

Abstract
Endocannabinoid (eCB) signaling mediates short-term and long-term synaptic depression (LTD) in many brain areas. In the ventral tegmental area (VTA) and striatum, D2 dopamine receptors cooperate with group I metabotropic glutamate receptors (mGluRs) to induce eCB-mediated LTD of glutamatergic excitatory and GABAergic inhibitory (I-LTD) synaptic transmission. Because D2 receptors and group I mGluR agonists are capable of inducing the release of eCBs, the predominant hypothesis is that the cooperation between these receptors to induce eCB-mediated synaptic depression results from the combined activation of type I cannabinoid (CB1) receptors by the eCBs. By determining the downstream effectors for D2 receptor and group I mGluR activation in VTA dopamine neurons, we show that group I mGluR activation contributes to I-LTD induction by enhancing eCB release and CB1 receptor activation. However, D2 receptor activation does not enhance CB1 receptor activation, but facilitates I-LTD induction via direct inhibition of cAMP-dependent protein kinase A (PKA) signaling. We further demonstrate that cAMP/PKA signaling pathway is the downstream effector for CB1 receptors and is required for eCB-mediated I-LTD induction. Our results suggest that D2 receptors and CB1 receptors target the same downstream effector cAMP/PKA signaling pathway to induce I-LTD and D2 receptor activation facilitates eCB-mediated I-LTD in dopamine neurons not by enhancing CB1 receptor activation, but by enhancing its downstream effects.
Keywords: endocannabinoid, long-term depression, synaptic plasticity, GABA, cAMP/PKA, dopamine
Introduction
Endocannabinoids (eCBs) are a new class of signaling molecules that mediate short-term and long-term synaptic depression (LTD) in many brain areas (Gerdeman and Lovinger, 2003; Alger, 2005; Chevaleyre et al., 2006). Unlike traditional neurotransmitters that are stored in synaptic vesicles, eCBs are produced and released “on demand” (Di Marzo et al., 1994). eCBs are released by depolarization-induced Ca2+ influx (Di Marzo et al., 1994, 1998; Stella et al., 1997) or the activation of certain G-protein-coupled receptors (GPCRs), such as group I metabotropic glutamate receptors (mGluRs) (Maejima et al., 2001; Jung et al., 2005) and D2 dopamine receptors (Giuffrida et al., 1999). In response to these stimuli, eCBs are released from postsynaptic neurons and travel across the synaptic cleft to activate type I cannabinoid receptors (CB1) on presynaptic terminals, resulting in retrograde depression of synaptic transmission.
We have shown that repetitive synaptic activation of D2 dopamine receptors and group I mGluRs induces eCB-mediated LTD of inhibitory transmission (I-LTD) in dopamine neurons of the ventral tegmental area (VTA) (Pan et al., 2008). Similar cooperation between D2 receptors and group I mGluRs to induce eCB-mediated excitatory synaptic depression has been described in the striatum (Kreitzer and Malenka, 2005; Yin and Lovinger, 2006). N-Arachidonylethanolamine (AEA) and 2-arachidonoylglycerol (2-AG) are two major eCBs that activate CB1 receptors in the brain (Di Marzo et al., 1998; Piomelli, 2003). Because D2 receptor agonists increase AEA production (Giuffrida et al., 1999; Patel et al., 2003; Centonze et al., 2004) and group I mGluR agonists increase 2-AG production (Jung et al., 2005), it has been hypothesized that the cooperation between D2 receptors and group I mGluRs to induce eCB-mediated synaptic depression results from the combined action of these two eCBs (Kreitzer and Malenka, 2005; Yin and Lovinger, 2006; Pan et al., 2008).
Both D2 receptors and CB1 receptors are Gi/o-protein-coupled receptors whose activation leads to the inhibition of adenylyl cyclase (AC), resulting in decreased cAMP accumulation and protein kinase A (PKA) activity (Piomelli, 2003; Neve et al., 2004). Recent studies have shown that the cAMP/PKA signaling pathway mediates eCB-mediated I-LTD in the hippocampus (Chevaleyre et al., 2007) and LTD in the striatum (Mato et al., 2007). These studies raise the possibility that D2 receptor activation facilitates eCB/CB1-receptor-mediated synaptic depression through direct inhibition of cAMP/PKA signaling. In this study, we distinguish these two possibilities by determining the downstream effectors that mediate group I mGluR- and D2 receptor-induced synaptic depression in VTA dopamine neurons. We show that group I mGluR activation induces synaptic depression via eCB/CB1-mediated inhibition of cAMP/PKA signaling, whereas D2 receptor activation induces synaptic depression via direct inhibition of cAMP/PKA signaling. Thus, D2 receptor activation facilitates I-LTD induction not by enhancing CB1 receptor activation, but by enhancing its downstream effects. Our study reveals a previously unrecognized mechanism for D2 receptor-induced facilitation of eCB-mediated synaptic depression.
Materials and Methods
Slice preparation.
Midbrain slices (250 μm) from male Sprague Dawley rats (P18–25), CB1-knock-out (CB1−/−) mice and wild-type (CB1+/+) mice of either sex (P16–20) were prepared as described previously (Pan et al., 2008). These mice were generated on a genetic background 129/SvJ (Ibrahim et al., 2003), and back-crossed unto an outbred ICR strain for at least nine generations. Slices were prepared at 4−6°C in a solution containing (in mm): 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgSO4, 26 NaHCO3, 25 glucose, 11.6 sodium ascorbate, and 3.1 sodium pyruvate. The slices were incubated in artificial CSF (ACSF) containing (in mm): 125 NaCl, 3 KCl, 2.5 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. The ACSF was saturated with 95% O2 and 5% CO2. Slices were recovered for at least 1 h at room temperature before recordings. All recordings were performed at 32 ± 1°C by using an automatic temperature controller (Warner Instrument).
Electrophysiology.
Whole-cell recordings were made from VTA dopamine neurons in the midbrain slices (Pan et al., 2008). Dopamine neurons were identified by the presence of large Ih currents, rhythmic firing at low frequency and prominent afterhyperpolarization (Johnson and North, 1992; Jones and Kauer, 1999; Liu et al., 2005). A recent study has clearly demonstrated that these electrophysiological characteristics do not exclusively belong to dopamine neurons (Margolis et al., 2006). Putative dopamine neurons in our study may contain a small number of nondopamine neurons, which should be randomly distributed in different experimental groups. IPSCs were evoked by a bipolar tungsten stimulation electrode (WPI) that was placed ∼150 μm rostral to the recorded neuron. For recording of evoked IPSCs, glass pipette was filled with a solution containing (in mm): K-gluconate 100, KCl 50, HEPES 10, EGTA 0.2, MgCl2 2, MgATP 4, Na2GTP 0.3, and Na2-phosphocreatine 10 at pH 7.2 (with KOH). In experiments with postsynaptic Ca2+ buffer, 10 mm K-gluconate was replaced by 10 mm EGTA (see Fig. 3F). Cs+-based intracellular solution used in Figure 2D consists of the following (in mm): Cs-methanesulfonate 100, CsCl 50, HEPES 10, EGTA 0.2, MgCl2 2, MgATP 4, Na2GTP 0.3, and Na2-phosphocreatine 10 at pH 7.2 (with CsOH). For recording of miniature IPSCs (mIPSCs), K-gluconate was replaced by KCl in internal solution and tetrodotoxin (TTX) was added in the ACSF to block action potentials. All recordings were made in the presence of glutamate receptor antagonists 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 20 μm) and d-2-amino-5-phosphonovaleric acid (d-AP-5, 50 μm). Neurons were voltage-clamped at −70 mV unless stated otherwise. Series resistance (15–30 MΩ) was monitored throughout the recordings and data were discarded if the resistance changed by >20%. Tetrahydrolipstatin (THL) and CNQX were obtained from Sigma-Aldrich; guanosine-5′-O-(2-thiodiphosphate) 3Li (GDP-β-S) was from Biomol, EGTA-AM was from Anaspec and all other drugs were from Tocris Bioscience.
Figure 3.
