptic connections with their target cells due to the failure to obtain adequate amounts of neurotrophic factors that are required for the neurons to survive (Raff et al., 1993). Adult neurogenesis may thus function to produce new neurons to compensate for this constant neuronal death in the brain. It is intriguing that neurogenesis occurs in the hippocampus, a primary area involved in learning and memory formation. Does this imply that a continuous learning process would entail an activity-dependent neurogenesis in the adult brain? Since adult neurogenic activity declines with age (Kuhn et al., 1996) and coincides with the frequently observed age-related global DNA hypomethylation (Wilson et al., 1987), loss of DNA methylation in the aging brain may reduce adult neurogenesis and lead to age-related declines in learning and memory performance.
Neural stem cells (NSCs) possess the ability to self-renew and maintain the ability to generate other CNS cell types including neurons, astrocytes and oligodendrocytes. NSCs were first isolated from the SVZ in the adult brain, which displays a high neurogenic activity (Reynolds and Weiss, 1992). NSCs are proposed to go through limited cycles of expansion through symmetric divisions before undergoing asymmetric divisions to initiate neurogenesis (Gage, 2000). Nestin is a stem cell marker originally described as an intermediate filament protein expressed in neuroepithelial stem cells. It is sharply down-regulated at the transition from proliferating stem cells to postmitotic neurons. The nestin gene contains two tissue-specific transcription elements: a cis-element in the first intron drives nestin expression in somatic muscle precursors, while an enhancer in the second intron directs expression to CNS precursor cells (Zimmerman et al., 1994). Sequence analysis of the nestin gene locus reveals a high CpG content in its promoter and around the first two introns and exons. The presence of such a CpG island indicates that its gene expression may well be subjected to DNA methylation control, although no studies have explored this possibility. The progressive decline of precursor cell proliferation during aging leads to decreased neurogenic activity. Future studies need to determine whether an age-related decrease in neurogenesis is the primary cause of age-related memory impairment.
3. DNA methylation control of neuronal gene expression
As aforementioned, Dnmt1 is highly expressed in neurons. Given the postmitotic status of neurons, it has been debated whether DNA methylation acts as a dynamic process in regulating gene activities in neurons. However, several recent studies on gene-specific methylation studies have demonstrated that neural gene promoters indeed undergo dynamic methylation changes in postmitotic neurons. This observation is best illustrated by studies on the gene encoding the brain-derived neurotrophic factor (BDNF), which is involved in neural plasticity including learning and memory (West et al., 2001). In addition to its role in neuronal survival, BDNF also modulates synaptic activity and is one of the best-studied activity-induced genes in response to neuronal activity (Poo, 2001). BDNF is categorized as an immediate-early gene, meaning that stimulus-induced transcriptional initiation of new BDNF mRNA occurs rapidly without the need for new protein synthesis (Lauterborn et al., 1996). BDNF is thought to be essential for converting transient stimuli into long-term changes in brain activity (Jones et al., 1994; Schwartz et al., 1997). The BDNF gene contains four well-characterized promoters that give rise to at least eight different mRNAs. The methylation status of BDNF promoters in neurons is shown to undergo dynamic changes in response to activity changes. A lower level of methylation generally correlates with a higher level of BDNF gene transcription, but this correlation also depends on the binding of the transcription repressor MeCP2 protein. MeCP2 binding to the methylated rat BDNF promoter III can repress its expression in resting neurons (Chen et al., 2003). Upon exposure to potassium chloride, which causes membrane depolarization and calcium influx, BDNF expression increases (Chen et al., 2003). This neural activity-dependent calcium influx is suggested to increase the phosphorylation of MeCP2 and thus prevent it from binding to the methylated BDNF promoter. In addition to MeCP2 phosphorylation, membrane depolarization can also cause BDNF promoter demethylation (Martinowich et al., 2003), which disassociates the MeCP2 transcription repressor complex from the BDNF promoter to allow its expression. Consistent with this observation, the amount of BDNF mRNA in Dnmt1 mutant mouse brain appears to be much higher than that in littermate controls (Martinowich et al., 2003). Together, these studies suggest that methylation plays a dynamic role in neurons by reversibly modulating key gene activities in response to external stimulus (Fig. 1).
Fig. 1.
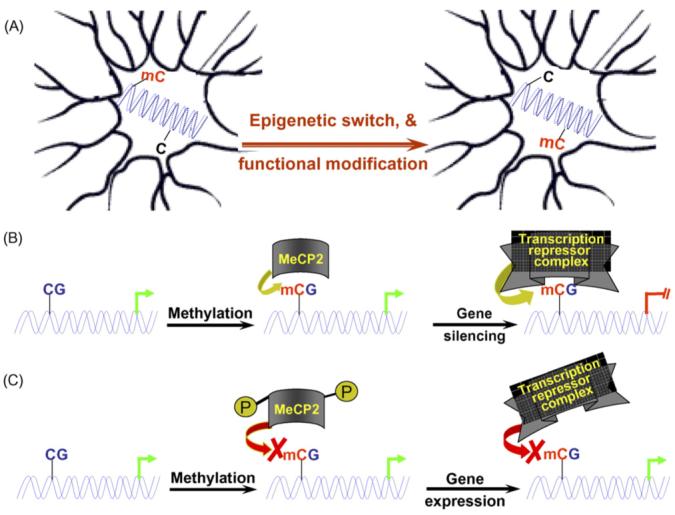
(A) A hypothetical model depicts that methylation changes of neuronal DNA in response to an external stimulus can alter the epigenotype of the neuron, which subsequently modulates the expression of a subset of genes encoding key neuronal proteins such as neurotransmitters/receptors. Such changes in neurotransmitter/receptor levels will in turn alter the neuron’s synaptic strength with its associated neurons, leading to a modified response upon exposure to the same stimulus later; (B and C) models illustrating methylation-dependent regulation of the activities of neuronal genes such as BDNF in neurons. In addition to the methylation status of the promoter per se, posttranslational modification of the MeCP2 protein alters how the MeCP2-associated transcription repressor complex reads the methylation codes to determine the expression status of key genes in response to external stimuli. C: Unmethylated cytosine; mC: methylated cytosine.
