

Abstract
Graft failure after liver transplantation may involve mitochondrial dysfunction. We examined whether prevention of mitochondrial injury would improve graft function. Orthotopic rat liver transplantation was performed after 18 hours' cold storage in University of Wisconsin solution and treatment with vehicle, minocycline, tetracycline, or N-methyl-4-isoleucine cyclosporin (NIM811) of explants and recipients. Serum alanine aminotransferase (ALT), necrosis, and apoptosis were assessed 6 hours after implantation. Mitochondrial polarization and cell viability were assessed by intravital microscopy. Respiration and the mitochondrial permeability transition (MPT) were assessed in isolated rat liver mitochondria. After transplantation with vehicle or tetracycline, ALT increased to 5242 U/L and 4373 U/L, respectively. Minocycline and NIM811 treatment decreased ALT to 2374 U/L and 2159 U/L, respectively (P < 0.01). Necrosis and terminal deoxynucleotidyl transferase-mediated nick-end labeling (TUNEL) also decreased from 21.4% and 21 cells/field, respectively, after vehicle to 10.1% and 6 cells/field after minocycline and to 8.7% and 5.2 cells/field after NIM811 (P < 0.05). Additionally, minocycline decreased caspase-3 activity in graft homogenates (P < 0.05). Long-term graft survival was 27% and 33%, respectively, after vehicle and tetracycline treatment, which increased to 60% and 70% after minocycline and NIM811 (P < 0.05). In isolated mitochondria, minocycline and NIM811 but not tetracycline blocked the MPT. Minocycline blocked the MPT by decreasing mitochondrial Ca2+ uptake, whereas NIM811 blocks by interaction with cyclophilin D. Intravital microscopy showed that minocycline and NIM811 preserved mitochondrial polarization and cell viability after transplantation (P < 0.05).
Conclusion
Minocycline and NIM811 attenuated graft injury after rat liver transplantation and improved graft survival. Minocycline and/or NIM811 might be useful clinically in hepatic surgery and transplantation.
Although liver transplantation is an established therapy for end-stage liver disease, poor initial graft function and graft failure still occur unpredictably. Graft dysfunction is caused and aggravated by prolonged cold ischemic storage, which leads to sinusoidal endothelial cell killing and Kupffer cell activation within minutes after reperfusion. Parenchymal cell death then follows after 3 to 6 hours. These events lead to graft dysfunction and failure.1,2 After transplantation, both necrotic and apoptotic cell death pathways have been described to coexist.3,4
In liver and other organs, the mitochondrial permeability transition (MPT) plays an important role in the pathogenesis of warm ischemia/reperfusion injury.3,5,6 Opening of permeability transition pores in the mitochondrial inner membrane causes the MPT.7 Permeability transition pores nonspecifically conduct low molecular weight solutes to cause mitochondrial depolarization, uncoupling of oxidative phosphorylation, and large-amplitude colloid osmotic swelling. Adenosine triphosphate (ATP) depletion after uncoupling produces necrotic cell killing (oncosis), whereas swelling leads to outer membrane rupture and release of proapoptotic proteins such as cytochrome c from the intermembrane space that activate apoptotic pathways.5 Thus, onset of the MPT within hepatocytes leads both to oncotic necrosis from ATP depletion and caspase-dependent apoptosis if ATP depletion does not occur fully.3,5 Because hepatocellular killing typically develops as a second wave beginning 3 to 6 hours after sinusoidal endothelial cell death and Kupffer cell activation after cold storage/reperfusion injury, parenchymal cell death may represent a second hit that extends and aggravates initial hepatic damage.2,8,9 In this respect, if the MPT is important to parenchymal cell death after reoxygenation, protective strategies directed to mitochondria might be beneficial.
Minocycline is a semisynthetic tetracycline antibiotic that is protective against neurodegenerative disease, trauma, and hypoxia–ischemia.10-18 Mechanisms by which minocycline may exert neuroprotection include inhibition of apoptotic pathways, decreased mitochondrial release of pro-apoptotic factors such as cytochrome c,18 and up-regulation of antiapoptotic Bcl-2 and inhibitor of apoptosis proteins.14 Furthermore, in cardiac and renal ischemia, minocycline inhibits caspase-1 and caspase-3 activation and suppresses cytochrome c release.10,13 A study of cold-stored rat livers also demonstrated decreased sinusoidal endothelial cell rounding and increased viability after minocycline pretreatment.12 In another study, minocycline protected against Fas-dependent fulminant hepatitis and death in mice.10 We investigated whether blockade of the MPT is a mechanism of minocycline cytoprotection and whether minocycline decreases injury and improves function and survival of transplanted liver grafts. Furthermore, we compared the effects of minocycline with those of N-methyl-4-isoleucine cyclosporin (NIM811), a nonimmunosuppressive derivative of cyclosporin A (CsA) that is a specific inhibitor of the MPT.
Materials and Methods
Chemicals and Reagents
NIM811 was the gift of Novartis (Basel, Switzerland). CsA was purchased from Calbiochem (La Jolla, CA), Fluo-5N from Molecular Probes (Eugene, OR), and minocycline, tetracycline, rhodamine 123, propidium iodide (PI), and other reagents from Sigma-Aldrich (St. Louis, MO).
Liver Harvest and Storage
Experiments involving animals were conducted using protocols approved by the Institutional Animal Care and Use Committee. Inbred male Lewis rats (Harlan, Indianapolis, IN) weighing 250 to 280 g were used as donors and recipients to exclude immunologic interference. Donor and recipient rats were fasted overnight before organ harvest. For the donor operation under ether anesthesia, the liver was freed, and the common bile duct was cannulated with 24-gauge polyethylene tubing and divided. Ice-cold University of Wisconsin solution (10 mL) was infused through the portal vein. The suprahepatic inferior vena cava, subhepatic inferior cava, portal vein, and celiac artery were divided at the level of the diaphragm, left renal vein, splenic vein, and splenic artery, respectively. The liver was excised and placed in ice-chilled University of Wisconsin solution, and plastic cuffs (14 G, Medex, Carlsbad, CA) were placed on the portal vein and subhepatic inferior cava before storage at 0°C to 1°C until implantation. For treating liver explants, either lactated Ringer's solution as vehicle, minocycline, tetracycline, or NIM811 was added to the perfusion, storage, and rinse solutions. For recipient treatment, compounds were injected intraperitoneally 1 hour before recipient surgery.
Liver Transplantation
Orthotopic rat liver transplantation was performed under ether anesthesia using an arterialized two-cuff method, as described previously with slight modification.19 In the recipient rat, the proper hepatic artery was divided at its hilar bifurcation. The stump was clamped at the gastroduodenal artery, and the bifurcation was cut to leave a funnel-shaped opening into which a 4-mm polyethylene splint (24 G, B. Braun Melsungen, Bethlehem, PA) was inserted.20 After dividing the bile duct at the hilum, the suprahepatic inferior cava, portal vein, and subhepatic inferior cava were clamped and divided, and the recipient liver was removed. The donor liver was then washed with 10 mL Ringer's solution at room temperature. Subsequently, the suprahepatic inferior cava was anastomosed with a 7–0 Prolene (Ethicon, Somerville, NJ) running suture. The portal vein and subhepatic inferior cava were connected in sequence by insertion of cuffs. Both the hepatic artery and the bile duct were anastomosed over the intraluminal polyethylene splints. The time of implantation averaged 52 minutes, and portal vein clamping averaged 15 minutes.
Alanine Aminotransferase
Blood samples to measure alanine aminotransferase (ALT) were collected from the inferior vena cava 6 hours after transplantation for analysis by standard methods.