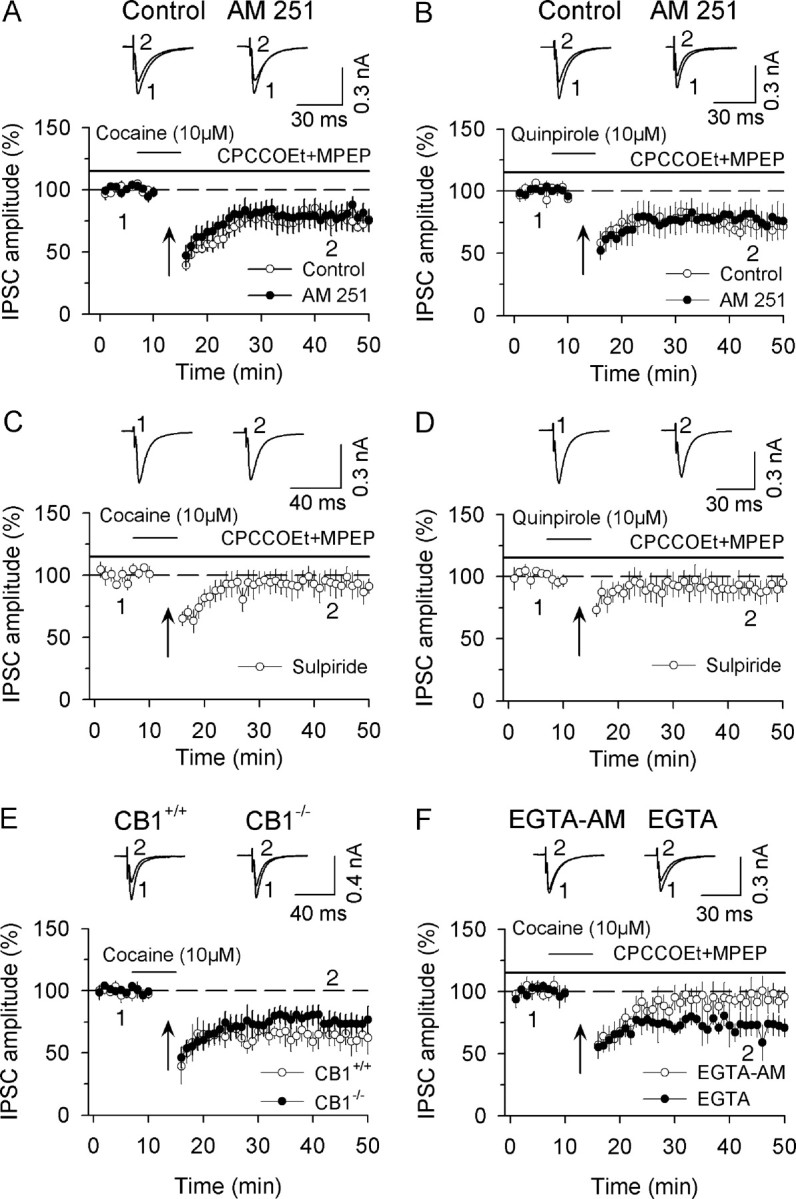
CB1 receptor activation is not responsible for D2-receptor-induced facilitation of I-LTD induction. A, In the presence of group I mGluR antagonists CPCCOEt (50 μm) and MPEP (10 μm), the combination of application of higher concentration of cocaine (10 μm) with the 10 Hz stimulation induced I-LTD (n = 7). CB1 receptor antagonist AM 251 (2 μm) had no significant effect on I-LTD induction (n = 8). B, Same as in A except D2 receptor agonist quinpirole (10 μm), instead of cocaine, was used (n = 6). AM 251 had no significant effect on I-LTD induced in the presence of quinpirole (n = 6). C, D, D2 receptor antagonist sulpiride (10 μm) blocked I-LTD that was induced in the presence of cocaine (C) or quinpirole (D) (n = 5 for each group). E, A combination of the 10 Hz stimulation with cocaine application (10 μm) induced I-LTD in dopamine neurons in slices prepared from CB1+/+ mice (n = 5) and CB1−/− mice (n = 5). F, Bath application of the membrane-permeable Ca2+ chelator EGTA-AM (100 μm) blocked I-LTD (n = 5) whereas intracellular loading of membrane-impermeable Ca2+ chelator EGTA (10 mm) into dopamine neurons did not have significant effect on I-LTD (n = 5).
Figure 2.

CB1 receptor activation is not responsible for D2 receptor-induced acute depression of IPSCs. A, Bath application of cocaine (10 μm) produced reversible depression of IPSCs (n = 5). In the presence of CB1 receptor antagonist AM 251 (2 μm), cocaine produced similar depression of IPSCs (n = 5). B, AM 251 had no significant effect on D2 receptor agonist quinpirole-induced depression of IPSCs (n = 5). C, Bath application of quinpirole induced similar depression of IPSCs in dopamine neurons in slices from CB1+/+ mice (n = 7) and CB1−/− mice (n = 5). D, Blocking postsynaptic K+ conductance with either Cs+-based internal solution (n = 6) or K+-based internal solution containing GDP-β-S (2 mm, n = 7) had no significant effect on quinpirole-induced depression of IPSCs. E, The effect of bath application of quinpirole (10 μm) on mIPSCs in dopamine neurons. F, Quinpirole decreased the mean frequency of mIPSCs (n = 6; p < 0.05). G, Quinpirole did not significantly shift the amplitude distribution of mIPSCs as shown by cumulative probability plots.
Statistics.
Data are presented as the mean ± SEM. I-LTD (%) was calculated as follows: 100 × [mean amplitude of IPSCs during the final 10 min of recording/mean amplitude of baseline IPSCs]. The acute depression of evoked IPSCs (%) was calculated as follows: 100 × [mean amplitude of 30 IPSCs after drug treatment/mean amplitude of baseline IPSCs]. Data sets were compared with either paired or unpaired Student's t test, or ANOVA followed by Tukey's post hoc analysis. Results were considered to be significant at p < 0.05.
Results
D2 receptors cooperate with group I mGluRs to induce eCB-mediated I-LTD in dopamine neurons
We previously reported that synaptic activation of group I mGluRs and D2 receptors leads to eCB-mediated I-LTD in dopamine neurons and that the presence of D2 receptor agonist quinpirole or dopamine uptake inhibitor cocaine during the repetitive synaptic stimulation is required for I-LTD induction (Pan et al., 2008). To systematically investigate the mechanism for the cooperation between group I mGluRs and D2 receptors to induce I-LTD, we began by corroborating and extending our previous observations. Consistent with our previous study (Pan et al., 2008), we found that neither the application of a low concentration of cocaine (3 μm) nor 10 Hz stimulation for 5 min significantly affected IPSCs (cocaine, 99.2 ± 6.9% of baseline, n = 5, p > 0.05; 10 Hz stimulation, 95.5 ± 6.1% of baseline, n = 6, p > 0.05). However, the combination of cocaine and 10 Hz stimulation induced I-LTD (68.7 ± 5.5% of baseline, n = 8, p < 0.01, Fig. 1A) (see Materials and Methods for the quantification of I-LTD magnitude).
Figure 1.

D2-like receptors cooperate with group I mGluRs to induce eCB-mediated I-LTD in VTA dopamine neurons. A, The presence of cocaine (3 μm) during the 10 Hz stimulation induced I-LTD in dopamine neurons (n = 8). The horizontal bar denotes the time of cocaine perfusion, and the arrows indicate the application of the 10 Hz stimulation. Sample IPSCs averaged from 20 consecutive trials before and after I-LTD induction are shown on the top. This I-LTD was not affected by D1-like antagonist SCH 23390 (10 μm, n = 6) but was blocked by D2-like antagonist sulpiride (10 μm, n = 7). All antagonists were present throughout the experiments. B, I-LTD was attenuated by selective D2 receptor antagonist L-741,626 (100 nm, n = 8) or selective D3 receptor antagonist GR 103691 (10 nm, n = 7), but was unaffected by selective D4 receptor antagonist L-745,870 (100 nm, n = 5). C, I-LTD was blocked by a combination of group I mGluR antagonists CPCCOEt (50 μm) and MPEP (10 μm, n = 7) or by CB1 receptor antagonist AM 251 (2 μm, n = 7). D, The presence of quinpirole (1 μm) during the 10 Hz stimulation enabled I-LTD induction (n = 9). This I-LTD was blocked by D2-like antagonist sulpiride (10 μm, n = 6) but was unaffected by D1-like antagonist SCH 23390 (10 μm, n = 5). E, I-LTD was blocked by the combination of group I mGluR antagonists CPCCOEt (50 μm) and MPEP (10 μm, n = 7). F, I-LTD was blocked by CB1 receptor antagonist AM 251 (2 μm, n = 6).
Cocaine at 3 μm is effective in inhibiting dopamine uptake in the VTA (Beckstead et al., 2004), whereas the repetitive synaptic stimulation (10 Hz, 5 min) likely activates dopaminergic and glutamatergic afferents. Indeed, it has been shown that neither group I mGluRs nor D2 dopamine receptors are activated by single synaptic stimulation but rather by repetitive synaptic stimulation (Batchelor and Garthwaite, 1997; Beckstead et al., 2004). Dopamine receptors consist of two subfamilies: D1-like (D1, D5) and D2-like (D2, D3, and D4) receptors (Neve et al., 2004; Pollack, 2004). For simplicity, we often use D1 and D2 receptors to represent D1- and D2-like receptors, respectively. We determined which subtypes of dopamine receptors mediate the induction of I-LTD.
Consistent with our previous study (Pan et al., 2008), we found that the I-LTD induced by the combination of 10 Hz stimulation with cocaine application was not affected by D1 receptor antagonist (R)-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH 23390) at 1 μm (72.3 ± 7.8 of baseline, n = 5, p > 0.05 vs I-LTD control) or at 10 μm (70.8 ± 8.2% of baseline, n = 6, p > 0.05 vs I-LTD control) (Fig. 1A). However, I-LTD was blocked by the D2 receptor antagonist sulpiride (10 μm, 93.2 ± 6.3% of baseline, n = 7, p < 0.05 vs I-LTD control; Fig. 1A). D1 receptor agonists selectively enhance GABAB-receptor-mediated IPSPs but have no significant effect on GABAA-receptor-mediated IPSPs in VTA dopamine neurons (Cameron and Williams, 1993). Consistent with the latter observation, we found that bath application of D1 receptor agonist SKF 38393 (10 μm) had no significant effect on IPSCs in VTA dopamine neurons (96.1 ± 7.8% of baseline, n = 5, p > 0.05) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus, D1 receptor activation is not involved in I-LTD induction in dopamine neurons.
Sulpiride is a D2-like receptor antagonist that blocks both D2 and D3 receptor subtypes (Seeman and Van Tol, 1994). D2 and D3 subtype receptors are expressed in the VTA (Diaz et al., 2000; Centonze et al., 2002b). D3 subtype receptor antagonists demonstrate considerable potential for therapeutic intervention in a number of mental disorders including cocaine addiction (Le Foll et al., 2005; Xi and Gardner, 2007). It is of interest to know which subtype receptor(s) mediate I-LTD induction in dopamine neurons. We found that I-LTD was significantly attenuated in the presence of selective D2 receptor antagonist L-741,626 (Pillai et al., 1998) (100 nm; Fig. 1B) (89.8 ± 6.1% of baseline, n = 8, p < 0.05 vs I-LTD control in Fig. 1A) or D3 receptor antagonist GR 103691 (Zapata and Shippenberg, 2002) (10 nm; Fig. 1B) (89.3 ± 7.1% of baseline, n = 7, p < 0.05 vs I-LTD control in Fig. 1A). However, selective D4 receptor antagonist L-745,870 (Pillai et al., 1998) had no significant effect on I-LTD induction (100 nm; Fig. 1B) (67.0 ± 7.9% of baseline, n = 5, p < 0.05 vs I-LTD control in Fig. 1A). These results indicate that synaptic activation of D2 and D3 receptor subtypes, but not D4 subtype, mediates the induction of I-LTD in dopamine neurons.