Reelin is an extracellular matrix protein that plays an important role in brain development. It is secreted by specific GABAergic interneurons from the telencephalon and hippocampus (Costa et al., 2002). Several studies have implicated that DNA methylation plays an important role in regulating reelin expression (Abdolmaleky et al., 2005; Dong et al., 2005; Grayson et al., 2005; Numachi et al., 2004). Reelin and glutamic acid decarboxylase (GAD67) are coexpressed in GABAergic neurons. Down-regulation of reelin and GAD67 is often detected in various cortical structures of postmortem brain from Schizophrenia patients (Costa et al., 2002; Impagnatiello et al., 1998). Comparative analysis of the reelin promoter methylation between postmortem brain DNA samples from male schizophrenic patients and that from normal controls reveals an inverse relationship between the level of DNA methylation and reelin gene expression (Abdolmaleky et al., 2005). Furthermore, reelin and GAD67 mRNAs are lower in GABAergic neurons with a high level of Dnmt1 activity (Veldic et al., 2004). Methionine treatment can reduce reelin and GAD67 expressions in both mouse brain and in vitro cultured neurons (Noh et al., 2005; Tremolizzo et al., 2002). Knockdown of Dnmt1 expression by antisense oligonucleotides, however, can diminish the methionine-induced down-regulation of reelin and GAD67 (Noh et al., 2005), implying that methylation is a key factor in regulating the expressions of reelin and GAD67. These findings collectively suggest that DNA methylation is actively involved in reversible gene expression control in postmitotic neurons.
Nicotinic acetylcholine receptors (nAChR) are important for memory performance and are implicated in a variety of disorders of the human central nervous system including Alzheimer’s disease, anxiety, autism, depression, epilepsy, Parkinson’s disease (Picciotto et al., 2000; Rusted et al., 2000; Woodruff-Pak, 2003). Neuronal nAChR subunit genes encompass a family of genes (Lukas, 1995). The major isoform of nAChR in the brain is made up of the α4 and β2 subunits and possesses a high affinity for nicotine. The sequence around the transcription initiation site and first exon of the α4 gene is very GC-rich, which predisposes this gene to DNA methylation modification. It has been shown that the GC-rich sequence of the α4 gene is methylated in a tissue-specific manner (Watanabe et al., 1998). More methylation is observed in non-neuronal tissues than in the brain, which may explain its neuronal-specific expression. Expression comparison between aged and young mice reveals that the expression of nAChR α4 and β2 decrease in key cholinergic regions during normal aging (Rogers et al., 1998; Tohgi et al., 1998), but it is unknown whether this is due to an age-dependent increase in α4 promoter methylation that causes its decreased expression. In addition to these nAChR subunit genes, the promoter of human mu-opioid receptor (MOR) gene is also under reversible methylation modification that alters MOR expression status in neurons (Andria and Simon, 1999). Many other neuron-specific genes appear to contain GC-rich sequences around their transcription initiation sites as well (Table 1). The methylation status of these GC-rich sequences has not been investigated in most cases. Whether promoter methylation of these neuronal-specific genes is a common mechanism underlying their neuron-specific expressions remains to be determined. Future methylation analysis of the promoters of these neuronal genes will shed light on gene regulatory mechanisms related to learning and memory that will help better understand the age-dependent decline in memory function in the elderly population.
Table 1.
Neural-specific genes that have CG-rich promoters with undetermined methylation status
| Gene | Protein function |
|---|---|
| Synapsin I | Nerve terminal-specific phosphoproteins involved in the regulation of neurotransmitter release |
| AChE | Acetyl cholinesterase |
| m4 muscarinic receptor | Membrane-bound acetylcholine receptors |
| NMDAR2C | N-Methyl-d-aspartate receptor subunit NR2C |
| NMDAR1 | N-Methyl-d-aspartate receptor R1 |
| Aldolase C | Fructose-1,6-bisphosphate aldolase (glycolytic isoenzyme) |
| MAO | Monoamine oxidase (metabolize catecholamines and xenobiotics) |
| NGF receptor | Nerve growth factor receptor |
| Kv1.4 | Voltage gated potassium channels |
| GAD67 | Glutamate decarboxylase 67 (GABA synthesis enzyme) |
| GRIK5 | Glutamate receptor ionotropic kainate-5 (kainate-preferring glutamate receptor subunit KA2) |
| GluR2 | AMPA receptor subunit |
| PGP9.5/UCHL1 | C-terminal ubiquitin hydrolase (disassembles polyubiquitin chains to increase the availability of free monomeric ubiquitin) |
| MOR | Mu-opioid receptor (opiate binding) |
| RYK | Orphan receptor containing a catalytically inactive tyrosine-kinase-related domain |
| Nestin | Intermediate filaments expressed in neural stem cells |
| NeuroD2 | Neurogenic differentiation factor and transcription factor |
Multiple studies have also examined gene regulation by DNA methylation during gliogenesis. The glial fibrillary acidic protein (GFAP) is an intermediate filament protein specific to the cytoskeleton of astrocytes in the CNS. The GFAP promoter contains a conserved cluster of CpG sites that are differently methylated among different tissues and cell types (Barresi et al., 1999; Condorelli et al., 1994; Teter et al., 1994). Teter et al. (1996) have shown that the GFAP promoter undergoes a dynamic demethyaltion/remethylation cycle during perinatal development. During early embryonic development, the GFAP promoter becomes demethylated to allow GFAP transcription. Increased promoter methylation follows at the GFAP promoter during postnatal development. Reporter gene analysis has confirmed that promoter methylation is inversely correlated with its activity (Teter et al., 1996), although a more comprehensive analysis revealed more complex functional relationship between promoter methylation and GFAP expression (Condorelli et al., 1994). Studies by Takizawa et al. (2001) suggest that the methylation status of a key CpG site in the GFAP promoter appears to interfere with the binding of the transcription activator STAT3. This provides a candidate mechanism underlying the regulation of lineage specification by DNA methylation in the CNS. Furthermore, a recent study by Fan et al. (2005) demonstrated that progeny cells of Dnmt1-/- neural precursors in culture were highly demethylated with increased demethylation of glial differentiation-related genes including Gfap, Stat1 and S100β. This demethylation process is proposed to play an important role for the switch from neurogenesis to gliogenesis during CNS development (Fan et al., 2005).