Histology
Histology was evaluated 6 hours after liver transplantation. Liver tissues were fixed by immersion in 4% buffered paraformaldehyde and embedded in paraffin. Sections (4 μm) were stained with hematoxylin-eosin. Ten random fields were assessed for necrosis by standard morphologic criteria (for example, loss of architecture, vacuolization, karyolysis, increased eosinophilia). Images were captured by an image analysis system (Olympus BH-2 Microscope; Micropublisher 5.0 RTV, Center Valley, PA), and the area percentage of necrosis was quantified using a computer program (BioQuant BQ Nova Prime 6.7, R&M Biometrics, Nashville, TN).
Immunohistochemistry
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) was performed on paraffin sections using an in situ cell death detection kit (Roche Diagnostics, Penzberg, Germany). TUNEL-positive parenchymal and nonparenchymal cells were counted by light microscopy in 10 random high-power fields (HPF).
Caspase-3
Liver tissue (approximately 100 mg) was homogenized (Polytron PT-MR2100, Kinematica, Luzern, Switzerland) in 1 mL lysis buffer containing 0.1% 3-[3-Cholamidopropyl) dimethylammonio]-1-propanesulfonate, 2 mM ethylene diamione tetra-acetic acid, 5 mM dithiothreitol, 1 mM Pefabloc, 10 ng/mL pepstatin A, 10 ng/mL aprotinin, 20 μg /mL leupeptin, and 10 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid) buffer, pH 7.4. The lysate was centrifuged at 21,000 g for 30 minutes. Activity of caspase-3 in the supernatant was determined using a Caspase-3 Colorimetric Assay Kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. Activity was normalized to protein concentration of each sample and expressed as fold increase compared with sham.
Survival Study
Treatment with vehicle, minocycline, tetracycline, and NIM811 was performed in a randomized, prospective fashion, and rats were followed 30 days after surgery. Rats dying or requiring euthanasia for presumed surgical complications within 6 hours of transplantation were excluded.
Plate Reader Assays of the MPT
Rat liver mitochondria were isolated in 250 mM sucrose, 2 mM K+-HEPES buffer, pH 7.4, as previously described.21 Using a ThermoMax 96-well plate reader (Molecular Devices, Sunnyvale, CA), mitochondrial swelling was assessed from the decrease of absorbance at 540 nm of 0.5 mg/mL rat liver mitochondria suspended in 0.2 M sucrose, 20 mM Tris, 20 mM HEPES, 5 mM succinate, 1 mM KH2PO4, 2 μM rotenone, and 1 μg/ml oligomycin at pH 7.2, 25°C, as described.22 Mitochondrial suspensions were treated with 0 to 50 μM minocycline, 0 to 50 μM tetracycline, 5 μM NIM811, or 1 μM CsA After 2 minutes of incubation, 250 μM CaCl2 was added to induce the MPT.
In other experiments, fluorescence of the Ca2+ indicator Fluo-5N (1 μM) was determined in a fluorescence plate reader (BMG LABTECH GmbH, Offenburg, Germany). Mitochondrial suspensions were treated with various compounds. After 2 minutes, aliquots of 50 μM CaCl2 were added every 5 minutes.
Oxygen Uptake
Oxygen consumption was measured with a Clark electrode (Hansatech Instruments, Norfolk, England) in medium containing 1 mg/mL rat liver mitochondria, 5 mM disodium succinate, 1 μM rotenone, 150 mM sucrose, 5 mM MgCl2, 7.5 mM KPi buffer, 25 mM K-HEPES buffer, pH 7.4, 23°C, as described.21 Minocycline (0-100 μM), 218 μM adenosine diphosphate (ADP), 250 μM CaCl2, 2 μM CsA, and 1 μM carbonyl cyanide m-chlorophenylhydrazone were added as indicated.
Intravital Microscopy
At 2, 4, and 6 hours after transplantation, rats were anesthetized with pentobarbital (50 mg/kg) and connected to a small animal ventilator via a tracheostomy and respiratory tube (14-gauge catheter) as described in a mouse model.23 Laparotomy was performed using the previous incision line, and a 32-gauge catheter was inserted into the distal part of the right colic vein. Using a syringe pump, membrane potential–indicating fluorophore rhodamine 123 (10 μmol/rat) was infused via the catheter over 10 minutes. Immediately afterward, cell death–indicating PI (0.5 μmol/rat) was infused over 3 minutes. After prone positioning of the rat, the liver was gently withdrawn from the abdominal cavity and placed over a glass coverslip on the stage of a Zeiss Axiovert 100 microscope (Thornwood, NY) equipped with a spinning disk confocal imaging system (Attofluor CARV Optical Module, BD Bioimaging Systems, San Jose, CA), a 12-bit cooled CCD camera (Hamamatsu, Bridgewater, NJ), and a 40× 1.2 NA water-immersion objective lens for imaging the green fluorescence of rhodamine 123 and red fluorescence of PI. In some experiments, rhodamine 123 and PI fluorescence was excited with 820-nm light from a Chameleon Ultra Ti-Sapphire pulsed laser (Coherent Inc., Santa Clara, CA) and imaged with a Zeiss LSM 510 NLO inverted laser scanning confocal microscope using a 63× 1.3 NA water-immersion objective lens. Green rhodamine 123 and red PI fluorescence was collected through 525 ± 25 nm band pass and 700 ± 25 nm band pass filters, respectively. During image acquisition, the respirator was turned off for approximately 5 seconds to eliminate breathing movement artifacts. In 20 fields per liver, parenchymal cells were scored for bright punctate rhodamine 123 fluorescence representing cells with polarized mitochondria or a dimmer diffuse cytosolic fluorescence representing cells with depolarized mitochondria. Nonviable PI-positive cells, indicated by bright nuclear fluorescence, were also counted in the same fashion. Image analysis was performed in a blinded fashion.
Statistical Analysis
Data are presented as means ± standard error, unless otherwise noted. Statistical analysis was performed by Student t test, analysis of variance plus Student-Newman-Keuls test, Fisher's exact test, or Kaplan-Meier test, as appropriate, using P < 0.05 as the criterion of significance.
Results
Decreased ALT Release and Graft Necrosis After Minocycline and NIM811
Rat livers were stored for 18 hours and transplanted. Liver explants or recipients were treated with drug or vehicle. At 6 hours postoperatively, sham-operated rats had serum ALT averaging 105 ± 15 U/L. After transplantation, ALT after vehicle treatment increased to 5242 ± 517 U/L, which decreased to 2374 ± 280 U/L after explant and recipient treatment with 18 μM minocycline plus 10 mg/kg minocycline 1 hour prior to surgery (P < 0.01 versus vehicle treatment). Identical treatment of explants and recipients with tetracycline failed to decrease serum ALT after transplantation (Fig. 1). Treatment of explants and recipients with 5 μM and 10 mg/kg NIM811, respectively, also decreased ALT levels to 2159 ± 374 U/L (P < 0.01 versus vehicle treatment). Although treatment of liver explants alone with 18 μM minocycline decreased ALT levels (3825 ± 457 U/L, P = 0.05), we used the combination of explant and recipient minocycline treatment to achieve better results for further experiments. Other doses of minocycline treatment to liver explants alone (2-200 μM) and minocycline treatment to recipients alone (10 mg/kg) did not show a benefit on ALT release (not shown).
Fig. 1.