Group I mGluRs consist of mGluR1 and mGluR5 subtypes (Conn and Pin, 1997). Both mGluR1 and mGluR5 receptor subtypes are known to be distributed on VTA dopamine neurons (Kane et al., 2005). Our previous study has shown that either mGluR1 antagonist 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt) or mGluR5 antagonist 6-methyl-2-(phenylethynyl)-pyridine (MPEP) significantly attenuated I-LTD induction (Pan et al., 2008). Consistent with this study, we found that a combination of the mGluR1 antagonist CPCCOEt (50 μm) and mGluR5 antagonist MPEP (10 μm) blocked I-LTD (Fig. 1C) (93.3 ± 8.1%, n = 7, p < 0.05 vs I-LTD control in Fig. 1A). In addition, I-LTD was also blocked in slices that were preincubated (≥1 h) and continuously superfused with the CB1 receptor antagonist N-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM 251, 2 μm) (Fig. 1C) (93.4 ± 8.7% of baseline, n = 7, p < 0.05 vs I-LTD control in Fig. 1A). These results confirm and extend our previous findings that I-LTD in response to cocaine and the repetitive stimulation requires the activation of D2 receptors, group I mGluRs and CB1 receptors in dopamine neurons (Pan et al., 2008).
We next determined whether the D2-like agonist quinpirole could mimic cocaine in facilitating I-LTD induction. Quinpirole is an unselective D2/D3 receptor agonist (Rosenzweig-Lipson and Barrett, 1995). Although bath application of quinpirole (1 μm) alone had no significant effect on evoked IPSCs (96.5 ± 6.1% of baseline, n = 5, p > 0.05), the presence of quinpirole (1 μm) during the 10 Hz stimulation induced I-LTD (70.4 ± 7.8% of baseline, n = 9, p < 0.01; Fig. 1D), which was not significantly different from I-LTD induced in the presence of cocaine (p > 0.05). I-LTD induced in the presence of quinpirole was not affected by D1 receptor antagonist SCH 23390 (10 μm; Fig. 1D) (61.7 ± 7.9%, n = 5, p > 0.05 vs I-LTD control in Fig. 1D), but was blocked by the D2 receptor antagonist sulpiride (10 μm; Fig. 1D) (97.8 ± 6.9%, n = 6, p < 0.05 vs I-LTD control in Fig. 1D), by the group I mGluR antagonists CPCCOEt (50 μm) and MPEP (10 μm; Fig. 1E) (92.7 ± 6% of baseline, n = 7, p < 0.05 vs I-LTD control in Fig. 1D), and by the CB1 receptor antagonist AM 251 (2 μm; Fig. 1F) (92.3 ± 7.1% of baseline, n = 6, p < 0.05 vs I-LTD control in Fig. 1D). Together, the above results support the hypothesis that D2 receptors cooperate with group I mGluRs to induce eCB-mediated I-LTD in dopamine neurons.
CB1 receptor activation is not responsible for D2-receptor-induced acute depression of IPSCs
To investigate the mechanism by which D2 receptors cooperate with group I mGluRs to induce eCB-mediated I-LTD in dopamine neurons, we examined the individual contributions of each of these receptors to I-LTD induction and determined their respective downstream effectors. We first determined how the D2 receptor activation leads to the depression of inhibitory synaptic transmission. Our previous study has shown that bath application of higher concentrations of cocaine (10 μm) or quinpirole (10 μm) to midbrain slices produced reversible depression of IPSCs in VTA dopamine neurons, and the cocaine-induced depression of IPSCs was not affected by D1 receptor antagonist SCH 23390, but was partially blocked by D2 receptor antagonist sulpiride (Pan et al., 2008). It has been shown that quinpirole and cocaine induce the release of AEA in the striatum and other forebrain regions via D2 receptor activation (Giuffrida et al., 1999; Patel et al., 2003; Centonze et al., 2004) and that quinpirole- and cocaine-induced acute depression of IPSCs can be partially blocked by a CB1 receptor antagonist in the striatum (Centonze et al., 2004). If cocaine or quinpirole depresses IPSCs in the VTA via the release of AEA and subsequent activation of CB1 receptors, the depression should be blocked by disrupting eCB/CB1 signaling. We examined whether CB1 receptor antagonist AM 251 affected cocaine- and quinpirole-induced depression of IPSCs. Consistent with our previous study (Pan et al., 2008), we found that bath application of 10 μm cocaine or 10 μm quinpirole reversibly depressed evoked IPSCs in rat VTA dopamine neurons (cocaine, 57.8 ± 10.4% of baseline, n = 5, p < 0.001, Fig. 2A) (quinpirole, 71.0 ± 7.2% of baseline, n = 9, p < 0.01, Fig. 2B). However, preincubation (≥1 h) and continuous superfusion of interleaved slices with CB1 receptor antagonist AM 251 (2 μm) did not significantly affect the depression of IPSCs produced by quinpirole and cocaine (cocaine, 64.9 ± 7.7% of baseline, n = 5; quinpirole, 73.5 ± 6.2% of baseline, n = 5; p > 0.05 vs corresponding controls) (Fig. 2A,B).
Cocaine is a local anesthetic (Grzybowski, 2008) that may depress IPSCs by blocking presynaptic Na+ conductance and action potential (AP) firing. Cocaine at 10 μm inhibited spontaneous action potential firing by activating D2 receptor-mediated increase in K+ conductance, and this effect was completely reversed by D2 receptor antagonist sulpiride (Lacey et al., 1990). Consistent with these findings, we found that in the continuous presence of sulpiride (1 μm), bath application of 10 μm cocaine had no significant effect on the frequency of spontaneous action potential firing in VTA dopamine neurons (baseline, 1.8 ± 0.2 Hz; cocaine 1.9 ± 0.2 Hz; n = 4, p > 0.05). It has been shown that cocaine at 1–300 μm has no significant effect on EPSCs in the striatum, whereas it produces a dose-dependent depression of IPSCs (Centonze et al., 2002a). These studies imply that, at the concentration used in the present study, cocaine-induced depression of IPSCs is not mediated by suppression of presynaptic action potential firing.
We next determined whether CB1 knock-out affects quinpirole-induced depression of IPSCs. We found that bath application of quinpirole (10 μm) produced a similar degree of IPSC depression in VTA dopamine neurons in midbrain slices prepared from CB1−/− and CB1+/+ mice (CB1+/+, 77.3 ± 5.8% of baseline, n = 7 from four mice; CB1−/−, 70.1 ± 9% of baseline, n = 5 from three mice; p > 0.05; Fig. 2C). Thus, neither CB1 blockade nor genetic deletion affected the D2-receptor-mediated acute depression of IPSCs in dopamine neurons.
D2 autoreceptors on the somatodendritic (postsynaptic) regions of midbrain dopamine neurons are associated with G-protein-coupled, inwardly rectifying K+ channels (GIRKs) whose activation leads to an outward K+ current or membrane hyperpolarization (Lacey et al., 1987; Centonze et al., 2002b; Beckstead et al., 2004). Indeed, we found that cocaine- and quinpirole-induced depression of IPSCs was accompanied by an outward shift of the baseline holding current (10 μm cocaine, 56.7 ± 11.6 pA, n = 5; 10 μm quinpirole, 45.9 ± 7.4 pA, n = 9), presumably via the activation of GIRKs. To investigate whether the change in the hold current contributes to cocaine- and quinpirole-induced depression of IPSCs, we used two independent approaches to block D2-receptor-mediated activation of GIRKs.
First, GDP-β-S (2 mm), an irreversible G-protein inhibitor shown to block GIRKs (Tamae et al., 2005), was loaded into dopamine neurons via whole-cell patch pipettes. In the presence of GDP-β-S, quinpirole (10 μm) induced significant depression of IPSCs (74.7 ± 6.1% of baseline, n = 7, p < 0.01 vs baseline; p > 0.05 vs control, i.e., in the absence of GDP-β-S) (Fig. 2D). There was no significant shift of the holding current (6.4 ± 9.9 pA, p > 0.05), indicating that postsynaptic GIRKs were blocked by GDP-β-S. Second, whole-cell recordings were made using Cs+-based internal solution (see Materials and Methods). Under this condition, quinpirole (10 μm) produced significant depression of IPSCs (69.2 ± 6.4% of baseline, n = 6, p < 0.01 vs baseline; p > 0.05 vs control group with K+-based internal solution) (Fig. 2D), although it did not significantly shift the holding current (5.7 ± 11.6 pA, p > 0.05). Together, these results suggest that D2-receptor-mediated activation of postsynaptic GIRKs does not contribute to cocaine- and quinpirole-induced depression of IPSCs.
Quinpirole can depress IPSCs by decreasing presynaptic GABA release or postsynaptic sensitivity to GABA. To determine the site at which quinpirole depressed IPSCs, we examined the effect of quinpirole on miniature spontaneous IPSCs (mIPSCs) recorded in the presence of TTX to block action potential firing. A change in mIPSC frequency indicates a presynaptic mechanism, whereas a change in mIPSC amplitude signifies a change in postsynaptic responsiveness (Van der Kloot, 1991). We found that bath application of quinpirole induced a significant decrease in the mean frequency of mIPSCs (control, 1.2 ± 0.2 Hz, quinpirole, 0.7 ± 0.2 Hz, n = 6, p < 0.05; Fig. 2E,F) without affecting the amplitude of mIPSCs, as shown in cumulative frequency plots of amplitude distribution (Fig. 2G) (Van der Kloot, 1991). Together, these data suggest that D2 receptor activation depresses IPSCs by activating presynaptic D2 receptors.
