

Abstract
Sphingosine 1-phosphate (S1P) is a lysophospholipid mediator that exerts numerous biological activities both as a receptor ligand and as an intracellular second messenger. In the present study, we explored roles of sphingosine kinase (SphK), an S1P-producing enzyme, in adipose tissue. We utilized mouse 3T3-L1 cells as an in vitro model of adipogenesis, using a mixture of insulin/dexamethasone/3-isobutyl-1-methylxanthine (IBMX) to induce differentiation. Real-time quantitative PCR (qRT-PCR) assays revealed that the expression levels of transcripts encoding both isoforms of SphK-1 and SphK-2 are up-regulated during adipogenesis (37.6- and 6.6-fold vs. basal, P < 0.05, respectively). Concomitantly, SphK-1/SphK-2 protein abundance and S1P contents of these cells increased at 3 days after hormonal stimulation. Loss-of-function approaches by pharmacological inhibition of SphK activity as well as by transfection with small interfering RNA (siRNA) against SphK-1 led to significant attenuation of lipid droplet accumulation and adipocyte marker gene expression. We detected marked elevation of SphK-1 mRNA in adipose tissue derived from 13-week-old ob/ob mice with obese phenotype than their lean littermates. These results suggest that increased expression of SphK, an S1P-producing enzyme, plays a significant role during adipogenesis, potentially providing a novel point of control in adipose tissue.
Keywords: sphingosine 1-phosphate, differentiation, adipose tissue
Sphingolipids have been emerging as multifunctional intra- and intercellular signal transducing molecules, in addition to serving their well-established roles as structural components of cellular lipid membrane bilayers (1). Sphingosine 1-phosphate (S1P) is a phosphorylated derivative of sphingosine, the core structure of this class of lipid. S1P is formed by an action of enzyme termed sphingosine kinase (SphK) that converts sphingosine and ATP to S1P (2, 3) and represents a key molecule among signaling sphingolipids (4). Two isoforms of SphK, SphK-1 and SphK-2, have been found in mammals. SphK enzyme activity is expressed in various cell types (5), and is dynamically regulated by various extracellular stimuli (as reviewed in Ref. 6).
S1P modulates many fundamental cellular responses such as proliferation, survival, migration, regulation of cell permeability, hypertrophic responses, among others (as reviewed in Ref. 7). Yet another interesting feature of S1P actions in the context of cellular physiology is that this lipid is capable of markedly influencing the processes of differentiation. An example of S1P-regulated differentiation derives from an earlier study by Hla and Maciag (8), who discovered that “angiogenic” differentiation of vascular endothelial cells is associated with an up-regulation of endothelial transcripts that encode endothelial differentiation gene-1 receptors. Subsequent studies established that S1P serves as a specific ligand for the G-protein coupled endothelial differentiation gene-1 receptors [EDG-1 receptors; now renamed as S1P1 receptors (as reviewed in Ref. 9)], and that S1P/S1P1 receptor system plays a crucial role in promoting angiogenesis by regulating differentiation of vascular endothelial cells (10, 11). S1P also promotes myogenic differentiation of mouse C2C12 skeletal myocyte cell line (12, 13) and modulates mouse osteoclast differentiation (14). However, roles of S1P and those of S1P-related molecules in differentiation of the other cell species remain largely unknown.
Obesity represents a key risk factor of cardiovascular diseases in industrialized countries, defined as an excessive deposit of adipose tissue in a human body. Adipogenesis is a process where preadipocytes undergo differentiation into mature adipocytes. Several adipocyte marker genes, including peroxisome proliferator-activated receptor-γ (PPARγ) and adipocyte fatty acid binding protein-2 (aP2), have been found to play pivotal roles during adipogenesis (15, 16), but proximal signal transduction machineries that transmit cues from extracellular stimuli to adipogenic transcription factors in the nucleus remain incompletely defined.