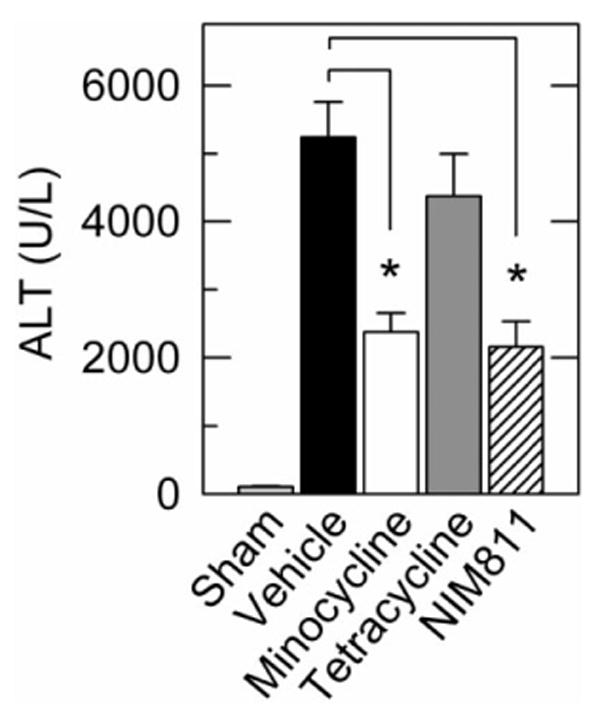
Minocycline and NIM811 decrease ALT release after rat liver transplantation. Serum ALT was assessed 6 hours after sham operation or rat liver transplantation. Explants and recipients were treated with vehicle (lactated Ringer's solution), minocycline (18 μM to explants, 10 mg/kg to recipients), tetracycline (18 μM to explants, 10 mg/kg to recipients), or NIM811 (5 μM to explants, 10 mg/kg to recipients). Group sizes were: sham, 4; vehicle, 21; minocycline, 10; tetracycline, 10; NIM811, 8. *P< 0.05.
Graft injury was also assessed histologically at 6 hours postoperatively. Liver histology was normal and indistinguishable in unoperated and sham-operated rats (Fig. 2A and data not shown). After vehicle treatment of both liver explants and transplant recipients, large areas of necrosis were present 6 hours postoperatively with a predominately pericentral and midzonal distribution (Fig. 2B, dashed line). Explant and recipient treatment with minocycline and NIM811 decreased hepatic necrosis from 21.4% ± 2.5% after vehicle to 10.1% ± 1.4% after minocycline and 8.7% ± 1.3% after NIM811 (P < 0.05, Fig. 2C-E). Thus, both minocycline and NIM811 treatment decreased hepatic necrosis by greater than 2-fold.
Fig. 2.
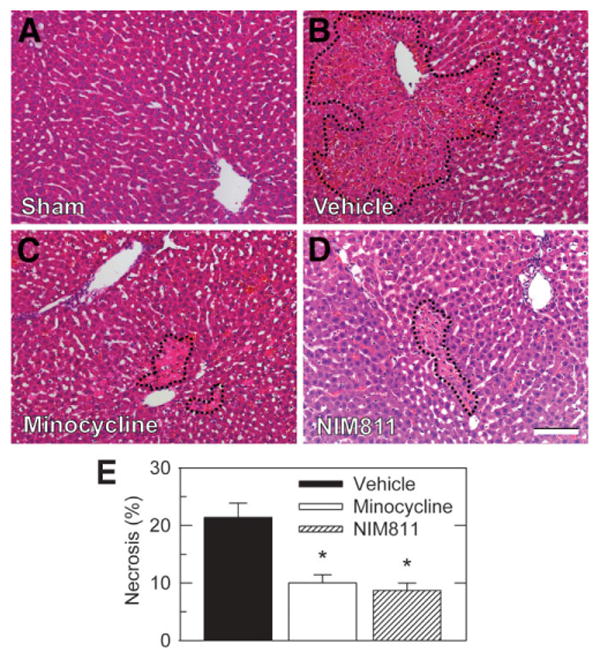
Minocycline and NIM811 decrease necrosis after liver transplantation. Rat livers were transplanted, as described in Fig. 1. At 6 hours postoperatively, necrosis was assessed by hematoxylin-eosin histology in sham-operated (A), vehicle-treated (B), minocycline-treated (C), and NIM811-treated (D) grafts. Dashed lines identify necrotic areas. (E) Necrosis as percent area in liver sections averaged from 5 livers per group. Necrosis in sham was absent and not plotted. Bar is 100 μm. *P < 0.05 versus vehicle.
Decreased Graft Apoptosis After Minocycline and NIM811
TUNEL was performed on tissue sections to assess double-stranded DNA breaks that are characteristic of apoptosis. Differentiation of parenchymal cells and nonparenchymal cells was made on the basis of nuclear morphology and location. TUNEL-positive parenchymal and nonparenchymal cells were rare after sham operation, averaging less than 1 cell/HPF (Fig. 3A). At 6 hours after transplantation with vehicle treatment, TUNEL of parenchymal cells and nonparenchymal cells in nonnecrotic areas increased to 21.2 ± 3.5 and 9.5 ± 2.5 cells/HPF, respectively, without apparent zonal localization (Fig. 3B). Treatment with minocycline of both explants and recipients decreased TUNEL by approximately two-thirds to 6.1 ± 1.7 cells/HPF in parenchymal cells and by approximately half to 5.3 ± 1.9 cells/HPF in nonparenchymal cells (P < 0.01 and P < 0.05, respectively, Fig. 3E, F). Treatment with NIM811 of both explants and recipients decreased TUNEL by three-fourths to 5.2 ± 2.4 cells/HPF in parenchymal cells (P < 0.01, Fig. 3E). A trend toward decreased TUNEL-positive nonparenchymal cells was also observed but was not statistically significant (P > 0.1). As a percentage of all parenchymal cells, the percentage of TUNEL was 2.7% ± 0.3% after transplantation with vehicle treatment versus 0.9% ± 0.2% and 0.8% ± 0.3% with minocycline and NIM811 treatment, respectively (P < 0.01). The percentage of nonparenchymal cell TUNEL could not be reliably determined because counterstaining was too weak to identify all nonparenchymal cells confidently.
Fig. 3.
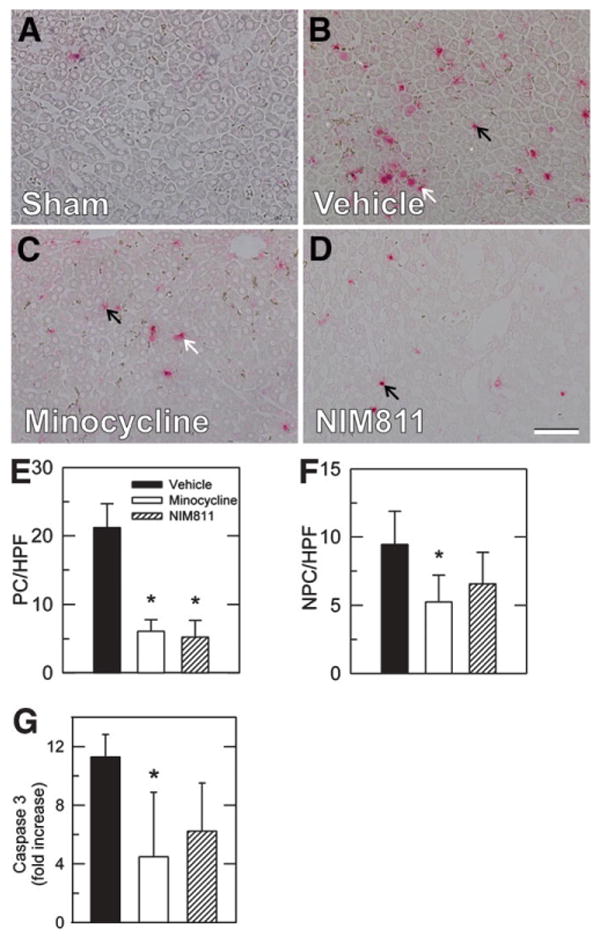
Minocycline and NIM811 decrease apoptosis after rat liver transplantation. Rat livers were transplanted, as described in Fig. 1. At 6 hours postoperatively, TUNEL was assessed in sections of sham-operated (A), vehicle-treated (B), minocycline-treated (C), and NIM811-treated (D) grafts. (E and F) TUNEL in parenchymal (PC) and nonparenchymal (NPC) cells. TUNEL for sham was virtually zero and not plotted. (G) Caspase 3 activity in liver homogenates at 6 hours after transplantation as fold increase over sham operation. Bar is 50 μm. *P < 0.05 versus vehicle.