We have shown previously that I-LTD was attenuated by D2 and D3 receptor antagonists, but not by a D4 receptor antagonist (Fig. 1B). We now show that D2/D3 agonist quinpirole depressed IPSCs by a presynaptic mechanism (Fig. 2E–G). However, there is no direct anatomical evidence for the presence of D2 subtype receptors on the inhibitory axonal terminals that innervate VTA dopamine neurons. Two isoforms of the D2 receptor, D2 long (D2L) and D2 short (D2S), are generated from the same gene by alternative splicing (Picetti et al., 1997). In the midbrain (VTA and substantia nigra), the immunoreactivity of the D2S is more prevalent, whereas D2L expression is sparse (Khan et al., 1998). Both electrophysiological and anatomical studies indicate that the D2S isoform is the somatodendritic D2 autoreceptor whose activation hyperpolarizes dopamine neurons (Khan et al., 1998; Centonze et al., 2002b). The D2L is strongly expressed on medium spiny neurons (MSNs) in the nucleus accumbens (Khan et al., 1998). MSNs are GABAergic neurons whose axonal terminals innervate VTA dopamine neurons and provide the major source of GABAergic inhibition to these neurons (Kalivas et al., 1993; Sano and Yokoi, 2007). We speculate that the D2L is the presynaptic D2 subtype receptor that mediates the depression of IPSCs in the VTA.
D2S and D2L have very similar pharmacological profiles, but display differential affinities for G-proteins. D2S is more efficient in inhibiting the adenylyl cyclase activity than D2L (Montmayeur et al., 1993). This finding, together with the finding that D2S is more prevalently expressed in the midbrain (Khan et al., 1998), could explain why a higher concentration of quinpirole is required for depressing IPSCs (the present study), whereas a lower concentration is sufficient to induce the hyperpolarization of dopamine neurons (Centonze et al., 2002b).
It has been shown that lesions of the nucleus accumbens induce large decreases in D3 receptor binding in the VTA, suggesting that D3 receptors are distributed on the axonal terminals of the MSNs of the nucleus accumbens that impinge on VTA neurons (Diaz et al., 2000). Together, the above findings provide a putative anatomical basis for D2/D3-receptor-mediated presynaptic depression of IPSCs in VTA dopamine neurons.
CB1 receptor activation is not responsible for D2-receptor-induced facilitation of I-LTD induction
Thus far, we have shown that cocaine- and quinpirole-induced depression of IPSCs is not affected by the disruption of eCB signaling. These findings suggest that increasing eCB release and CB1 receptor activation is not responsible for D2-receptor-induced facilitation of I-LTD induction. However, an important caveat needs to be considered. It has been shown that eCB/CB1-mediated synaptic depression is activity-dependent, and coincident presynaptic activity exerts a powerful influence on the depression (Földy et al., 2006; Singla et al., 2007; Heifets et al., 2008). It is possible that D2 receptor activation induces eCB release when it occurs simultaneously with repetitive presynaptic activity. We therefore investigated whether the combination of repetitive synaptic stimulation (10 Hz, 5 min) with cocaine or quinpirole (10 μm each) application induced eCB-mediated synaptic depression.
Our previous study indicates that repetitive synaptic stimulation activates group I mGluRs, and the activation of these receptors could increase the release of 2-AG to activate CB1 receptors (Pan et al., 2008). We therefore used group I mGluR antagonists CPCCOEt (50 μm) and MPEP (10 μm) throughout this set of experiments to block mGluR-induced eCB release. We found that the combination of 10 Hz stimulation with application of cocaine (10 μm) or quinpirole (10 μm) induced I-LTD (cocaine, 72.6 ± 7.8% of baseline, n = 7; Fig. 3A) (quinpirole, 65.2 ± 8.5% of baseline, n = 6; Fig. 3B; p < 0.01). This I-LTD was not significantly affected by CB1 receptor antagonist AM 251 (2 μm) (cocaine, 76.0 ± 8.5% of baseline, n = 8; quinpirole, 68.7 ± 10.3% of baseline, n = 6; p > 0.05 vs corresponding controls; Fig. 3A,B), but was blocked by D2 receptor antagonist sulpiride (10 μm) (cocaine, 92.4 ± 9.6% of baseline, n = 5; quinpirole, 90.7 ± 8.9% of baseline, n = 5; p < 0.01 vs corresponding controls) (Fig. 3C,D).
We next determined whether CB1 knock-out affected the I-LTD induction. We found that the combination of 10 Hz stimulation with application of cocaine (10 μm) induced I-LTD in VTA dopamine neurons in slices prepared from CB1-deficient (CB1−/−) and wild-type (CB1+/+) mice. Although the magnitude of I-LTD from CB1-deficient mice appeared somewhat less than that of I-LTD from wild-type mice, the difference was not statistically significant (CB1−/−, 63.5 ± 9.7% of baseline, n = 5 from two mice; CB1+/+, 74.7 ± 7.9% of baseline, n = 5 from two mice; p > 0.05) (Fig. 3E). Thus, both the pharmacological blockade and genetic deletion studies indicate that D2 receptor activation does not induce detectable eCB-mediated synaptic depression.
We have shown that bath application of higher concentration (i.e., 10 μm) of cocaine or quinpirole alone produces only transient depression of IPSCs (Fig. 2A,B). Although the 10 Hz stimulation alone has no significant effect on IPSCs, the combination of 10 Hz stimulation with application of cocaine or quinpirole 10 μm induces long-lasting depression of IPSCs (I-LTD) (Fig. 3A,B). But why is repetitive presynaptic stimulation required for the induction of I-LTD?
Repetitive presynaptic stimulation induces Ca2+ entry into presynaptic axonal terminals. Activity-induced change in presynaptic Ca2+ level is critical for the induction of group II mGluR-mediated LTD at hippocampal mossy fiber synapses (Tzounopoulos et al., 1998) and eCB/CB1-mediated I-LTD at hippocampal CA1 interneuron-pyramidal cell synapses (Heifets et al., 2008). We suspect that similar mechanisms underlie the requirement of the repetitive presynaptic stimulation for cocaine- and quinpirole-induced I-LTD in dopamine neurons. To test this possibility, we used the membrane-permeable Ca2+ chelator EGTA-AM to dampen activity-induced presynaptic Ca2+ increase. Previous studies have shown that this approach reduces, but does not block presynaptic transmitter release (Castillo et al., 1996; Tzounopoulos et al., 1998; Heifets et al., 2008). Consistent with these findings, we found that bath application of EGTA-AM (100 μm) for 30 min produced a gradual decrease in the amplitude of IPSCs in rat dopamine neurons (61.5 ± 10.0% of baseline at 20–30 min, n = 3, p < 0.001) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
We next determined whether EGTA-AM blocked I-LTD induction. Slices were preincubated with 100 μm EGTA-AM for 30 min and then continuously perfused with the same concentration of EGTA-AM and group I mGluR antagonists CPCCOEt (50 μm) and MPEP (10 μm) throughout this experiment. As shown in Figure 3F, the combination of the 10 Hz stimulation with cocaine application (10 μm) induced transient depression of IPSCs rather than I-LTD (94.7 ± 9.7% of baseline at 40–50 min, n = 5, p < 0.05 vs control in Fig. 3A). However, when EGTA (10 mm) was added into intracellular solution and loaded into dopamine neurons via whole-cell recordings, I-LTD was induced (70.8 ± 8.9% of baseline at 40–50 min, n = 5, p > 0.05 vs control in Fig. 3A). Thus, in addition to the D2 receptor activation, activity-induced increase in presynaptic Ca2+ level is required for I-LTD induction. Together, these results suggest that CB1 receptor activation is not responsible for D2 receptor-induced acute depression of IPSCs and facilitation of I-LTD induction in dopamine neurons.
cAMP/PKA signaling mediates D2-receptor-induced depression of IPSCs and facilitation of I-LTD induction
The above findings raise the question of how the D2 receptor activation facilitates eCB-mediated I-LTD induction in dopamine neurons. Recent studies have shown that cAMP/PKA signaling is required for eCB-mediated LTD in the striatum and I-LTD in the hippocampus (Chevaleyre et al., 2007; Mato et al., 2008). Both D2 receptors and CB1 receptors are Gi/o-protein-coupled receptors whose activation leads to the inhibition of adenylyl cyclase (AC), resulting in a decrease in cAMP/PKA activity (Neve et al., 2004; Howlett, 2005). We hypothesized that D2 receptor activation facilitates eCB-mediated I-LTD via the converged inhibition of cAMP/PKA activity. To test this hypothesis, we examined whether disruption of cAMP/PKA signaling blocked the D2-receptor-induced acute depression of IPSCs and facilitation of I-LTD in dopamine neurons.
It has been shown that activating cAMP/PKA signaling enhances neurotransmitter release at many excitatory and inhibitory synapses (Chavez-Noriega and Stevens, 1994; Capogna et al., 1995; Chen and Regehr, 1997; Kaneko and Takahashi, 2004), whereas inhibiting cAMP/PKA signaling depresses neurotransmitter release (Marty et al., 1996; Price et al., 2005). We first determined whether disruption of cAMP/PKA signaling blocked D2 receptor agonist quinpirole-induced depression of IPSCs in dopamine neurons. Bath application of forskolin (10 μm), which activates AC to increase intracellular cAMP levels, significantly increased the amplitude of IPSCs (126.7 ± 5.9% of baseline, n = 5, p < 0.01). In the continuous presence of forskolin, bath application of quinpirole (10 μm) for 10 min had no significant effect on IPSCs (99.3 ± 7.8% of prequinpirole levels, n = 5, p > 0.05; Fig. 4A). Because cAMP can exert its action on IPSCs through PKA-dependent and PKA-independent mechanisms (Seino and Shibasaki, 2005), we examined whether PKA is involved in quinpirole-induced depression of IPSCs through the use of PKA inhibitors. Slices were incubated (≥1 h) and continuously superfused with PKA inhibitor H89 (10 μm) or PKI 14-22 (10 μm) (Chevaleyre et al., 2007). In the presence of the PKA inhibitors (Fig. 4B,C), quinpirole (10 μm) had no significant effect on the amplitude of IPSCs (H89, 97.0 ± 3.1% of prequinpirole levels, n = 5; PKI 14-22, 96.4 ± 7.6% of prequinpirole levels, n = 6; p < 0.01 vs quinpirole alone shown in Fig. 2B). Finally, we investigated whether presynaptic or postsynaptic cAMP/PKA signaling mediates the D2-receptor-induced depression of IPSCs. The cell-impermeable PKA inhibitor, PKI 6-22 amide (1 μm), was loaded into dopamine neurons via whole-cell recording for at least 15 min (Chevaleyre et al., 2007). In the presence of PKI 6-22 amide, bath application of quinpirole (10 μm) induced significant depression of IPSCs (71.5 ± 7.8% of prequinpirole levels, n = 5, p < 0.01) (Fig. 4D), which is not significantly different from that in the absence of PKI 6-22 amide (p > 0.05). Together with the data shown earlier that postsynaptic loading of GDP-β-S did not affect quinpirole-induced depression of IPSCs (Fig. 2D) and that quinpirole decreased the mean frequency, but not the amplitude distribution of mIPSCs (Fig. 2E–G), these results suggest that quinpirole activates presynaptic D2 receptors to decrease cAMP/PKA activity, resulting in presynaptic depression of IPSCs.