4. DNA methylation aberrancies in neurological disorders
Many types of human neurological disorders, including mental retardation (MR) syndromes and neurodegenerative diseases, display cognitive defects as reflected by impaired learning and memory abilities. Genetic studies in the past have uncovered many genetic mutations, deletions and translocations that can lead to mental retardation and learning disorders. Well-established genetic causes of neurological disorders include chromosomal anomalies and monogenic MR diseases. Like many other diseases with complex profiles, the underlying causes of most neural diseases are often heterogeneous and involve the interplays among different genes and environmental factors. Although genetic components underlying neurodegenerative diseases are supported by overwhelming evidence, epigenetic mechanisms are gaining new perspectives and are being explored in different forms of neurological diseases and psychiatric disorders (Tsankova et al., 2007). Cocaine addiction, for example, is related to the fact that cocaine exposure can alter chromatin conformations at specific neural gene promoters and lead to the binding of FosB, a transcription factor that facilitates the transition to addiction (Kelz et al., 1999; Kumar et al., 2005). Increased global DNA methylation is observed in alcoholic patients (Bonsch et al., 2004), which is consistent with the observation of promoter-specific hypermethylation of the α-synuclein gene, an important regulator of dopamine function and craving for alcohol (Bonsch et al., 2005). In a rat stress model study, it has been demonstrated that the level of maternal nurturing behavior in early life can affect promoter methylation and subsequent expression of the glucocorticoid receptor (GR) gene in a lasting but reversible manner (Weaver et al., 2005). Pups from a less nurturing mother display a low level of GR expression due to increased CpG methylation of the GR promoter in the hippocampus (Weaver et al., 2005). In addition, there is mounting evidence that aberrant DNA methylation events are associated with the pathogenesis of schizophrenia as discussed above. Taken together, these recent studies have demonstrated the functional significance of DNA methylation in neurological and psychiatric disorders. Below we will focus on Rett Syndrome and Alzheimer’s disease (AD) to exemplify how disrupted DNA methylation processes contribute to neurological disorders in detail. Rett Syndrome represents an excellent example of combined genetic and epigenetic neural disorder (Bienvenu and Chelly, 2006), whereas AD represents a polygenic disease with a complex profile that is often linked with various environmental factors (Zawia and Basha, 2005).
Rett syndrome is largely a genetic disorder that results from a faulty MeCP2 gene that is located on the X-chromosome. Approximately 70-90% of Rett patients are associated with mutations in MeCP2 (Amir et al., 1999; Shahbazian et al., 2002), a methyl CpG binding protein that mediates the gene-silencing effect by DNA methylation via recruiting transcription co-repressors to methylated gene regulatory elements (Bienvenu and Chelly, 2006). However, most Rett cases are sporadic and seldom inherited or passed from one generation to the next. Similar to Dnmt1, MeCP2 is widely expressed but is especially abundant in the brain (Nan et al., 1997). Its levels increase in cortical neurons throughout development (Shahbazian et al., 2002). During postnatal development, rat brain regions having more mature neurons exhibit the strongest MeCP2 expression; whereas late developing structures such as the cortex exhibit the most significant increases in MeCP2 expression (Mullaney et al., 2004). The degree of MeCP2 expression in the cortex and hippocampus appears to correlate with the synaptogenesis in both regions (Mullaney et al., 2004). Comparative analyses of the elementary properties of synaptic transmission between cultured hippocampal neurons from MeCP2 knockout and wild-type control mice confirm that the loss of MeCP2 decreases the frequency of spontaneous excitatory synaptic transmission in hippocampal neurons (Nelson et al., 2006). These studies suggest that MeCP2 is important in the maintenance or modulation of synaptic maturity and plasticity, which is likely mediated by its epigenetic control of target gene activity. Furthermore, conditional knockout of the MeCP2 gene in postnatal mouse forebrain is shown to be sufficient to produce behavioral abnormalities similar to RTT phenotypes (Gemelli et al., 2006). Conversely, transgene-based overexpression of MeCP2 appears to have neurotoxic effects including profound motor dysfunction, the development of seizures and hypoactivity that leads to early deaths (Collins et al., 2004; Luikenhuis et al., 2004). To explore the therapeutic potential of MeCP2 in curing Rett, a recent study employed an elegant transgenic approach that allows conditional reactivation of the silenced endogenous MeCP2 gene (Guy et al., 2007). The results suggest that a gradual MeCP2 reactivation can progressively reverse the Rett-related symptoms without causing significant neurotoxicity-related deaths. This study confirms that the functional impact due to the absence of MeCP2 in early CNS development can be largely recovered by restoring MeCP2 function later, opening new possibilities for future treatment of Rett-related neurological disorders.
Alzheimer’s disease (AD) is a progressive age-related neurodegenerative disorder, characterized by memory loss and confusion at the clinical level. Genetic causes of AD include genetic mutations of the presenilin and amyloid precursor protein (APP) genes (Lippa, 1999), which are thought to contribute largely to the familial form of the AD disease (FAD) that usually manifests around the 40 s in age. However, FAD accounts for only a small proportion of AD, and the majority of the AD cases are known to occur sporadically. The sporadic nature of most AD cases strongly argues for an environmental origin that drives AD pathogenesis. APP is a transmembrane protein. Its proteolytic processing generates a 39-42 residue peptide referred to as β-amyloid (Aβ). Aβ peptides are the primary constituents of amyloid deposits found in the aging brain, and the aggregation of Aβ into extraneuronal amyloid plaques is one of the pathological hallmarks of AD. Overexpression of the APP gene is found in brain regions from Down’s Syndrome and AD patients. APP over-expression in transgenic mice leads to amyloid deposition and cognition impairment at a late age (Hsiao et al., 1996; Rumble et al., 1989; West et al., 1995). Two GC-rich elements in the APP promoter are shown to regulate its transcriptional activity (Pollwein et al., 1992). Tissue-specific patterns of methylation have been observed in the upstream sequences of the APP promoter that is consistent with its variable levels of expression in different human tissues including the brain (Rogaev et al., 1994). Hypomethylation of the APP promoter occurs with age in the cerebral cortex of human brain (Tohgi et al., 1999), and is also observed in brain tissues from AD patients (West et al., 1995). APP mRNA expression is lower in female mouse brain than in male mouse brain, which is inversely correlated with the level of APP promoter methylation (Mani and Thakur, 2006). In addition to the regulation of APP expression by DNA methylation, the activity of other genes such as BACE (β-secretase) and Presenilin1 (PS1, γ-secretase) have also been implicated to be modulated by DNA methylation (Fuso et al., 2005). Since BACE and PS1 are involved in the production of Aβ from APP, methylation control of these gene activities adds additional complexity to the polygenic features of AD pathogenesis.
5. Nutritional impact on memory function via DNA methylation
The relation between nutrition and cognition is widely recognized (Gordon, 1997; Kretchmer et al., 1996; Lieberman, 2003; Scott et al., 2006). Dietary components that are known to affect brain development and cognition include alcohol consumption (Espeland et al., 2006), vitamins A (Misner et al., 2001), B6 and B12 (Haan et al., 2007; Miller, 2006), iron (Jorgenson et al., 2005), fatty acids (Laurin et al., 2003), folate (Craciunescu et al., 2004; Ramos et al., 2005) and choline (Mellott et al., 2007; Niculescu et al., 2006; Teather and Wurtman, 2005). Although the underlying molecular mechanisms remain to be determined, most of these nutrients are implicated to interact with biological DNA methylation (Fuso et al., 2005; Liu et al., 2003; Niculescu et al., 2006). We will mainly focus on dietary folate and choline because they are two of the most extensively investigated substrates that are linked with biological DNA methylation and the pathogenesis of various diseases.