To further investigate the extent of apoptosis after minocycline treatment, caspase-3 activity was measured in liver extracts at 6 hours after transplantation with vehicle, minocycline, or NIM811 in comparison with sham-operated livers (Fig. 3G). After sham operation, caspase activity was nearly undetectable. After transplantation with vehicle treatment, caspase-3 activity increased 11-fold, which decreased to 4.5-fold after minocycline (P < 0.05 compared with vehicle, Fig. 3G) and also trended lower after NIM811 (P = 0.2).
Improved Graft Survival After Minocycline and NIM811 Treatment
Graft survival was assessed by mortality out to 30 days. After transplantation with vehicle, 8 of 11 rats died within 36 hours, and 30-day survival was 27% (Fig. 4). After minocycline treatment, 30-day survival was 60% (P< 0.05 compared with vehicle, Fig. 4). Furthermore, graft loss was delayed, because 3 of the 4 rats died 48 hours or more after surgery. Similarly, NIM811 treatment led to 70% survival after 30 days (P < 0.05 compared with vehicle, Fig. 4). After tetracycline treatment, 30-day survival was 33% with all deaths occurring within 48 hours, which was not different from vehicle treatment (Fig. 4).
Fig. 4.
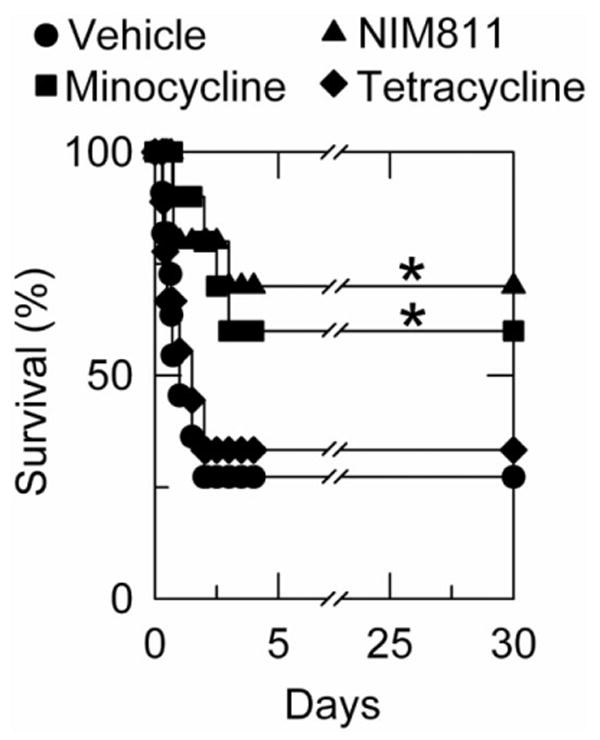
Minocycline and NIM811 improve survival after liver transplantation. Thirty-day survival was assessed in rats subjected to liver transplantation after treatments with vehicle, minocycline, NIM811, or tetracycline, as described in Fig. 1. Size of individual groups was 9 to 11. *P < 0.05 compared with vehicle and tetracycline.
Inhibition by Minocycline and NIM811 But Not Tetracycline of the Mitochondrial Permeability Transition in Isolated Rat Liver Mitochondria
The potential for minocycline to block the MPT was investigated in isolated rat liver mitochondria. Addition of 250 μM calcium to mitochondria induced MPT-dependent swelling, as shown by a large decrease in absorbance at 540 nm that was blocked by 1 μM CsA, an MPT inhibitor (Fig. 5). Minocycline also blocked calcium-induced mitochondrial swelling in a dose-dependent fashion, with nearly complete inhibition occurring at 50 μM. By contrast, tetracycline in the same dose range had no effect on calcium-induced mitochondrial swelling (Fig. 5). NIM811, a non-immunosuppressive CsA analog, also blocked swelling (data not shown, see also Fig. 6), as described previously.24
Fig. 5.
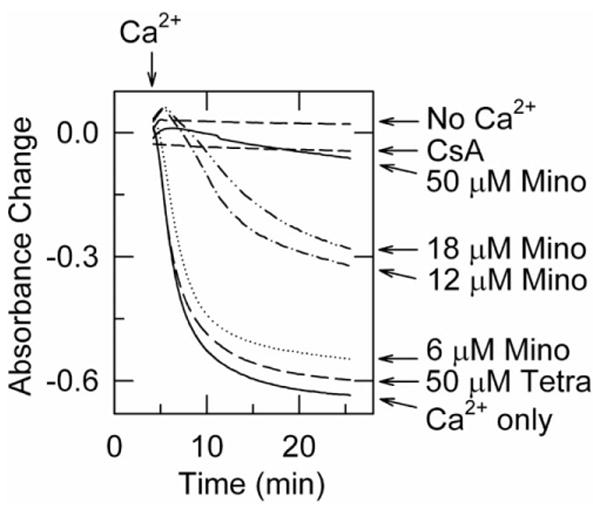
Minocycline inhibits calcium-induced swelling of isolated mitochondria. Swelling of rat liver mitochondria was monitored by absorbance, as described in Materials and Methods. CaCl2 (250 μM) was added at arrow except for tracing marked No Ca2+. Two minutes before CaCl2 addition, mitochondria were treated with minocycline (Mino), tetracycline (Tetra), or 1 μM CsA.
Fig. 6.
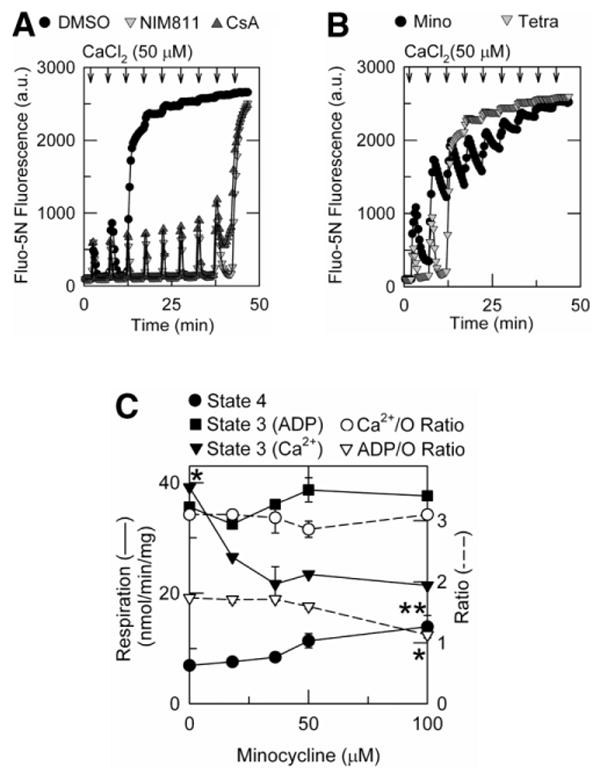
Minocycline inhibits calcium uptake without altering oxidative phosphorylation or energetic efficiency of calcium accumulation. Free Ca2+ was measured from Fluo-5N fluorescence, and oxygen consumption was measured polarographically, as described in Materials and Methods. (A) Mitochondria were preincubated with dimethylsulfoxide, 5 μM NIM811, or 1 μM CsA. After 2 minutes, 50 μM CaCl2 was added every 5 minutes (arrows). (B) Mitochondria were preincubated for 2 minutes with 18 μM minocycline or 18 μM tetracycline, and CaCl2 was added as in (A). (C) State 4 and state 3 respiration were measured before and after addition of 218 μM ADP or 250 μM CaCl2, as described in Materials and Methods. ADP/O and Ca2+/O ratios were calculated from total oxygen consumed during state 3 respiration from 3 or more measurements. *P < 0.05, **P < 0.05 versus no minocycline.