Figure 4.

cAMP/PKA signaling is required for D2-receptor-induced acute depression of IPSCs and facilitation of I-LTD induction. A, Bath application of AC activator forskolin (FSK, 10 μm) increased the amplitude of IPSCs and blocked quinpirole-induced depression of IPSCs (n = 5). B, Bath application of PKA inhibitor H89 (10 μm) blocked quinpirole-induced depression of IPSCs (n = 5). C, Bath application of PKA inhibitor PKI 14-22 (10 μm) blocked quinpirole-induced depression of IPSCs (n = 6). D, Intracellular loading of cell-impermeable PKA inhibitor PKI 6-22 amide (1 μm) had no significant effect on quinpirole-induced depression of IPSCs (n = 5). E, In the presence of group mGluR antagonists CPCCOEt (50 μm) and MPEP (10 μm), the combination of application of higher concentration of cocaine (10 μm) with the 10 Hz stimulation induced I-LTD (n = 7). This I-LTD was blocked by PKA inhibitor H89 (10 μm) (n = 7). F, Same as in E except D2 receptor agonist quinpirole (10 μm), instead of cocaine, was used (n = 6). H89 also blocked I-LTD induced in the presence of quinpirole (n = 6).
We showed in Figure 3 that in the presence of group I mGluR antagonists CPCCOEt and MPEP, the combination of quinpirole (10 μm) or cocaine (10 μm) with the 10 Hz stimulation induced I-LTD that was not affected by the CB1 receptor antagonist AM 251. To determine whether cAMP/PKA signaling mediates this I-LTD, we examined the effects of PKA inhibitor H89 on I-LTD induction in dopamine neurons. We found that H89 blocked I-LTD induced in the presence of either cocaine (93.8 ± 7.8% of baseline, n = 7, p < 0.05 vs corresponding control recorded in the absence of H89; Fig. 4E) or quinpirole (92.7 ± 8.3% of baseline, n = 6, p < 0.05 vs corresponding control recorded in the absence of H89; Fig. 4F). Together, the above results indicate that D2 receptor activation facilitates I-LTD induction via direct inhibition of cAMP/PKA activity.
CB1-receptor-mediated acute depression of IPSCs and I-LTD depend on cAMP/PKA signaling
We hypothesized that D2 receptors and CB1 receptors target the same downstream effector cAMP/PKA signaling pathway to induce I-LTD in dopamine neurons. Having shown that D2 receptor activation facilitates I-LTD via cAMP/PKA signaling, we next investigated whether eCB-mediated I-LTD in VTA dopamine neurons is mediated by the same downstream signaling mechanism. It has been shown that CB1 receptor agonist (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de)-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN 55,212-2) depresses EPSCs in the striatum and IPSCs in the hippocampus by inhibiting cAMP/PKA signaling (Huang et al., 2002; Chevaleyre et al., 2007). We first examined whether CB1 receptor agonist WIN 55,212-2 depressed IPSCs via cAMP/PKA signaling in dopamine neurons. Consistent with previous studies (Szabo et al., 2002; Pan et al., 2008), we found that bath application of CB1 receptor agonist WIN 55,212-2 (2 μm) produced a gradual depression of IPSCs (72.3 ± 8.9% of baseline, n = 5, p < 0.01), which was reversed by subsequent addition of the CB1 receptor antagonist AM 251 (4 μm, Fig. 5A). Bath application of the AC activator forskolin (10 μm) increased the amplitude of evoked IPSCs (132.3 ± 7.1%, n = 5, p < 0.01) and prevented WIN 55,212-2-induced depression of IPSCs (97.1 ± 7.5% of pre-WIN 55,212-2 levels, n = 5, p > 0.05) (Fig. 5B). In the continuous presence of PKA inhibitor H89 (10 μm), WIN55,212-2 had no significant effect on evoked IPSCs (94.9 ± 8.8% of baseline control, n = 5, p > 0.05) (Fig. 5C). These results suggest that CB1 receptor agonist WIN 55,212-2 depresses IPSCs in dopamine neurons by inhibiting cAMP/PKA activity.
Figure 5.

cAMP/PKA signaling is required for CB1-receptor-mediated acute depression of IPSCs and I-LTD induction. A, Bath application of CB1 receptor agonist WIN 55,212-2 (2 μm) decreased the amplitude of evoked IPSCs, and this depression was reversed by CB1 receptor antagonist AM251 (4 μm, n = 5). B, Bath application of AC activator forskolin (FSK, 10 μm) increased the amplitude of IPSCs and blocked WIN 55,212-2-induced depression of IPSCs (n = 5). C, Bath application of PKA inhibitor H89 (10 μm) blocked WIN 55,212-2-induced depression of IPSCs (n = 5). D, The presence of cocaine (3 μm) during the 10 Hz stimulation induced I-LTD. The trace was taken from Figure 1A for the purpose of comparison. E, In the presence of forskolin, the combined application of cocaine (3 μm) with the 10 Hz stimulation did not induce I-LTD (n = 5). F, Bath application of H89 blocked I-LTD induced in the presence of cocaine (n = 5). G, The presence of quinpirole (1 μm) during the 10 Hz stimulation induced I-LTD. The trace was taken from Figure 1D for the purpose of comparison. H, In the presence of forskolin, the combined application of quinpirole (1 μm) with the 10 Hz stimulation did not induce I-LTD (n = 5). I, Bath application of H89 blocked I-LTD induced in the presence of quinpirole (n = 5).
We have shown that the combination of cocaine (3 μm) or quinpirole (1 μm) with the 10 Hz stimulation induced eCB-mediated I-LTD induction (Fig. 1). We first examined whether I-LTD induced in the presence of cocaine and the 10 Hz stimulation was blocked by disrupting cAMP/PKA signaling. As shown before, the combination of cocaine (3 μm) with the 10 Hz stimulation induced I-LTD in dopamine neurons (68.7 ± 5.5% of baseline, n = 8, p < 0.01; Fig. 5D). AC activator forskolin (10 μm) increased the amplitude of IPSCs (140.8 ± 8.2%, n = 5, p < 0.01) and prevented I-LTD induction (95.3 ± 6.5% of pre-10 Hz stimulation levels, n = 5, p > 0.05; Fig. 5E). This I-LTD was blocked by PKA inhibitor H89 (10 μm, 97.5 ± 7.9% of baseline, n = 5, p > 0.05; Fig. 5F). We next examined whether I-LTD induced in the presence of quinpirole and the 10 Hz stimulation was blocked by disrupting cAMP/PKA signaling. The combination of quinpirole (1 μm) with the 10 Hz stimulation induced I-LTD (70.4 ± 7.8% of baseline, n = 9, p < 0.01) (Fig. 5G). Forskolin (10 μm) increased the amplitude of IPSCs (137.0 ± 6.2%, n = 5, p < 0.01) and prevented I-LTD induction (98.4 ± 7.4% of pre-10 Hz stimulation levels, n = 5, p > 0.05; Fig. 5H). This I-LTD was also blocked by PKA inhibitor H89 (10 μm, 94.3 ± 7.2% of baseline, n = 5, p > 0.05; Fig. 5I). These results indicate that eCB-mediated I-LTD in the VTA also requires cAMP/PKA signaling. Together, these data support the hypothesis that D2 receptor activation facilitates eCB-mediated I-LTD through converged inhibition of cAMP/PKA signaling.
Group I mGluR activation triggers eCB release to induce I-LTD
We have shown that D2 receptors cooperate with group I mGluRs to induce I-LTD in dopamine neurons and this I-LTD was blocked by CB1 receptor antagonist AM 251 (Fig. 1). Because D2 receptor activation does not induce eCB release in dopamine neurons (Fig. 2), it is very likely that group I mGluR activation induces eCB release to contribute to I-LTD induction. To test this possibility, we used pharmacological manipulations to enhance group I mGluR activation and examined whether group I mGluR-induced synaptic depression could be blocked by disrupting eCB signaling. Group I mGluR activation was enhanced by glutamate uptake inhibitor dl-threo-β-benzyloxyaspartic acid (TBOA). We found that bath application of TBOA (40 μm) had no significant effect on the amplitude of evoked IPSCs (87.5 ± 7.5% of baseline, n = 6, p > 0.05) (Fig. 6A), suggesting that glutamate accumulation during low-frequency synaptic stimulation does not induce significant synaptic depression. Although the 10 Hz stimulation alone did not have a significant effect on IPSCs (95.5 ± 6.1% of baseline, n = 6, p > 0.05), the combination of TBOA application with the 10 Hz stimulation resulted in I-LTD induction (68.2 ± 7.4% of baseline, n = 7, p < 0.01) (Fig. 6B). The TBOA-enabled I-LTD was blocked by group I mGluR antagonists CPCCOEt (50 μm) and MPEP (10 μm) (92.1 ± 8.4% of baseline, n = 6, p > 0.05) (Fig. 6C). Thus, when glutamate uptake is compromised, repetitive synaptic stimulation at 10 Hz induces significant increase in extracellular glutamate, which activates group I mGluRs to induce I-LTD. We also examined the effect of D2 receptor antagonist sulpiride on I-LTD induction. As shown in Figure 6D, sulpiride (10 μm) had no significant effect on I-LTD induction (68.9 ± 8.9% of baseline, n = 7; p > 0.05 vs I-LTD in Fig. 6B). Thus, D2 receptor activation does not make significant contribution to I-LTD induced in the presence of glutamate uptake inhibitor TBOA.