Folate is an important substrate in one-carbon metabolism (Fig. 2). Folate provides the dietary source of methyl group for biological methylation. It is required for the conversion of homocysteine to methionine and the formation of S-adenosylmethionine (SAM). SAM participates in biological methylation reactions, which generates S-adenosylhomocysteine (SAH) that subsequently forms homocysteine. Folate-depletion can cause genomic DNA hypomethylation, which can be reversed upon dietary folate restoration (Davis and Uthus, 2003; Sibani et al., 2002). Folate depletion can also lead to cellular accumulation of SAH and dramatically increase blood homocysteine levels. SAH is a potent inhibitor of Dnmt activity through the product inhibition pathway and can lead to genome hypomethylation (Lee and Zhu, 2006). The disruption of homocysteine/folate metabolism can adversely affect both the developing and the adult brain. Serum folate concentrations are shown to be significantly lower in subjects with brain infarcts than in subjects without infarcts (Snowdon et al., 2000). In vitro, the disruption of one-carbon metabolism can repress the proliferation of cultured multipotent neuroepithelial progenitor cells and alter cell cycle distribution (Kruman et al., 2005). In vivo studies show that folic acid deficiency dramatically reduces the number of proliferating cells in the dentate gyrus of the hippocampus in adult mice (Kruman et al., 2005). Since neurogenesis in the adult hippocampus is possibly pivotal in learning and memory and in recovery from injury, these results suggest that dietary folate deficiency can affect neurogenesis via inhibiting the proliferation of neuronal progenitor cells in the adult brain.
Fig. 2.
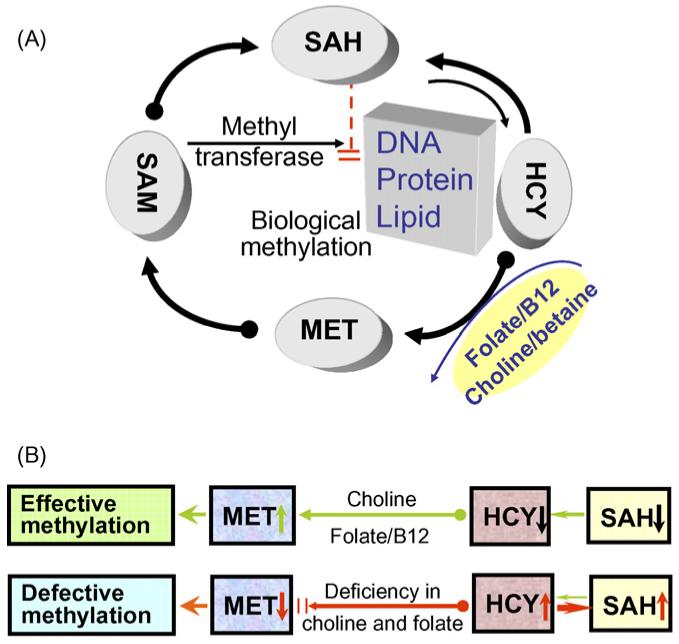
(A) A simplified map of one-carbon metabolism and biological DNA methylation. MET, methionine; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; HCY: homocysteine. (B) Expected effects of folate/B12 or choline deficiency include: (1) impairing the conversion of HCY into MET (and resulting in a low level of SAM); (2) increasing the level of HCY; (3) reversing the reaction dynamics between HCY and SAH that favors SAH synthesis. These effects will eventually lead to defective genomic methylation, whereas sufficient folate/B12 or choline supplies can maintain effective genomic methylation.
Choline and folate are interrelated metabolically. Choline is a substrate in methyl group metabolism that methylates homocysteine via its metabolite betaine to form methionine, a methyl group donor for most biological methylation reactions including DNA methylation (Zeisel, 2006; Zeisel and Blusztajn, 1994) (Fig. 2). Multiple studies in rodent models suggest that choline is critical for normal cognition development. Maternal dietary choline intake during late pregnancy can modulate mitosis and apoptosis in progenitor cells of the fetal hippocampus and septum (Albright et al., 1999; Craciunescu et al., 2003). Choline deficiency during the second half of pregnancy in rats leads to offspring with diminished memory function in adulthood (Meck and Williams, 1997), whereas long-term dietary choline supplementation can ameliorate hippocampal-dependent memory impairment caused by impoverished environmental conditions (Teather and Wurtman, 2005). A recent study reported that prenatal choline-deficiency decreases global DNA methylation in neuroepithelium of the fetal brain (Niculescu et al., 2006), which provides the first important evidence to support the epigenetic causes underlying these choline-dependent effects on cognitive functions. The same study further demonstrated that gene-specific promoter demethylation of the cell-cycle regulator gene Cdkn3 correlates with an increased Cdkn3 expression (Niculescu et al., 2006). The Pemt gene encodes the enzyme for de novo synthesis of choline in vivo. Deletion of the Pemt gene can paradoxically increase stem cell proliferation and DNA methylation in mouse embryonic hippocampus (Zhu et al., 2004). This may be attributed to an elevated level of SAM as observed in the Pemt-/- mice, which might compensate for choline deficiency in biological DNA methylation in the developing brain. These data suggest that dietary choline plays an important role in brain development and memory function by modulating biological methylation process.
For more than a decade, elevated plasma concentration of homocysteine (hyperhomocysteinemia) has been recognized as an independent risk factor for various vascular diseases and cognitive diseases (Lehmann et al., 1999; McCaddon et al., 1998; Refsum et al., 1998; Ulrey et al., 2005). Homocysteine can be remethylated to methionine to reenter the one-carbon metabolism cycle. Elevated plasma homocysteine is one of the primary consequences of folate deficiency. In recognition of the biological significance, folic acid fortification in grain products has been implemented in the US since 1996 to reduce the risk of neural-tube defects in newborns. This has proven to be successful in reducing both the prevalence of folate deficiency in the general population and the incidence of neural tube birth defects in newborns (Honein et al., 2001; Jacques et al., 1999). A recent study reports that low folate status is associated with poor cognitive function and dementia in a Latino population despite folic acid fortification (Ramos et al., 2005). A possible contributing factor of this discrepancy may be due to the age factor of the examined population because the elderly population in general has a high prevalence of vitamin B12 deficiency that renders folic acid fortification less effective. Furthermore, aging in the adult population is often associated with poor dietary intake, reduced nutrient absorption, and less efficient utilization of nutrients. These factors may also attenuate the effect of folic acid fortification, which should be evaluated prior to reassessing the amount of folic acid in the food supply.