Next, we compared the effects of minocycline, tetracycline, CsA, and NIM811 on calcium uptake and retention by isolated mitochondria using Fluo-5N as a fluorescent indicator of extramitochondrial free calcium. In vehicle (dimethylsulfoxide)-treated mitochondria, calcium uptake was so rapid after the first 50 μM pulse of CaCl2 that increased extramitochondrial Ca2+ was detectable only in the first fluorescence measurement, after which Fluo-5N fluorescence returned to baseline (Fig. 6A). This pattern was repeated after a second pulse of CaCl2, but after a third pulse calcium was not taken up into the mitochondria. Rather, calcium previously taken up was released, an event signifying onset of the MPT. When mitochondria were treated with CsA or NIM811, calcium retention was greatly increased, and 400 μM CaCl2 was required to induce calcium release and onset of the MPT. In this particular experiment, 5 μM NIM811 was employed to match the concentration used in liver storage versus 1 μM for CsA, but previous studies showed that NIM811 is equipotent to CsA in blocking the MPT.24
Although minocycline, like CsA and NIM811, suppressed onset of the MPT, minocycline had a very different effect on calcium uptake and release. In the presence of 18 μM minocycline, mitochondrial calcium uptake after a CaCl2 pulse was inhibited an estimated 65% compared with vehicle-treated mitochondria (Fig. 6B). With successive pulses, Fluo-5N fluorescence increased progressively and failed to return to baseline between pulses. After the accumulated addition of 350 μM CaCl2, no recovery of Fluo-5N fluorescence occurred. By contrast, tetracycline-treated mitochondria were indistinguishable from vehicle-treated mitochondria.
ADP- and Ca2+-Stimulated Respiration in Minocycline-Treated Mitochondria
Ca2+/O and ADP/O ratios were determined polarographically.21 After addition of mitochondria, an initial burst of oxygen consumption (respiration) occurred, which was followed by a slower constant rate of oxygen consumption (state 4). After addition of 218 μM ADP alone or 250 μM CaCl2 in the presence of 2 μM CsA to prevent the MPT, respiration increased immediately (state 3) because of energy-dependent ADP phosphorylation or the electrophoretic uptake of calcium. Because ADP was exhausted by conversion to ATP or calcium was completely taken up into mitochondria, respiration returned to the original “idling” rate of state 4. By dividing the amount of ADP or CaCl2 added by the amount of oxygen consumed during state 3, ADP/O and Ca2+/O ratios of 1.74 and 3.1, respectively, were calculated (Fig. 6C). With increasing minocycline (0-50 μM), ADP-stimulated state 3 respiration and ADP/O ratios were unchanged. At still higher minocycline, ADP/O decreased 35%. This was attributable to increased state 4 respiration because state 3 respiration was not changed. By contrast, minocycline (0-100 μM) dose-dependently decreased Ca2+-stimulated state 3 respiration without affecting Ca2+/O ratios. Together with the Fluo-5N results, these findings indicate that minocycline (0-50 μM) has negligible effects on the rate and efficiency of oxidative phosphorylation. Rather, minocycline inhibits calcium uptake without altering the energetic efficiency of calcium accumulation.
Mitochondrial Dysfunction In Vivo After Liver Transplantation: Protection by Minocycline and NIM811
At 4 hours after sham operation, intravital multiphoton microscopy showed bright fluorescence of rhodamine 123 in hepatocytes whose punctuate pattern denoted polarization of individual mitochondria (Fig. 7A). Cytosolic and nuclear areas had little fluorescence. PI labeling of nuclei after sham operation was very rare, indicating the absence of cell death. By contrast, at 4 hours after liver transplantation with vehicle treatment, rhodamine 123 staining became diffuse and dim in many hepatocytes (Fig. 7B, white arrows). Additionally, many PI-positive red-fluorescing nuclei were observed after transplantation with vehicle treatment (Fig. 7B, yellow arrows). PI labeling occurred both in the round nuclei of parenchymal cells and the flattened irregular nuclei of nonparenchymal cells. Overlays of the green and red channels showed that all PI-labeled cells also had diffuse or absent rhodamine 123 staining (Fig. 7B, yellow arrows). Thus, all nonviable cells had depolarized mitochondria. However, some parenchymal cells exhibited diffuse and dim rhodamine 123 staining but did not label with PI, indicating mitochondrial depolarization before onset of cell death (Fig. 7B, white arrows).
Fig. 7.
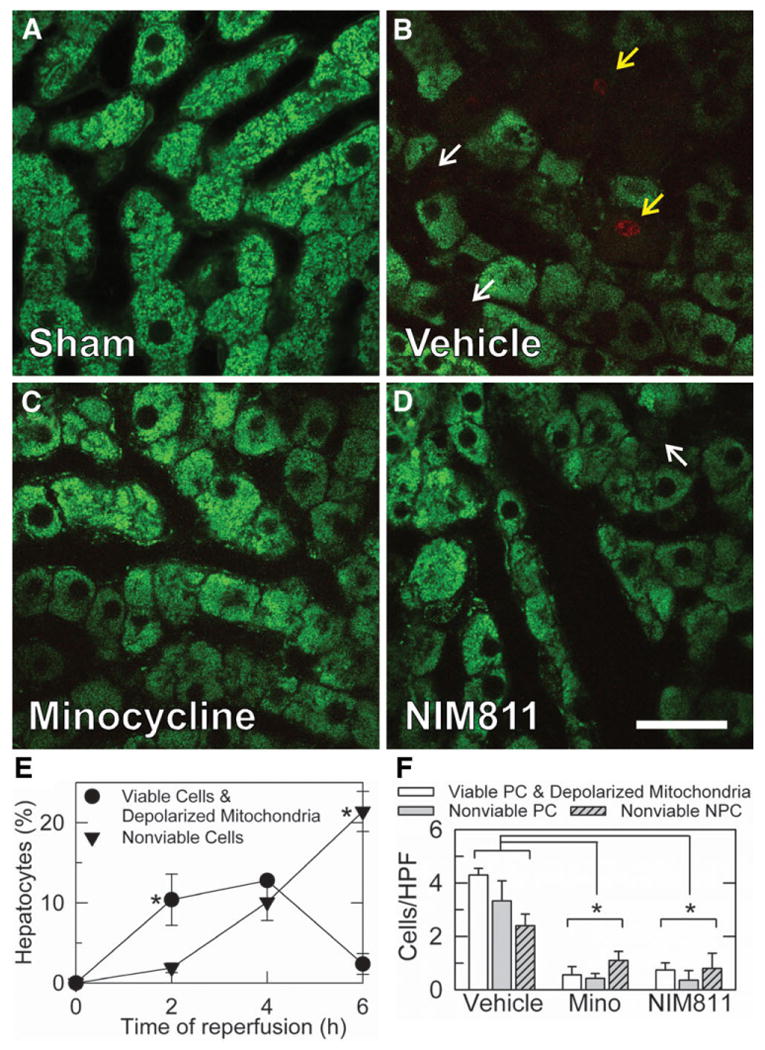
Minocycline and NIM811 decrease mitochondrial depolarization and cell death after rat liver transplantation. Donor livers and graft recipients were treated with vehicle, minocycline, or NIM811, as described in Fig. 1. At 4 hours postoperatively, liver grafts were visualized by intravital multiphoton microscopy of rhodamine 123 and PI fluorescence. Shown are representative overlay images of green rhodamine 123 and red PI fluorescence collected from livers after sham operation (A) and liver grafts after vehicle (B), minocycline (C), and NIM811 (D) treatment. Punctate staining of rhodamine 123 denoted polarization of individual mitochondria, whereas dim diffuse cellular staining indicated mitochondrial depolarization (white arrows). PI nuclear staining signified loss of cell viability (yellow arrows). (E) Quantification of the time course of mitochondrial depolarization and cell death after liver transplantation as a percentage of hepatocytes using intravital spinning disk confocal microscopy performed after sham operation (0 hours) and at 2-6 hours postoperatively. Individual group sizes were 3-4 rats per time-point. *P < 0.05 versus other group. (F) Quantification of numbers per HPF of viable parenchymal cells (PC) with depolarized mitochondria, nonviable PC, and nonviable nonparenchymal cells (NPC) at 4 hours after transplantation from intravital multiphoton microscopy. Size of individual groups was 5. Bar is 30 μm. *P < 0.05.