Figure 6.
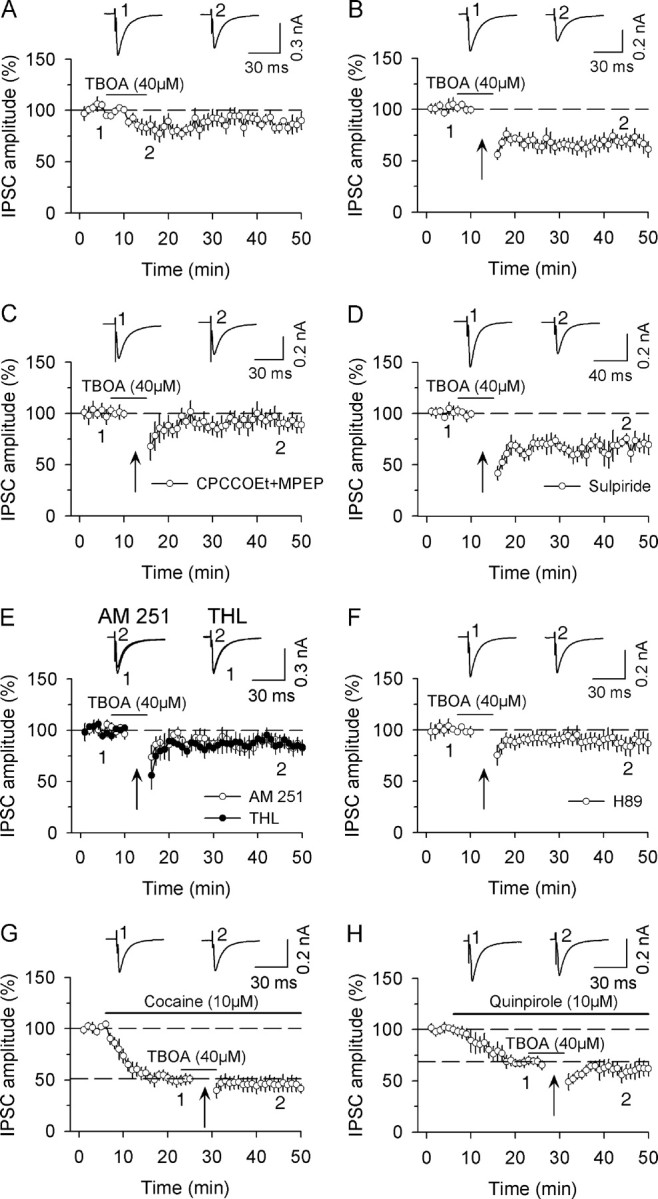
Group I mGluR activation enables I-LTD induction by inducing eCB release. A, Bath application of glutamate transporter inhibitor TBOA (40 μm) had no significant effect on evoked IPSCs (n = 6). B, The combination of TBOA application with the 10 Hz stimulation induced I-LTD (n = 7). C, Bath application of a combination of group mGluR antagonists CPCCOEt (50 μm) and MPEP (10 μm) blocked I-LTD induction (n = 6). D, D2 receptor antagonist sulpiride had no significant effect on I-LTD (n = 7). E, Bath application of CB1 receptor antagonist AM 251 (2 μm, n = 7) or intracellular loading of DAG lipase inhibitor THL (10 μm, n = 6) blocked I-LTD induction. F, Bath application of PKA inhibitor H89 (10 μm) blocked I-LTD induction (n = 7). G, H, Bath application of 10 μm cocaine (G) or quinpirole (H) decreased the amplitude of IPSCs and occluded I-LTD induced by the combination of 10 Hz stimulation with 40 μm TBOA (n = 5–6).
Group 1 mGluR activation induces the release of 2-AG, an eCB, in hippocampal slices (Jung et al., 2005). To test whether eCB signaling is involved in group I mGluR-mediated I-LTD, we first examined whether CB1 receptor antagonist AM 251 blocked this I-LTD induction. In slices were preincubated (≥1 h) and continuously superfused with CB1 receptor AM 251 (2 μm), I-LTD was significantly attenuated (87.4 ± 6.5% of baseline, n = 7, p < 0.05 vs I-LTD control in Fig. 6B; see Fig. 6E). We next examined whether disruption of 2-AG synthesis blocked I-LTD induction. Group 1 mGluRs are positively coupled to phospholipase C, which cleaves phosphatidylinositol 1,4,5-bisphosphate into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate, and DAG is subsequently converted into 2-AG by DAG lipase (Di Marzo et al., 1998; Piomelli, 2003). The DAG lipase inhibitor tetrahydrolipstatin (THL, 10 μm) was loaded into dopamine neurons via whole-cell recordings and its effect on I-LTD was examined. In the presence of THL, the combination of TBOA application with 10 Hz stimulation did not induce I-LTD (87.6 ± 6.9% of baseline, n = 6, p < 0.05 vs I-LTD control in Fig. 6B; see Fig. 6E). These results suggest that synaptic activation of group I mGluRs induces the release of 2-AG, which subsequently activates CB1 receptors to induce I-LTD.
Because CB1 receptor agonist WIN 55,212-2 induces synaptic depression via the inhibition of cAMP/PKA activity (Fig. 5), we examined whether PKA inhibitor H89 blocked group I mGluR-mediated I-LTD. As shown in Figure 6F, H89 (10 μm) blocked I-LTD induction (91.1 ± 9.1% of baseline, n = 7, p > 0.05 vs I-LTD control in Fig. 6B). Thus, repetitive synaptic activation of group I mGluRs initiates a cascade of events to induce I-LTD, which include the release of 2-AG, activation of CB1 receptors and inhibition of cAMP/PKA activity.
Thus far we have used a number of antagonists to demonstrate that D2 receptors and group I mGluRs converge on cAMP/PKA signaling pathway to induce I-LTD in dopamine neurons. In addition to the pharmacological blockade, occlusion is another powerful means to demonstrate that two biological processes converge on a specific signaling pathway. We have shown previously that I-LTD induced by the combination of 10 Hz stimulation and cocaine application was occluded by group I mGluR agonist DHPG (Pan et al., 2008). In the present study, we determined whether I-LTD induced by the combination of 10 Hz stimulation and TBOA application was occluded by cocaine or quinpirole. Bath application of cocaine or quinpirole (10 μm) induced rapid depression of IPSCs [cocaine, 50.7 ± 4.3% of baseline at 20–25 min, n = 5, p < 0.001 vs baseline, (Fig. 6G); quinpirole, 68.3 ± 4.4% of baseline at 20–25 min, n = 6, p < 0.001 vs baseline (Fig. 6H)]. In the continuous presence of cocaine or quinpirole, the combination of 10 Hz stimulation and TBOA application did not induce further depression [cocaine, 45. 5 ± 7.1% of baseline at 40–50 min, n = 5, p > 0.05 vs prestimulation level at 20–25 min (Fig. 6G); quinpirole, 60.8 ± 7.6% of baseline at 40–50 min, n = 6, p > 0.05 vs prestimulation level at 20–25 min (Fig. 6H)]. These results provide further evidence that D2 receptors and group I mGluRs converge on a common signaling pathway to induce I-LTD in dopamine neurons.
Discussion
D2 receptors cooperate with group I mGluRs to induce eCB-mediated synaptic depression in the striatum and VTA (Kreitzer and Malenka, 2005; Yin and Lovinger, 2006; Pan et al., 2008). Based on findings that D2 receptor activation induces the release of AEA (Giuffrida et al., 1999) and group I mGluR activation induces the release of 2-AG (Jung et al., 2005), we and others hypothesized that coactivation of CB1 receptors underlies the cooperation between D2 receptors and group I mGluRs to induce eCB-mediated synaptic depression (Kreitzer and Malenka, 2005; Yin and Lovinger, 2006; Pan et al., 2008). Although this hypothesis (Fig. 7A) provides an explanation for the mechanisms responsible, experimental data supporting this hypothesis have been indirect. In this study, we investigated mechanism by which D2 receptors cooperate with group I mGluRs to induce eCB-mediated I-LTD in VTA dopamine neurons. We show that group I mGluR activation facilitates I-LTD induction via the enhancement of eCB signaling and subsequent inhibition of cAMP/PKA activity, whereas D2 receptor activation facilitates I-LTD induction via direct inhibition of cAMP/PKA activity. Based on these results, we propose a refined model to explain the cooperation between group I mGluRs and D2 receptors to induce eCB-mediated synaptic depression (Fig. 7B).
Figure 7.

Signaling mechanisms underlying the cooperation between D2 receptors and group I mGluRs to induce I-LTD in dopamine neurons. A, In this model, D2 receptor activation induces the release of AEA and group I mGluR activation induces the release of 2-AG from dopamine neurons. These two eCBs travel across the synapses and activate presynaptic CB1 receptors, resulting in the inhibition of cAMP/PKA activity and I-LTD induction. B, In this model, group I mGluR activation induces the release of 2-AG, which activates presynaptic CB1 receptors to inhibit cAMP/PKA activity, whereas D2 receptor activation directly inhibits cAMP/PKA activity. D2 receptors and CB1 receptors target the same downstream effector cAMP/PKA signaling pathway to induce I-LTD.