6. Conclusions and future perspectives
Understanding the molecular basis underlying learning and memory remains one of the most challenging research topics. Previous physiological and genetic studies have generated some useful glimpses into how learning and memory function through different neural connections, but the entire picture is still far from being clear, partly due to the inherent complexity of the brain. The growing prevalence of memory dysfunction in the general population associated with normal aging and various disease conditions calls for more effective approaches to prevent or assist those affected individuals in living an improved quality of life. Epigenetic studies in cancer have revolutionized our perceptions of the etiology and progression of such complex diseases, and have led to the development of several epigenetic drugs to ameliorate the severity of such devastating diseases. Despite some individual efforts focusing on epigenetic studies of several forms of neurological disorders, there lacks a general appreciation of the role of DNA methylation among neurobiologists studying cognitive functions. Recent advances in epigenetic research have generated multiple high throughput technologies for global DNA methylation analysis. Such technical platforms can be readily applied to cognitive research to explore the epigenetic basis of learning and memory in the future. Dissection of the epigenetic basis of neurological disorders will eventually facilitate the application of epigenetic drugs in the treatment of patients with learning and memory dysfunction.
Acknowledgements
We would like to thank the Evelyn F. McKnight Brain Institute for grant support as well as the American Institute for Cancer Research, the NCI and the UAB Center for Aging.
Footnotes
Conflict of interest
The authors declare no actual or any potential conflict of interests.
References
- Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, Shafa R, Glatt SJ, Nguyen G, Ponte JF, Thiagalingam S, Tsuang MT. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005;134:60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Albright CD, Friedrich CB, Brown EC, Mar MH, Zeisel SH. Maternal dietary choline availability alters mitosis, apoptosis and the localization of TOAD-64 protein in the developing fetal rat septum. Brain Res. Dev. Brain Res. 1999;115:123–129. doi: 10.1016/s0165-3806(99)00057-7. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Andria ML, Simon EJ. Localization of promoter elements in the human mu-opioid receptor gene and regulation by DNA methylation. Brain Res. Mol. Brain Res. 1999;70:54–65. doi: 10.1016/s0169-328x(99)00126-6. [DOI] [PubMed] [Google Scholar]
- Barresi V, Condorelli DF, Giuffrida Stella AM. GFAP gene methylation in different neural cell types from rat brain. Int. J. Dev. Neurosci. 1999;17:821–828. doi: 10.1016/s0736-5748(99)00059-3. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsch D, Lenz B, Kornhuber J, Bleich S. DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport. 2005;16:167–170. doi: 10.1097/00001756-200502080-00020. [DOI] [PubMed] [Google Scholar]
- Bonsch D, Lenz B, Reulbach U, Kornhuber J, Bleich S. Homocysteine associated genomic DNA hypermethylation in patients with chronic alcoholism. J. Neural. Transm. 2004;111:1611–1616. doi: 10.1007/s00702-004-0232-x. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Brooks PJ, Marietta C, Goldman D. DNA mismatch repair and DNA methylation in adult brain neurons. J. Neurosci. 1996;16:939–945. doi: 10.1523/JNEUROSCI.16-03-00939.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, McKay RD. Restoring production of hippocampal neurons in old age. Nat. Neurosci. 1999;2:894–897. doi: 10.1038/13197. [DOI] [PubMed] [Google Scholar]
- Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell. Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Cheung P, Lau P. Epigenetic regulation by histone methylation and histone variants. Mol. Endocrinol. 2005;19:563–573. doi: 10.1210/me.2004-0496. [DOI] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David SJ, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Nicoletti VG, Barresi V, Caruso A, Conticello S, de Vellis J, Giuffrida Stella AM. Tissue-specific DNA methylation patterns of the rat glial fibrillary acidic protein gene. J. Neurosci. Res. 1994;39:694–707. doi: 10.1002/jnr.490390610. [DOI] [PubMed] [Google Scholar]
- Costa E, Davis J, Pesold C, Tueting P, Guidotti A. The heterozygote reeler mouse as a model for the development of a new generation of antipsychotics. Curr. Opin. Pharmacol. 2002;2:56–62. doi: 10.1016/s1471-4892(01)00121-7. [DOI] [PubMed] [Google Scholar]
- Craciunescu CN, Albright CD, Mar MH, Song J, Zeisel SH. Choline availability during embryonic development alters progenitor cell mitosis in developing mouse hippocampus. J. Nutr. 2003;133:3614–3618. doi: 10.1093/jn/133.11.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciunescu CN, Brown EC, Mar MH, Albright CD, Nadeau MR, Zeisel SH. Folic acid deficiency during late gestation decreases progenitor cell proliferation and increases apoptosis in fetal mouse brain. J. Nutr. 2004;134:162–166. doi: 10.1093/jn/134.1.162. [DOI] [PubMed] [Google Scholar]
- Davis CD, Uthus EO. Dietary folate and selenium affect dimethylhydrazine-induced aberrant crypt formation, global DNA methylation and one-carbon metabolism in rats. J. Nutr. 2003;133:2907–2914. doi: 10.1093/jn/133.9.2907. [DOI] [PubMed] [Google Scholar]
- Dong E, Agis-Balboa RC, Simonini MV, Grayson DR, Costa E, Guidotti A. Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine-induced mouse model of schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 2005;102:12578–12583. doi: 10.1073/pnas.0505394102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat. Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Espeland MA, Coker LH, Wallace R, Rapp SR, Resnick SM, Limacher M, Powell LH, Messina CR. Association between alcohol intake and domain-specific cognitive function in older women. Neuroepidemiology. 2006;27:1–12. doi: 10.1159/000093532. [DOI] [PubMed] [Google Scholar]
- Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, Trumpp A, Poon C, Wilson CB, Jaenisch R. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J. Neurosci. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Martinowich K, Chin MH, He F, Fouse SD, Hutnick L, Hattori D, Ge W, Shen Y, Wu H, ten Hoeve J, Shuai K, Sun YE. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development. 2005;132:3345–3356. doi: 10.1242/dev.01912. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- Feng J, Chang H, Li E, Fan G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res. 2005;79:734–746. doi: 10.1002/jnr.20404. [DOI] [PubMed] [Google Scholar]
- Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S. S-Adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol. Cell. Neurosci. 2005;28:195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol. Psychiatry. 2006;59:468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Gordon N. Nutrition and cognitive function. Brain Dev. 1997;19:165–170. doi: 10.1016/s0387-7604(96)00560-8. [DOI] [PubMed] [Google Scholar]
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E. Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haan MN, Miller JW, Aiello AE, Whitmer RA, Jagust WJ, Mungas DM, Allen LH, Green R. Homocysteine, B vitamins, and the incidence of dementia and cognitive impairment: results from the Sacramento Area Latino Study on Aging. Am. J. Clin. Nutr. 2007;85:511–517. doi: 10.1093/ajcn/85.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan YI, Zempleni J. Epigenetic regulation of chromatin structure and gene function by biotin. J. Nutr. 2006;136:1763–1765. doi: 10.1093/jn/136.7.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B, Bird A. Mammalian methyltransferases and methyl-CpG-binding domains: proteins involved in DNA methylation. Curr. Top. Microbiol. Immunol. 2000;249:55–74. doi: 10.1007/978-3-642-59696-4_4. [DOI] [PubMed] [Google Scholar]
- Holliday R. Is there an epigenetic component in long-term memory? J. Theor. Biol. 1999;200:339–341. doi: 10.1006/jtbi.1999.0995. [DOI] [PubMed] [Google Scholar]
- Holliday R. Francis Crick (1916-2004) Cell. 2004;119:1–2. doi: 10.1016/j.cell.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Honein MA, Paulozzi LJ, Mathews TJ, Erickson JD, Wong LY. Impact of folic acid fortification of the US food supply on the occurrence of neural tube defects. JAMA. 2001;285:2981–2986. doi: 10.1001/jama.285.23.2981. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macro-molecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Huang YY, Pittenger C, Kandel ER. A form of long-lasting, learning-related synaptic plasticity in the hippocampus induced by heterosynaptic low-frequency pairing. Proc. Natl. Acad. Sci. U.S.A. 2004;101:859–864. doi: 10.1073/pnas.2237201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M, Smith MM. Functional consequences of histone modifications. Curr. Opin. Genet. Dev. 2003;13:154–160. doi: 10.1016/s0959-437x(03)00020-0. [DOI] [PubMed] [Google Scholar]
- Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, Uzunov DP, Smalheiser NR, Davis JM, Pandey GN, Pappas GD, Tueting P, Sharma RP, Costa E. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 1998;95:15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inano K, Suetake I, Ueda T, Miyake Y, Nakamura M, Okada M, Tajima S. Maintenance-type DNA methyltransferase is highly expressed in post-mitotic neurons and localized in the cytoplasmic compartment. J. Biochem. (Tokyo) 2000;128:315–321. doi: 10.1093/oxfordjournals.jbchem.a022755. [DOI] [PubMed] [Google Scholar]
- Jacques PF, Selhub J, Bostom AG, Wilson PW, Rosenberg IH. The effect of folic acid fortification on plasma folate and total homocysteine concentrations. N. Engl. J. Med. 1999;340:1449–1454. doi: 10.1056/NEJM199905133401901. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Laird PW. Cancer epigenetics comes of age. Nat. Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Jorgenson LA, Sun M, O’Connor M, Georgieff MK. Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area CA1 of the developing rat hippocampus. Hippocampus. 2005;15:1094–1102. doi: 10.1002/hipo.20128. [DOI] [PubMed] [Google Scholar]
- Kelz MB, Chen J, Carlezon WA, Jr., Whisler K, Gilden L, Beckmann AM, Steffen C, Zhang YJ, Marotti L, Self DW, Tkatch T, Baranauskas G, Surmeier DJ, Neve RL, Duman RS, Picciotto MR, Nestler EJ. Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature. 1999;401:272–276. doi: 10.1038/45790. [DOI] [PubMed] [Google Scholar]
- Kim M, Campeau S, Falls WA, Davis M. Infusion of the non-NMDA receptor antagonist CNQX into the amygdala blocks the expression of fear-potentiated startle. Behav. Neural. Biol. 1993;59:5–8. doi: 10.1016/0163-1047(93)91075-x. [DOI] [PubMed] [Google Scholar]
- Kretchmer N, Beard JL, Carlson S. The role of nutrition in the development of normal cognition. Am. J. Clin. Nutr. 1996;63:997S–1001S. doi: 10.1093/ajcn/63.6.997. [DOI] [PubMed] [Google Scholar]
- Kruman II, Mouton PR, Emokpae R, Jr., Cutler RG, Mattson MP. Folate deficiency inhibits proliferation of adult hippocampal progenitors. Neuroreport. 2005;16:1055–1059. doi: 10.1097/00001756-200507130-00005. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Landsberger N, Wolffe AP. Remodeling of regulatory nucleoprotein complexes on the Xenopus hsp70 promoter during meiotic maturation of the Xenopus oocyte. EMBO J. 1997;16:4361–4373. doi: 10.1093/emboj/16.14.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurin D, Verreault R, Lindsay J, Dewailly E, Holub BJ. Omega-3 fatty acids and risk of cognitive impairment and dementia. J. Alzheimers Dis. 2003;5:315–322. doi: 10.3233/jad-2003-5407. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Rivera S, Stinis CT, Hayes VY, Isackson PJ, Gall CM. Differential effects of protein synthesis inhibition on the activity-dependent expression of BDNF transcripts: evidence for immediate-early gene responses from specific promoters. J. Neurosci. 1996;16:7428–7436. doi: 10.1523/JNEUROSCI.16-23-07428.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Kim JJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J. Neurosci. 1998;18:8444–8454. doi: 10.1523/JNEUROSCI.18-20-08444.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WJ, Zhu BT. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis. 2006;27:269–277. doi: 10.1093/carcin/bgi206. [DOI] [PubMed] [Google Scholar]
- Lehmann M, Gottfries CG, Regland B. Identification of cognitive impairment in the elderly: homocysteine is an early marker. Dement. Geriatr. Cogn. Disord. 1999;10:12–20. doi: 10.1159/000017092. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Choi S, Lee SY, Cao YA, Ahn HJ, Worley KC, Pizzi M, Liou HC, Sweatt JD. A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel. Neurosci. 2004a;24:3933–3943. doi: 10.1523/JNEUROSCI.5646-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004b;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Lieberman HR. Nutrition, brain function and cognitive performance. Appetite. 2003;40:245–254. doi: 10.1016/s0195-6663(03)00010-2. [DOI] [PubMed] [Google Scholar]