In order to assess the time course of mitochondrial dysfunction and cell death in vivo after reperfusion, intravital microscopy was performed at different times after transplantation. For these experiments we used intravital spinning disk confocal microscopy of rhodamine 123 and PI at 0 hours (sham), 2 hours, 4 hours, and 6 hours after liver transplantation. Quantification was performed for each liver, and hepatocytes were scored for PI and rhodamine 123 staining (Fig. 7E). In sham-operated livers, virtually no hepatocytes contained depolarized mitochondria, and none were nonviable by PI labeling. At 2 hours after transplantation, 10.4% ± 3.2% of hepatocytes contained depolarized mitochondria but were still viable (excluded PI), whereas 1.9% ± 0.6% of hepatocytes were both depolarized and nonviable (PI positive) (Fig. 7E). At 4 hours, 12.8% ± 0.6% of hepatocytes contained depolarized mitochondria but were viable, and another 10.1% ± 2.3% were nonviable (and depolarized). At 6 hours, viable cells with depolarized mitochondria were rare (2.4% ± 1.3%), and another 21.4% ± 2.5% were nonviable (Fig. 7E). These results illustrate a progression of mitochondrial depolarization followed by cell death.
After transplantation with minocycline treatment, fewer hepatocytes were seen with depolarized mitochondria (Fig. 7C). Most hepatocytes demonstrated bright, punctate staining by rhodamine 123, indicative of their content of polarized mitochondria. PI labeling in both hepatocytes and nonparenchymal cells also decreased after minocycline treatment. Overlays again showed that all nonviable PI-labeled cells had diffuse or absent rhodamine 123 staining, signifying mitochondrial depolarization, but not all cells with depolarized mitochondria were labeled with PI. NIM811 treatment also decreased mitochondrial depolarization and loss of viability of parenchymal and nonparenchymal cells to a similar extent as minocycline treatment (Fig. 7D). By contrast, mitochondrial depolarization and loss of cell viability after tetracycline treatment were indistinguishable from vehicle-treated liver grafts (data not shown).
At 4 hours after transplantation, cells were scored and counted for PI and rhodamine 123 staining for each liver (Fig. 7F). PI-labeled nonviable cells were identified as parenchymal or nonparenchymal cells on the basis of nuclear morphology and location adjacent to sinusoids. In sham-operated livers, 0.05 ± 0.002 hepatocytes/HPF contained depolarized mitochondria, but no parenchymal or nonparenchymal cells were nonviable by the criterion of PI labeling. After transplantation with vehicle treatment, 4.3 ± 0.3 hepatocytes/HPF contained depolarized mitochondria but were still viable, whereas nonviable hepatocytes and nonparenchymal cells were 3.3 ± 0.8 and 2.4 ± 0.4 cells/HPF, respectively (Fig. 7F). After transplantation with minocycline treatment, viable hepatocytes with depolarized mitochondria decreased to 0.6 ± 0.3 hepatocytes/HPF (P< 0.05 versus vehicle, Fig. 7F), and nonviable parenchymal and nonparenchymal cells decreased to 0.4 ± 0.2 and 1.1 ± 0.3 cells/HPF, respectively (P< 0.05 versus vehicle, Fig. 7F). After transplantation with NIM811 treatment, viable hepatocytes with depolarized mitochondria also decreased to 0.7 ± 0.3 cells/HPF (P< 0.05 versus vehicle, Fig. 7F), and nonviable parenchymal and nonparenchymal cells decreased to 0.4 ± 0.4 and 0.8 ± 0.6 cells/HPF, respectively (P< 0.05 versus vehicle, Fig. 7F). Thus, minocycline and NIM811 conferred similar protection.
Discussion
Damage to liver caused by ischemia/reperfusion can be a limiting factor in clinical settings of liver surgery and transplantation. The aim of this study was to evaluate the hypothesis that mitochondrial dysfunction is a key event in liver graft injury. Specifically, we tested the hypothesis that minocycline, a tetracycline previously shown to have cytoprotective properties, decreases storage/reperfusion injury after rat liver transplantation by inhibition of the MPT. Additionally, we investigated the effects of NIM811, a nonimmunosuppressive CsA derivative that blocks the MPT. Our findings showed that minocycline and NIM811 decreased necrosis and apoptosis in liver grafts after rat liver transplantation and improved graft survival to a similar extent. Protection by both minocycline and NIM811 appeared to be attributable to blockade of the MPT and prevention of mitochondrial depolarization, a consequence of the MPT.
In addition to antimicrobial actions of minocycline, previous studies demonstrated its cytoprotective potential in a variety of settings, including ischemic cardiac, renal, and central nervous system disease.10-18 Relevant to our study, improved sinusoidal cell lining viability during cold storage of rat liver grafts was reported previously after minocycline pretreatment, an effect attributed to suppression of angiogenic factor production by Kupffer cells that may cause sinusoidal endothelial cell sloughing and detachment during preservation.12 However, the effect of minocycline on graft function and survival after transplantation has not previously been determined. Here, our results demonstrated a more than 2-fold increase in 30-day graft survival after liver transplantation with perioperative minocycline treatment (Fig. 4). Similarly, a recent study showed that minocycline protects against cell death and improves survival in a mouse model of Fas ligation-mediated fulminant liver failure.10 Importantly, minocycline administered up to 30 minutes after anti-Fas antibody treatment delays mortality kinetics and rescues mice from death. After Fas ligation, cell death is predominantly apoptotic through a mitochondrial pathway.25 Thus, protection implies that minocycline suppresses death receptor–mediated and mitochondria-dependent apoptotic signaling, which previous studies show to involve the MPT.26,27
In our model, minocycline decreased both necrosis and apoptosis. Necrosis assessed by ALT and histology decreased by half at 6 hours after transplantation with minocycline treatment (Figs. 1, 2). In hepatocytes and nonparenchymal cells, apoptosis assessed by TUNEL decreased 71% and 45%, respectively, whereas caspase-3 activity in whole liver extracts decreased 69% (Fig. 3). NIM811 afforded similar protection with regard to parenchymal TUNEL-positive cells (Fig. 3E), but only nonsignificant trends toward protection were seen with regard to nonparenchymal cells (Fig. 3F, G). Necrosis represented the predominant mode of cell death in our studies, because after liver transplantation with vehicle treatment, necrosis involved 21% of the parenchyma, whereas TUNEL occurred in only 2.7% of hepatocytes. These findings support earlier assessments that apoptosis contributes much less to graft storage/reperfusion injury than necrosis.3,28 However, both modes of cell death can occur through a mitochondrial pathway involving the MPT, a phenomenon of necrapoptosis.5. Because minocycline and NIM811 protected against both necrosis and apoptosis, graft injury and cell death after storage/reperfusion would appear to be largely a necrapoptotic phenomenon.