CB1 receptors are not a downstream effector for D2 receptor activation
Gas chromatography/mass spectrometry studies have revealed that quinpirole and cocaine increase tissue contents of AEA in the striatum and other forebrain regions (Giuffrida et al., 1999; Patel et al., 2003; Centonze et al., 2004). Furthermore, quinpirole- and cocaine-induced acute depression of IPSCs was partially blocked by a CB1 receptor antagonist in the striatum (Centonze et al., 2004). It is temping to hypothesize that D2 receptor activation facilitates I-LTD induction via eCB signaling in VTA dopamine neurons. However, we show that the D2 receptor-mediated depression of IPSCs induced by bath application of quinpirole and cocaine was not affected by CB1 receptor blockade or knock-out. It has been shown that coincident presynaptic activity exerts a powerful influence on the depression (Földy et al., 2006; Singla et al., 2007; Heifets et al., 2008). We show here that the combination of D2 receptor activation with repetitive synaptic stimulation at 10 Hz induced a form of I-LTD that was also insensitive to CB1 blockade or knock-out if group I mGluR antagonists were present (Fig. 3). Thus, eCB signaling is not involved in D2 receptor-induced depression of IPSCs and facilitation of I-LTD induction in dopamine neurons.
D2 receptors could recruit different downstream effectors to induce synaptic depression in different brain areas. The synthesis of AEA involves the hydrolysis of N-arachidonoyl-phosphatidylethanolamine (NAPE) by NAPE-hydrolyzing phospholipase D (NAPE-PLD) (Okamoto et al., 2007). A potential mechanism for D2-receptor-induced release of AEA is via the activation of NAPE-PLD (Senogles, 2000). Neuroanatomical studies indicate region-specific distribution of NAPE-PLD. For example, NAPE-PLD is densely expressed on mossy fiber of granule cells in the hippocampus, but is absent in several other brain areas (Egertová et al., 2008; Nyilas et al., 2008). The lack of NAPE-PLD or the uncoupling between D2 receptor and NAPE-PLD in the VTA could explain why eCB signaling is not involved in D2 receptor-mediated depression of IPSCs and facilitation of I-LTD induction.
eCB-mediated I-LTD and D2-receptor-induced facilitation of I-LTD induction require cAMP/PKA signaling
Both D2 and CB1 receptors are coupled to Gi/o proteins whose activation leads to the inhibition of cAMP/PKA activity (Neve et al., 2004; Howlett, 2005). Our results indicate that eCB-mediated I-LTD and D2 receptor-induced facilitation of I-LTD induction both require cAMP/PKA signaling. Indeed, disruption of normal cAMP/PKA signaling with either the AC activator forskolin or PKA inhibitors H89 and PKI 14-22 abrogated both D2 and CB1 agonist-induced depression of IPSCs and eCB-mediated I-LTD in dopamine neurons (Figs. 4, 5). The latter findings are consistent with recent studies showing that cAMP/PKA signaling is required for eCB-mediated I-LTD in the hippocampus and LTD in the striatum (Chevaleyre et al., 2007; Mato et al., 2007).
Interestingly, the cAMP/PKA signaling pathway is also involved in the induction of non-eCB-mediated LTD. For example, the activation of mGluR II/III induces LTD through the inhibition of cAMP/PKA activity in several brain regions (Tzounopoulos et al., 1998; Robbe et al., 2002; Huang et al., 2007). We find that, when group I mGluRs were blocked, the combination of 10 Hz stimulation with quinpirole or cocaine at a higher concentration (10 μm) induced a form of I-LTD that was not affected by CB1 receptor antagonist or CB1 knock-out, but was blocked by PKA inhibitors (Figs. 3, 4). Thus, inhibition of cAMP/PKA activity appears to be a common signal transduction mechanism by which eCB- and non-eCB-mediated LTD can be induced.
D2 receptors cooperate with group I mGluRs to induce I-LTD
We show that group I mGluR activation facilitates I-LTD induction via the enhancement of eCB release and CB1R-mediated inhibition of cAMP/PKA signaling (Fig. 6). Thus, convergence at inhibition of cAMP/PKA signaling represents a mechanism by which D2 receptors cooperate with group I mGluRs to induce I-LTD. Our data support a model in which a threshold level of cAMP/PKA activity controls I-LTD induction. When group I mGluRs or D2 receptors are modestly activated (i.e., with 3 μm cocaine or 1 μm quinpirole, without TBOA, Fig. 1), the activation of both receptors is required to decrease cAMP/PKA activity to the threshold for induction of I-LTD. However, stronger activation of either group I mGluRs (i.e., with TBOA, Fig. 6) or D2 receptors (i.e., with 10 μm cocaine or quinpirole, Figs. 3, 4) alone can decrease cAMP/PKA sufficiently to induce I-LTD. Previous studies have shown that 10 μm cocaine produces greater D2 receptor-mediated responses than 3 μm cocaine (Lacey et al., 1990; Beckstead et al., 2004). Therefore, it is likely that 10 μm cocaine produces a significantly greater decrease in cAMP/PKA activity than cocaine at 3 μm. As a result, I-LTD can be induced by 10 μm cocaine or quinpirole alone without the participation of group I mGluRs and CB1 receptors. Nevertheless, the cooperation between D2 receptors and group I mGluRs allows I-LTD induction by pathophysiologically relevant stimuli such as in vivo cocaine exposure (Pan et al., 2008).
In addition to a decrease in cAMP/PKA level, repetitive presynaptic activity is also required for I-LTD induction. We find that D2 receptor-mediated I-LTD was blocked by the membrane-permeable Ca2+ chelator EGTA-AM, but was not affected by EGTA loading into postsynaptic neurons (Fig. 3). Thus, activity-induced changes in presynaptic Ca2+ levels are critical for I-LTD induction in dopamine neurons. Such changes in presynaptic Ca2+ levels are also required for LTD and I-LTD in the hippocampus (Tzounopoulos et al., 1998; Heifets et al., 2008). Importantly, calcineurin has been identified as the downstream target that is activated by presynaptic Ca2+ increase during I-LTD induction (Heifets et al., 2008). Similar mechanisms may underlie the requirement of the change of presynaptic Ca2+ levels for D2 receptor-mediated I-LTD induction in VTA dopamine neurons.
It remains unclear whether the mechanism discovered here underlies the cooperation between D2 receptors and group I mGluRs to induce eCB-mediated frequency-specific depression and LTD of EPSCs in the striatum (Kreitzer and Malenka, 2005; Yin and Lovinger, 2006). D2 receptor and group I mGluR agonists are capable of induce AEA and 2-AG, respectively (Giuffrida et al., 1999; Jung et al., 2005). However, there is evidence that AEA and 2-AG do not always act cooperatively to enhance eCB-mediated synaptic depression. AEA is also an endogenous ligand for transient receptor potential vanilloid 1 (TRPV1) channels (Starowicz et al., 2007). A recent study has shown that the group I mGluR agonist DHPG depressed IPSCs in the striatum via the release of 2-AG and subsequent activation of CB1 receptors, bath application of AEA or a stable AEA analog attenuated 2-AG production and DHPG-induced depression of IPSCs via the activation of TRPV1 (Maccarrone et al., 2008). Whether these two eCBs also have similar antagonistic effects on EPSCs in the striatum remains unknown. Nevertheless, this finding leaves open the possibility that the converged inhibition of cAMP/PKA signaling also participates in the cooperation between D2 receptors and group I mGluRs to induce eCB-mediated depression in the striatum.
In summary, the present study reveals a novel mechanism by which D2 receptor activation facilitates eCB-mediated I-LTD in VTA dopamine neurons. Although the prevailing hypothesis was that D2 receptor activation facilitates I-LTD by enhancing eCB release and subsequent CB1 receptor activation, our study has shown that D2 receptors and CB1 receptors inhibit the same downstream effector adenylyl cyclase, and this combined inhibition results in additive effects that dampen cAMP/PKA signaling, leading to I-LTD induction.
Footnotes
This work was supported by National Institutes of Health Grants DA024741 (Q.S.L) and DA09155 (C.J.H.). We thank Jon Hansen for critical reading of this manuscript.