- Lippa CF. Familial Alzheimer’s disease: genetic influences on the disease process (review) Int. J. Mol. Med. 1999;4:529–536. doi: 10.3892/ijmm.4.5.529. [DOI] [PubMed] [Google Scholar]
- Liu L, Wylie RC, Andrews LG, Tollefsbol TO. Aging, cancer and nutrition: the DNA methylation connection. Mech. Ageing Dev. 2003;124:989–998. doi: 10.1016/j.mad.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Long-distance neuronal migration in the adult mammalian brain. Science. 1994;264:1145–1148. doi: 10.1126/science.8178174. [DOI] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc. Natl. Acad. Sci. U.S.A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas RJ. Diversity and patterns of regulation of nicotinic receptor subtypes. Ann. N.Y. Acad. Sci. 1995;757:153–168. doi: 10.1111/j.1749-6632.1995.tb17471.x. [DOI] [PubMed] [Google Scholar]
- Luskin MB. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 1993;11:173–189. doi: 10.1016/0896-6273(93)90281-u. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- Mani ST, Thakur MK. In the cerebral cortex of female and male mice, amyloid precursor protein (APP) promoter methylation is higher in females and differentially regulated by sex steroids. Brain Res. 2006;1067:43–47. doi: 10.1016/j.brainres.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- McCaddon A, Davies G, Hudson P, Tandy S, Cattell H. Total serum homocysteine in senile dementia of Alzheimer type. Int. J. Geriatr. Psychiatry. 1998;13:235–239. doi: 10.1002/(sici)1099-1166(199804)13:4<235::aid-gps761>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Meck WH, Williams CL. Simultaneous temporal processing is sensitive to prenatal choline availability in mature and aged rats. Neuroreport. 1997;8:3045–3051. doi: 10.1097/00001756-199709290-00009. [DOI] [PubMed] [Google Scholar]
- Mellott TJ, Follettie MT, Diesl V, Hill AA, Lopez-Coviella I, Krzysztof Blusztajn J. Prenatal choline availability modulates hippocampal and cerebral cortical gene expression. FASEB J. 2007 doi: 10.1096/fj.06-6597com. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Miller JW. Assessing the association between vitamin B-12 status and cognitive function in older adults. Am. J. Clin. Nutr. 2006;84:1259–1260. doi: 10.1093/ajcn/84.6.1259. [DOI] [PubMed] [Google Scholar]
- Misner DL, Jacobs S, Shimizu Y, de Urquiza AM, Solomin L, Perlmann T, De Luca LM, Stevens CF, Evans RM. Vitamin A deprivation results in reversible loss of hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11714–11719. doi: 10.1073/pnas.191369798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullaney BC, Johnston MV, Blue ME. Developmental expression of methyl-CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience. 2004;123:939–949. doi: 10.1016/j.neuroscience.2003.11.025. [DOI] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr. Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Kandel ER. Brief theta-burst stimulation induces a transcription-dependent late phase of LTP requiring cAMP in area CA1 of the mouse hippocampus. Learn. Mem. 1997;4:230–243. doi: 10.1101/lm.4.2.230. [DOI] [PubMed] [Google Scholar]
- Nicholson JM, Wood CM, Reynolds CD, Brown A, Lambert SJ, Chantalat L, Baldwin JP. Histone structures: targets for modifications by molecular assemblies. Ann. N.Y. Acad. Sci. 2004;1030:644–655. doi: 10.1196/annals.1329.075. [DOI] [PubMed] [Google Scholar]
- Niculescu MD, Craciunescu CN, Zeisel SH. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB J. 2006;20:43–49. doi: 10.1096/fj.05-4707com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh JS, Sharma RP, Veldic M, Salvacion AA, Jia X, Chen Y, Costa E, Guidotti A, Grayson DR. DNA methyltransferase 1 regulates reelin mRNA expression in mouse primary cortical cultures. Proc. Natl. Acad. Sci. U.S.A. 2005;102:1749–1754. doi: 10.1073/pnas.0409648102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numachi Y, Yoshida S, Yamashita M, Fujiyama K, Naka M, Matsuoka H, Sato M, Sora I. Psychostimulant alters expression of DNA methyltransferase mRNA in the rat brain. Ann. N.Y. Acad. Sci. 2004;1025:102–109. doi: 10.1196/annals.1316.013. [DOI] [PubMed] [Google Scholar]
- Peterson CL, Laniel MA. Histones and histone modifications. Curr. Biol. 2004;14:R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Caldarone BJ, King SL, Zachariou V. Nicotinic receptors in the brain. Links between molecular biology and behavior. Neuropsychopharmacology. 2000;22:451–465. doi: 10.1016/S0893-133X(99)00146-3. [DOI] [PubMed] [Google Scholar]
- Pollwein P, Masters CL, Beyreuther K. The expression of the amyloid precursor protein (APP) is regulated by two GC-elements in the promoter. Nucleic Acids. Res. 1992;20:63–68. doi: 10.1093/nar/20.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Ramos MI, Allen LH, Mungas DM, Jagust WJ, Haan MN, Green R, Miller JW. Low folate status is associated with impaired cognitive function and dementia in the Sacramento Area Latino Study on Aging. Am. J. Clin. Nutr. 2005;82:1346–1352. doi: 10.1093/ajcn/82.6.1346. [DOI] [PubMed] [Google Scholar]
- Refsum H, Ueland PM, Nygard O, Vollset SE. Homocysteine and cardiovascular disease. Annu. Rev. Med. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Sweatt JD. A biochemical blueprint for long-term memory. Learn. Mem. 1999;6:381–388. [PMC free article] [PubMed] [Google Scholar]
- Rogaev EI, Lukiw WJ, Lavrushina O, Rogaeva EA, St George-Hyslop PH. The upstream promoter of the beta-amyloid precursor protein gene (APP) shows differential patterns of methylation in human brain. Genomics. 1994;22:340–347. doi: 10.1006/geno.1994.1393. [DOI] [PubMed] [Google Scholar]
- Rogers SW, Gahring LC, Collins AC, Marks M. Age-related changes in neuronal nicotinic acetylcholine receptor subunit alpha4 expression are modified by long-term nicotine administration. J. Neurosci. 1998;18:4825–4832. doi: 10.1523/JNEUROSCI.18-13-04825.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, Martins R, Hockey A, Montgomery P, Beyreuther K, Masters CL. Amyloid A4 protein and its precursor in Down’s syndrome and Alzheimer’s disease. N. Engl. J. Med. 1989;320:1446–1452. doi: 10.1056/NEJM198906013202203. [DOI] [PubMed] [Google Scholar]