Minocycline and NIM811 protected against cell death to both nonparenchymal and parenchymal cells after liver transplantation. In agreement with earlier studies,2,9,29 intravital microscopy and histology suggested that endothelial cell killing occurring at early time points after reperfusion led to parenchymal cell damage at later time points (data not shown). Indeed, later injury to hepatocytes was more strongly protected than the earlier injury to sinusoidal cells (Fig. 7F).
Although NIM811 is a well-established inhibitor of the MPT, the mechanism of minocycline inhibition has been unclear. Our findings here indicate that minocycline cytoprotection like that of NIM811 was mediated by suppression of the MPT. The MPT causes mitochondrial depolarization with consequent uncoupling of oxidative phosphorylation, mitochondrial swelling, and cell death by necrosis and apoptosis. Intravital confocal imaging showed directly mitochondria depolarization after liver transplantation after a time of cold ischemic storage leading to graft injury and failure (Fig. 7). This depolarization signifying the MPT preceded onset of necrotic cell death. Minocycline like NIM811 substantially prevented mitochondrial depolarization, and in isolated liver mitochondria minocycline blocked onset of CsA-sensitive mitochondrial swelling induced by Ca2+ (Fig. 5). Tetracycline, which did not decrease graft failure and ALT release after transplantation, did not block MPT onset in isolated mitochondria and did not prevent mitochondrial depolarization assessed by intravital confocal microscopy after liver transplantation. Hence, protection by minocycline seems most likely attributable to inhibition of the MPT.
Although NIM811 and minocycline both blocked the MPT, the mechanism of blockade by the 2 agents was different. NIM811 has been shown previously to block the MPT through its interaction with cyclophilin D.30 By contrast, we show here that minocycline blocked the MPT by inhibiting mitochondrial calcium uptake as monitored by the calcium indicator Fluo-5N (Fig. 6). Calcium uptake into the mitochondrial matrix space is a necessary step for the Ca2+-induced MPT, and calcium uptake inhibitors such as ruthenium red potently block the MPT.31,32 Simultaneous treatment with minocycline and NIM811 did not confer additional protection against the MPT in isolated mitochondria beyond either agent alone (data not shown). Respiratory measurements in mitochondria showed that inhibition of mitochondrial Ca2+ uptake by minocycline was not attributable to respiratory inhibition or uncoupling, because ADP-stimulated state 3 respiration and ADP/O and Ca2+/O ratios were not changed with as much as 50 μM minocycline (Fig. 6C). Rather, minocycline specifically inhibited Ca2+-stimulated state 3 respiration consistent with inhibition of calcium uptake. Recently, high doses (75-150 μM) of minocycline were reported to stimulate state 4 respiration and inhibit ADP-stimulated state 3 respiration in rat brain mitochondria.32 These findings are consistent with our observation of a decreased ADP/O ratio at 100 μM minocycline (Fig. 6C) and with another report of complex II/III activity inhibition at the same dose.18,31
In rat liver transplantation, we observed best protection when both explants and recipient animals were treated with minocycline (Figs. 1-4 and data not shown). Preliminary experiments indicated that explant treatment with 18 μM minocycline yielded the greatest protection and that no protection was observed at higher concentrations of minocycline (data not shown). These observations are somewhat in contrast with our measurements in isolated rat liver mitochondria in which 18 μM minocycline partially blocked the MPT, whereas 50 μM protected nearly completely (Fig. 5). Still higher concentrations of minocycline caused partial uncoupling and respiratory inhibition in isolated mitochondria (Fig. 6C). Minocycline is a semisynthetic tetracycline analog with enhanced lipophilicity and tissue penetration.33 Such properties may promote tissue uptake and serve to concentrate minocycline intracellularly, which might explain the increased efficacy of minocycline in vivo compared with isolated mitochondria in vitro.
Although previous studies implicated the MPT in warm ischemia/reperfusion injury, involvement of the MPT in graft dysfunction and failure after cold organ preservation has not been previously shown. After warm ischemia/reperfusion, specific MPT blockers, such as CsA, NIM811, and sanglifehrin A, protect against injury.24,31,34-38 In various models, CsA protects against both apoptosis and necrosis.36 However, the toxic and immunosuppressive effects of CsA limit its usefulness in a clinical setting of ischemia/reperfusion. Even in a transplantation setting, CsA is being supplanted by newer less toxic immunosuppressants.39 In this regard, NIM811, a nonimmunosuppressive derivative of CsA that inhibits MPT pore opening,24 may be a more useful cytoprotective agent than CsA. In addition to protecting against storage/reperfusion injury (Figs. 1-4), NIM811 attenuates myocardial apoptosis and necrosis when used at reperfusion after acute myocardial ischemia34 and also decreases reperfusion injury after small-for-size rat liver transplantation.40
The danger of bacterial infection necessitates prophylactic antibiotic use in most gastrointestinal surgery. To decrease the incidence of posttransplant infection, many centers supplement parenteral broad-spectrum antibiotics with regimens of oral antimicrobial agents.41 Antibiotics, including penicillin, streptomycin, and others, are also routinely added to the University of Wisconsin storage solution.42 Thus, treatment with minocycline of stored livers and graft recipients could be consistent with current clinical practice and have the additional benefit of decreasing storage/reperfusion injury. Future studies will be needed to determine what benefit, if any, minocycline and other MPT inhibitors might have in human clinical liver transplantation.
Acknowledgments
Supported in part by Grants DK37034, and C06 RR015455 from the National Institutes of Health and Grant DFG TH1328/1-1 from the Deutsche Forschungsgemeinschaft. Dr. Theruvath is the recipient of an American Liver Foundation Postdoctoral Research Fellowship Award.
Abbreviations
- ADP
adenosine diphosphate
- ALT
alanine aminotransferase
- ATP
adenosine triphosphate
- CsA
cyclosporin A
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPF
high power field
- MPT
mitochondrial permeability transition
- NIM811
N-methyl-4-isoleucine cyclosporin
- PI
propidium iodide
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
Footnotes
Presented in part at the Digestive Disease Week conference, Chicago, Illinois, May 14-19, 2005.
Potential conflict of interest: Nothing to report.