References
- Alger BE. Endocannabinoid identification in the brain: studies of breakdown lead to breakthrough, and there may be NO hope. Sci STKE. 2005;2005:pe51. doi: 10.1126/stke.3092005pe51. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J. Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature. 1997;385:74–77. doi: 10.1038/385074a0. [DOI] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- Capogna M, Gähwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. J Neurosci. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Salin PA, Weisskopf MG, Nicoll RA. Characterizing the site and mode of action of dynorphin at hippocampal mossy fiber synapses in the guinea pig. J Neurosci. 1996;16:5942–5950. doi: 10.1523/JNEUROSCI.16-19-05942.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Picconi B, Baunez C, Borrelli E, Pisani A, Bernardi G, Calabresi P. Cocaine and amphetamine depress striatal GABAergic synaptic transmission through D2 dopamine receptors. Neuropsychopharmacology. 2002a;26:164–175. doi: 10.1016/S0893-133X(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Centonze D, Usiello A, Gubellini P, Pisani A, Borrelli E, Bernardi G, Calabresi P. Dopamine D2 receptor-mediated inhibition of dopaminergic neurons in mice lacking D2L receptors. Neuropsychopharmacology. 2002b;27:723–726. doi: 10.1016/S0893-133X(02)00367-6. [DOI] [PubMed] [Google Scholar]
- Centonze D, Battista N, Rossi S, Mercuri NB, Finazzi-Agrò A, Bernardi G, Calabresi P, Maccarrone M. A critical interaction between dopamine D2 receptors and endocannabinoids mediates the effects of cocaine on striatal gabaergic Transmission. Neuropsychopharmacology. 2004;29:1488–1497. doi: 10.1038/sj.npp.1300458. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Südhof TC, Purpura DP, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Diaz J, Pilon C, Le Foll B, Gros C, Triller A, Schwartz JC, Sokoloff P. Dopamine D3 receptors expressed by all mesencephalic dopamine neurons. J Neurosci. 2000;20:8677–8684. doi: 10.1523/JNEUROSCI.20-23-08677.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- Egertová M, Simon GM, Cravatt BF, Elphick MR. Localization of N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) expression in mouse brain: A new perspective on N-acylethanolamines as neural signaling molecules. J Comp Neurol. 2008;506:604–615. doi: 10.1002/cne.21568. [DOI] [PubMed] [Google Scholar]
- Földy C, Neu A, Jones MV, Soltesz I. Presynaptic, activity-dependent modulation of cannabinoid type 1 receptor-mediated inhibition of GABA release. J Neurosci. 2006;26:1465–1469. doi: 10.1523/JNEUROSCI.4587-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Lovinger DM. Emerging roles for endocannabinoids in long-term synaptic plasticity. Br J Pharmacol. 2003;140:781–789. doi: 10.1038/sj.bjp.0705466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodríguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Grzybowski A. Cocaine and the eye: a historical overview. Ophthalmologica. 2008;222:296–301. doi: 10.1159/000140625. [DOI] [PubMed] [Google Scholar]
- Heifets BD, Chevaleyre V, Castillo PE. Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc Natl Acad Sci U S A. 2008;105:10250–10255. doi: 10.1073/pnas.0711880105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC. Cannabinoid receptor signaling. Handb Exp Pharmacol. 2005;168:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- Huang CC, Chen YL, Lo SW, Hsu KS. Activation of cAMP-dependent protein kinase suppresses the presynaptic cannabinoid inhibition of glutamatergic transmission at corticostriatal synapses. Mol Pharmacol. 2002;61:578–585. doi: 10.1124/mol.61.3.578. [DOI] [PubMed] [Google Scholar]
- Huang CC, Yang PC, Lin HJ, Hsu KS. Repeated cocaine administration impairs group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. J Neurosci. 2007;27:2958–2968. doi: 10.1523/JNEUROSCI.4247-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, Vanderah TW, Lai J, Porreca F, Makriyannis A, Malan TP., Jr Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci U S A. 2003;100:10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Kauer JA. Amphetamine depresses excitatory synaptic transmission via serotonin receptors in the ventral tegmental area. J Neurosci. 1999;19:9780–9787. doi: 10.1523/JNEUROSCI.19-22-09780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KM, Mangieri R, Stapleton C, Kim J, Fegley D, Wallace M, Mackie K, Piomelli D. Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol Pharmacol. 2005;68:1196–1202. doi: 10.1124/mol.105.013961. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Churchill L, Klitenick MA. GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience. 1993;57:1047–1060. doi: 10.1016/0306-4522(93)90048-k. [DOI] [PubMed] [Google Scholar]
- Kane JK, Hwang Y, Konu O, Loughlin SE, Leslie FM, Li MD. Regulation of Homer and group I metabotropic glutamate receptors by nicotine. Eur J Neurosci. 2005;21:1145–1154. doi: 10.1111/j.1460-9568.2005.03945.x. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Takahashi T. Presynaptic mechanism underlying cAMP-dependent synaptic potentiation. J Neurosci. 2004;24:5202–5208. doi: 10.1523/JNEUROSCI.0999-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan ZU, Mrzljak L, Gutierrez A, de la Calle A, Goldman-Rakic PS. Prominence of the dopamine D2 short isoform in dopaminergic pathways. Proc Natl Acad Sci U S A. 1998;95:7731–7736. doi: 10.1073/pnas.95.13.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Actions of cocaine on rat dopaminergic neurones in vitro. Br J Pharmacol. 1990;99:731–735. doi: 10.1111/j.1476-5381.1990.tb12998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll B, Goldberg SR, Sokoloff P. The dopamine D3 receptor and drug dependence: effects on reward or beyond? Neuropharmacology. 2005;49:525–541. doi: 10.1016/j.neuropharm.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, Gasperi V, Prosperetti C, Bernardi G, Finazzi-Agrò A, Cravatt BF, Centonze D. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci. 2008;11:152–159. doi: 10.1038/nn2042. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty A, Glitsch M, Kondo S, Llano I. Cyclic AMP-regulated GABA release at inhibitory synapses in rat cerebellar slices. J Physiol Paris. 1996;90:327–328. doi: 10.1016/s0928-4257(97)87909-0. [DOI] [PubMed] [Google Scholar]
- Mato S, Lafourcade M, Robbe D, Bakiri Y, Manzoni OJ. Role of the cyclic-AMP/PKA cascade and of P/Q-type Ca2+ channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology. 2008;54:87–94. doi: 10.1016/j.neuropharm.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Montmayeur JP, Guiramand J, Borrelli E. Preferential coupling between dopamine D2 receptors and G-proteins. Mol Endocrinol. 1993;7:161–170. doi: 10.1210/mend.7.2.7682286. [DOI] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Nyilas R, Dudok B, Urbán GM, Mackie K, Watanabe M, Cravatt BF, Freund TF, Katona I. Enzymatic machinery for endocannabinoid biosynthesis associated with calcium stores in glutamatergic axon terminals. J Neurosci. 2008;28:1058–1063. doi: 10.1523/JNEUROSCI.5102-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Wang J, Morishita J, Ueda N. Biosynthetic pathways of the endocannabinoid anandamide. Chem Biodivers. 2007;4:1842–1857. doi: 10.1002/cbdv.200790155. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci. 2008;28:1385–1397. doi: 10.1523/JNEUROSCI.4033-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Rademacher DJ, Hillard CJ. Differential regulation of the endocannabinoids anandamide and 2-arachidonylglycerol within the limbic forebrain by dopamine receptor activity. J Pharmacol Exp Ther. 2003;306:880–888. doi: 10.1124/jpet.103.054270. [DOI] [PubMed] [Google Scholar]
- Picetti R, Saiardi A, Abdel Samad T, Bozzi Y, Baik JH, Borrelli E. Dopamine D2 receptors in signal transduction and behavior. Crit Rev Neurobiol. 1997;11:121–142. doi: 10.1615/critrevneurobiol.v11.i2-3.20. [DOI] [PubMed] [Google Scholar]
- Pillai G, Brown NA, McAllister G, Milligan G, Seabrook GR. Human D2 and D4 dopamine receptors couple through betagamma G-protein subunits to inwardly rectifying K+ channels (GIRK1) in a Xenopus oocyte expression system: selective antagonism by L-741,626 and L-745,870 respectively. Neuropharmacology. 1998;37:983–987. doi: 10.1016/s0028-3908(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Pollack A. Coactivation of D1 and D2 dopamine receptors: in marriage, a case of his, hers, and theirs. Sci STKE. 2004;2004:pe50. doi: 10.1126/stke.2552004pe50. [DOI] [PubMed] [Google Scholar]
- Price CJ, Karayannis T, Pál BZ, Capogna M. Group II and III mGluRs-mediated presynaptic inhibition of EPSCs recorded from hippocampal interneurons of CA1 stratum lacunosum moleculare. Neuropharmacology. 2005;49(Suppl 1):45–56. doi: 10.1016/j.neuropharm.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Robbe D, Bockaert J, Manzoni OJ. Metabotropic glutamate receptor 2/3-dependent long-term depression in the nucleus accumbens is blocked in morphine withdrawn mice. Eur J Neurosci. 2002;16:2231–2235. doi: 10.1046/j.1460-9568.2002.02273.x. [DOI] [PubMed] [Google Scholar]
- Rosenzweig-Lipson S, Barrett JE. K-channel blockers attenuate the presynaptic effects of the D2/D3 agonist quinpirole in monkeys. Pharmacol Biochem Behav. 1995;51:843–848. doi: 10.1016/0091-3057(95)00056-3. [DOI] [PubMed] [Google Scholar]
- Sano H, Yokoi M. Striatal medium spiny neurons terminate in a distinct region in the lateral hypothalamic area and do not directly innervate orexin/hypocretin- or melanin-concentrating hormone-containing neurons. J Neurosci. 2007;27:6948–6955. doi: 10.1523/JNEUROSCI.0514-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Van Tol HH. Dopamine receptor pharmacology. Trends Pharmacol Sci. 1994;15:264–270. doi: 10.1016/0165-6147(94)90323-9. [DOI] [PubMed] [Google Scholar]
- Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- Senogles SE. The D2s dopamine receptor stimulates phospholipase D activity: a novel signaling pathway for dopamine. Mol Pharmacol. 2000;58:455–462. doi: 10.1124/mol.58.2.455. [DOI] [PubMed] [Google Scholar]
- Singla S, Kreitzer AC, Malenka RC. Mechanisms for synapse specificity during striatal long-term depression. J Neurosci. 2007;27:5260–5264. doi: 10.1523/JNEUROSCI.0018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starowicz K, Nigam S, Di Marzo V. Biochemistry and pharmacology of endovanilloids. Pharmacol Ther. 2007;114:13–33. doi: 10.1016/j.pharmthera.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Szabo B, Siemes S, Wallmichrath I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci. 2002;15:2057–2061. doi: 10.1046/j.1460-9568.2002.02041.x. [DOI] [PubMed] [Google Scholar]
- Tamae A, Nakatsuka T, Koga K, Kato G, Furue H, Katafuchi T, Yoshimura M. Direct inhibition of substantia gelatinosa neurones in the rat spinal cord by activation of dopamine D2-like receptors. J Physiol (Lond) 2005;568:243–253. doi: 10.1113/jphysiol.2005.091843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Janz R, Südhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W. The regulation of quantal size. Prog Neurobiol. 1991;36:93–130. doi: 10.1016/0301-0082(91)90019-w. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Gardner EL. Pharmacological actions of NGB 2904, a selective dopamine D3 receptor antagonist, in animal models of drug addiction. CNS Drug Rev. 2007;13:240–259. doi: 10.1111/j.1527-3458.2007.00013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Lovinger DM. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc Natl Acad Sci U S A. 2006;103:8251–8256. doi: 10.1073/pnas.0510797103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata A, Shippenberg TS. D(3) receptor ligands modulate extracellular dopamine clearance in the nucleus accumbens. J Neurochem. 2002;81:1035–1042. doi: 10.1046/j.1471-4159.2002.00893.x. [DOI] [PubMed] [Google Scholar]