- Rusted JM, Newhouse PA, Levin ED. Nicotinic treatment for degenerative neuropsychiatric disorders such as Alzheimer’s disease and Parkinson’s disease. Behav. Brain Res. 2000;113:121–129. doi: 10.1016/s0166-4328(00)00207-2. [DOI] [PubMed] [Google Scholar]
- Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA. Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron. 1997;19:269–281. doi: 10.1016/s0896-6273(00)80938-1. [DOI] [PubMed] [Google Scholar]
- Scott TM, Peter I, Tucker KL, Arsenault L, Bergethon P, Bhadelia R, Buell J, Collins L, Dashe JF, Griffith J, Hibberd P, Leins D, Liu T, Ordovas JM, Patz S, Price LL, Qiu WQ, Sarnak M, Selhub J, Smaldone L, Wagner C, Wang L, Weiner D, Yee J, Rosenberg I, Folstein M. The Nutrition, Aging, and Memory in Elders (NAME) Study: design and methods for a study of micronutrients and cognitive function in a homebound elderly population. Int. J. Geriatr. Psychiatry. 2006;21:519–528. doi: 10.1002/gps.1503. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Sibani S, Melnyk S, Pogribny IP, Wang W, Hiou-Tim F, Deng L, Trasler J, James SJ, Rozen R. Studies of methionine cycle intermediates (SAM, SAH), DNA methylation and the impact of folate deficiency on tumor numbers in Min mice. Carcinogenesis. 2002;23:61–65. doi: 10.1093/carcin/23.1.61. [DOI] [PubMed] [Google Scholar]
- Snowdon DA, Tully CL, Smith CD, Riley KP, Markesbery WR. Serum folate and the severity of atrophy of the neocortex in Alzheimer disease: findings from the Nun study. Am. J. Clin Nutr. 2000;71:993–998. doi: 10.1093/ajcn/71.4.993. [DOI] [PubMed] [Google Scholar]
- Squire LR, 2nd, Emanuel CA, Davis HP, Deutsch JA. Inhibitors of cerebral protein synthesis: dissociation of aversive and amnesic effects. Behav. Biol. 1975;14:335–341. doi: 10.1016/s0091-6773(75)90467-8. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell. 2001;1:749–758. doi: 10.1016/s1534-5807(01)00101-0. [DOI] [PubMed] [Google Scholar]
- Tawa R, Ono T, Kurishita A, Okada S, Hirose S. Changes of DNA methylation level during pre- and postnatal periods in mice. Differentiation. 1990;45:44–48. doi: 10.1111/j.1432-0436.1990.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Teather LA, Wurtman RJ. Dietary CDP-choline supplementation prevents memory impairment caused by impoverished environmental conditions in rats. Learn. Mem. 2005;12:39–43. doi: 10.1101/lm.83905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teter B, Osterburg HH, Anderson CP, Finch CE. Methylation of the rat glial fibrillary acidic protein gene shows tissue-specific domains. J. Neurosci. Res. 1994;39:680–693. doi: 10.1002/jnr.490390609. [DOI] [PubMed] [Google Scholar]
- Teter B, Rozovsky I, Krohn K, Anderson C, Osterburg H, Finch C. Methylation of the glial fibrillary acidic protein gene shows novel biphasic changes during brain development. Glia. 1996;17:195–205. doi: 10.1002/(SICI)1098-1136(199607)17:3<195::AID-GLIA2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Utsugisawa K, Yoshimura M, Nagane Y, Mihara M. Age-related changes in nicotinic acetylcholine receptor subunits alpha4 and beta2 messenger RNA expression in postmortem human frontal cortex and hippocampus. Neurosci. Lett. 1998;245:139–142. doi: 10.1016/s0304-3940(98)00205-5. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Utsugisawa K, Nagane Y, Yoshimura M, Genda Y, Ukitsu M. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res. Mol. Brain Res. 1999;70:288–292. doi: 10.1016/s0169-328x(99)00163-1. [DOI] [PubMed] [Google Scholar]
- Tremolizzo L, Carboni G, Ruzicka WB, Mitchell CP, Sugaya I, Tueting P, Sharma R, Grayson DR, Costa E, Guidotti A. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc. Natl. Acad. Sci. U.S.A. 2002;99:17095–17100. doi: 10.1073/pnas.262658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Ulrey CL, Liu L, Andrews LG, Tollefsbol TO. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005;14(Spec No 1):R139–R147. doi: 10.1093/hmg/ddi100. [DOI] [PubMed] [Google Scholar]
- Veldic M, Caruncho HJ, Liu WS, Davis J, Satta R, Grayson DR, Guidotti A, Costa E. DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc. Natl. Acad. Sci. U.S.A. 2004;101:348–353. doi: 10.1073/pnas.2637013100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Zoli M, Changeux JP. Promoter analysis of the neuronal nicotinic acetylcholine receptor alpha4 gene: methylation and expression of the transgene. Eur. J. Neurosci. 1998;10:2244–2253. doi: 10.1046/j.1460-9568.1998.00235.x. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ, Szyf M. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J. Neurosci. 2005;25:11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RL, Lee JM, Maroun LE. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 1995;6:141–146. doi: 10.1007/BF02736773. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyl-deoxycytidine decreases with age. J. Biol. Chem. 1987;262:9948–9951. [PubMed] [Google Scholar]
- Woodruff-Pak DS. Mecamylamine reversal by nicotine and by a partial alpha7 nicotinic acetylcholine receptor agonist (GTS-21) in rabbits tested with delay eyeblink classical conditioning. Behav. Brain Res. 2003;143:159–167. doi: 10.1016/s0166-4328(03)00039-1. [DOI] [PubMed] [Google Scholar]
- Yonelinas AP, Kroll NE, Quamme JR, Lazzara MM, Sauve MJ, Widaman KF, Knight RT. Effects of extensive temporal lobe damage or mild hypoxia on recollection and familiarity. Nat. Neurosci. 2002;5:1236–1241. doi: 10.1038/nn961. [DOI] [PubMed] [Google Scholar]
- Zawia NH, Basha MR. Environmental risk factors and the developmental basis for Alzheimer’s disease. Rev. Neurosci. 2005;16:325–337. doi: 10.1515/revneuro.2005.16.4.325. [DOI] [PubMed] [Google Scholar]
- Zeisel SH, Blusztajn JK. Choline and human nutrition. Annu. Rev. Nutr. 1994;14:269–296. doi: 10.1146/annurev.nu.14.070194.001413. [DOI] [PubMed] [Google Scholar]
- Zeisel SH. Choline: critical role during fetal development and dietary requirements in adults. Annu. Rev. Nutr. 2006;26:229–250. doi: 10.1146/annurev.nutr.26.061505.111156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Mar MH, Song J, Zeisel SH. Deletion of the Pemt gene increases progenitor cell mitosis, DNA and protein methylation and decreases calretinin expression in embryonic day 17 mouse hippocampus. Brain Res. Dev. Brain Res. 2004;149:121–129. doi: 10.1016/j.devbrainres.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 1994;12:11–24. doi: 10.1016/0896-6273(94)90148-1. [DOI] [PubMed] [Google Scholar]