References
- 1.Caldwell-Kenkel JC, Currin RT, Tanaka Y, Thurman RG, Lemasters JJ. Kupffer cell activation and endothelial cell damage after storage of rat livers: effects of reperfusion. Hepatology. 1991;13:83–95. [PubMed] [Google Scholar]
- 2.Takei Y, Marzi I, Gao WS, Gores GJ, Lemasters JJ, Thurman RG. Leukocyte adhesion and cell-death following orthotopic liver-transplantation in the rat. Transplantation. 1991;51:959–965. doi: 10.1097/00007890-199105000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 4.Rudiger HA, Graf R, Clavien PA. Liver ischemia: apoptosis as a central mechanism of injury. J Invest Surg. 2003;16:149–159. [PubMed] [Google Scholar]
- 5.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304:463–470. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 6.Halestrap AP. The mitochondrial permeability transition: its molecular mechanism and role in reperfusion injury. Biochem Soc Symp. 1999;66:181–203. doi: 10.1042/bss0660181. [DOI] [PubMed] [Google Scholar]
- 7.Di Lisa F, Canton M, Menabo R, Dodoni G, Bernardi P. Mitochondria and reperfusion injury: the role of permeability transition. Basic Res Cardiol. 2003;98:235–241. doi: 10.1007/s00395-003-0415-x. [DOI] [PubMed] [Google Scholar]
- 8.Currin RT, Caldwell-Kenkel JC, Lichtman SN, Bachmann S, Takei Y, Kawano S, et al. Protection by Carolina rinse solution, acidotic pH, and glycine against lethal reperfusion injury to sinusoidal endothelial cells of rat livers stored for transplantation. Transplantation. 1996;62:1549–1558. doi: 10.1097/00007890-199612150-00004. [DOI] [PubMed] [Google Scholar]
- 9.Huet PM, Nagaoka MR, Desbiens G, Tarrab E, Brault A, Bralet MP, et al. Sinusoidal endothelial cell and hepatocyte death following cold ischemia-warm reperfusion of the rat liver. Hepatology. 2004;39:1110–1119. doi: 10.1002/hep.20157. [DOI] [PubMed] [Google Scholar]
- 10.Chu HC, Lin YL, Sytwu HK, Lin SH, Liao CL, Chao YC. Effects of minocycline on Fas-mediated fulminant hepatitis in mice. Br J Pharmacol. 2005;144:275–282. doi: 10.1038/sj.bjp.0706079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedlander RM. Mechanisms of disease: apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 12.Gao W, Washington MK, Bentley RC, Clavien PA. Antiangiogenic agents protect liver sinusoidal lining cells from cold preservation injury in rat liver transplantation. Gastroenterology. 1997;113:1692–1700. doi: 10.1053/gast.1997.v113.pm9352874. [DOI] [PubMed] [Google Scholar]
- 13.Kelly KJ, Sutton TA, Weathered N, Ray N, Caldwell EJ, Plotkin Z, et al. Minocycline inhibits apoptosis and inflammation in a rat model of ischemic renal injury. Am J Physiol Renal Physiol. 2004;287:F760–F766. doi: 10.1152/ajprenal.00050.2004. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Wei Q, Wang CY, Hill WD, Hess DC, Dong Z. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004;279:19948–19954. doi: 10.1074/jbc.M313629200. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Zhu S, Drozda M, Zhang WH, Stavrovskaya IG, Cattaneo E, et al. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:10483–10487. doi: 10.1073/pnas.1832501100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wells JEA, Hurlbert RJ, Fehlings MG, Yong VW. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126:1628–1637. doi: 10.1093/brain/awg178. [DOI] [PubMed] [Google Scholar]
- 17.Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3:744–751. doi: 10.1016/S1474-4422(04)00937-8. [DOI] [PubMed] [Google Scholar]
- 18.Zhu S, Stavrovskaya IG, Drozda M, Kim BYS, Ona V, Li MW, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 19.Steffen R, Ferguson DM, Krom RA. A new method for orthotopic rat liver transplantation with arterial cuff anastomosis to the recipient common hepatic artery. Transplantation. 1989;48:166–168. doi: 10.1097/00007890-198907000-00044. [DOI] [PubMed] [Google Scholar]
- 20.Gao W, Lemasters JJ, Thurman RG. Development of a new method for hepatic rearterialization in rat orthotopic liver transplantation: reduction of liver injury and improvement of surgical outcome by arterialization. Transplantation. 1993;56:19–24. doi: 10.1097/00007890-199307000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Lemasters JJ. The ATP-to-oxygen stoichiometries of oxidative phosphorylation by rat liver mitochondria: an analysis of ADP-induced oxygen jumps by linear nonequilibrium thermodynamics. J Biol Chem. 1984;259:13123–13130. [PubMed] [Google Scholar]
- 22.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- 23.Theruvath TP, Zhong Z, Currin RT, Ramshesh VK, Lemasters JJ. Endothelial nitric oxide synthase protects transplanted mouse livers against storage/reperfusion injury: role of vasodilatory and innate immunity pathways. Transplant Proc. 2006;38:3351–3357. doi: 10.1016/j.transproceed.2006.10.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- 25.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, et al. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806–809. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- 26.Hatano E, Bradham CA, Stark A, Iimuro Y, Lemasters JJ, Brenner DA. The mitochondrial permeability transition augments Fas-induced apoptosis in mouse hepatocytes. J Biol Chem. 2000;275:11814–11823. doi: 10.1074/jbc.275.16.11814. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Y, Ding WX, Qian T, Watkins S, Lemasters JJ, Yin XM. Bid activates multiple mitochondrial apoptotic mechanisms in primary hepatocytes after death receptor engagement. Gastroenterology. 2003;125:854–867. doi: 10.1016/s0016-5085(03)01066-7. [DOI] [PubMed] [Google Scholar]
- 28.Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: apoptosis or necrosis? Hepatology. 2001;33:397–405. doi: 10.1053/jhep.2001.22002. [DOI] [PubMed] [Google Scholar]
- 29.Lemasters JJ, Thurman RG. Reperfusion injury after liver preservation for transplantation. Annu Rev Pharmacol Toxicol. 1997;37:327–338. doi: 10.1146/annurev.pharmtox.37.1.327. [DOI] [PubMed] [Google Scholar]
- 30.Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem. 2003;10:1485–1506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez-Gomez FJ, Galindo MF, Gomez-Lazaro M, Gonzalez-Garcia C, Cena V, Aquirre N, et al. Involvement of mitochondrial potential and calcium buffering capacity in minocycline cytoprotective actions. Neuroscience. 2005;133:959–967. doi: 10.1016/j.neuroscience.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 32.Mansson R, Hansson MJ, Morota S, Uchino H, Ekdahl CT, Elmer E. Re-evaluation of mitochondrial permeability transition as a primary neuroprotective target of minocycline. Neurobiol Dis. 2007;25:198–205. doi: 10.1016/j.nbd.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Jonas M, Cunha BA. Minocycline. Ther Drug Monit. 1982;4:137–145. [PubMed] [Google Scholar]
- 34.Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J, Robert D, et al. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol. 2005;38:367–374. doi: 10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 35.Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549:513–524. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. 2003;124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury: modulating effects of FK506 and cyclosporine. Transplantation. 1993;55:1265–1272. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 38.Qian T, Herman B, Lemasters JJ. The mitochondrial permeability transition mediates both necrotic and apoptotic death of hepatocytes exposed to Br-A23187. Toxicol Appl Pharmacol. 1999;154:117–125. doi: 10.1006/taap.1998.8580. [DOI] [PubMed] [Google Scholar]
- 39.Fung J, Kelly D, Kadry Z, Patel-Tom K, Eghtesad B. Immunosuppression in liver transplantation: beyond calcineurin inhibitors. Liver Transpl. 2005;11:267–280. doi: 10.1002/lt.20373. [DOI] [PubMed] [Google Scholar]
- 40.Zhong Z, Theruvath TP, Currin RT, Waldmeier PC, Lemasters JJ. NIM811, a mitochondrial permeability transition inhibitor, prevents mitochondrial depolarization in small-for-size rat liver grafts. Am J Transplant. 2007;7:1103–1111. doi: 10.1111/j.1600-6143.2007.01770.x. [DOI] [PubMed] [Google Scholar]
- 41.Wiesner RH. Selective decontamination for infection prophylaxis in liver transplantation patients. Transplant Proc. 1991;23:1927–1928. [PubMed] [Google Scholar]
- 42.Belzer FO, Southard JH. Principles of solid-organ preservation by cold storage. Transplantation. 1988;45:673–676. doi: 10.1097/00007890-198804000-00001. [DOI] [PubMed] [Google Scholar]