In the present study, we examined a hypothesis that SphK are expressed in adipocytes/adipose tissue and modulate their functions. We provide evidence that adipogenesis of cultured mouse 3T3-L1 preadipocytes is associated with increases in SphK mRNA/protein abundance and S1P content. We further show that pharmacological or genetic inhibition of SphK leads to markedly attenuated magnitudes of adipogenesis.
MATERIALS AND METHODS
Reagents
FBS was purchased from JRH bioscience (Lenexa, KS). SuperScript RNase H- reverse transcriptase and Lipofectamine 2000 were from Invitrogen Life Technologies (Carlsbad, CA). Taq DNA polymerase was from Promega (Madison, WI). OPA (o-phthalaldehyde) was from Wako (Osaka, Japan). Primer oligonucleotides for RT-PCR assays were synthesized by Hokkaido System Science (Hokkaido, Japan). Small interfering RNA (siRNA) was from Qiagen (Valencia, CA). S1P, D-erythro-N,N,-dimethylsphingosine (DMS) and DL-threo-dihydrosphingosine (DHS) were from BioMol (Plymouth Meeting, PA). Other reagents were from Sigma (St. Louis, MO) unless otherwise noted.
Cell culture
Mouse 3T3-L1 fibroblasts were obtained from JHSF (Osaka, Japan) and were maintained in “Medium A” comprising DMEM containing 1.0 g/L D-glucose, 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 4 mM L-glutamine, and 3.7 g/L NaHCO3 at 37°C in 5% CO2. To induce differentiation, at 2 days postconfluence (day 0), culture medium was switched to “Medium B”, which substitutes Medium A with 4.5 g/L of D-glucose, supplemented with insulin (1 μg/ml), dexamethasone (1 μM), and 3-isobutyl-1-methylxanthine (IBMX) (0.5 mM). Two days later, medium was changed to “Medium C”, an equivalent of Medium B except supplemented only with insulin but without dexamethasone or IBMX. Cells were incubated to develop full differentiation by changing Medium C every second day. Fully differentiated adipocytes were identified by the characteristic morphology of multilocular lipid droplets, which are stained red by Oil Red O solution.
In some experiments, primary preadipocytes derived from rat subcutaneous adipose tissues were utilized instead of mouse 3T3-L1 cells. These cells were commercially purchased from Primary Cell (Hokkaido, Japan). They reach confluence at day 3 of culture, and then spontaneously start differentiating into adipocytes when cultured in Adipocyte Differentiation Medium (supplied) (17, 18).
RNA preparation and amplification by qRT-PCR
Total RNA was isolated from 3T3-L1 cells using the RNeasy mini column (Qiagen) following manufacturer's protocol. One microgram of total RNA was transcribed into cDNA using random hexamer and reverse transcriptase in a total volume of 20 μL. The reverse transcription was performed at 37°C for 90 min and then at 70°C for 15 min.
Resulting templates were subjected to real-time PCR analyses according to the manufacturer's procedure (Roche Diagnostics, Tokyo, Japan). Briefly, real-time quantitative RT-PCR (qRT-PCR) was performed by using TaqMan Universal PCR Master Mix and Universal Probe Library Probe TaqMan Gene Expression Assay Mix (19). Fluorescence-labeled TaqMan MGB probes (200 nM) were used for data collection during the log linear phase of the reaction. Predesigned primers and probe reagents for mouse SphK-1, SphK-2, and 18S were commercially obtained from Roche Diagnostics. Sequences of the primers and TaqMan probe were as follows: SphK-1 forward primer, 5′-TGT GAA CCA CTA TGC TGG GTA-3′; reverse primer, 5′-CAG CCC AGA AGC AGT GTG-3′; Universal ProbeLibrary probe no. 62; SphK-2, forward primer, 5′-AGA CGG GCT GCT TTA CGA G -3′; reverse primer 5′-CAG GGG AGG ACA CCA ATG-3′; Universal ProbeLibrary probe no. 88; 18S forward primer, 5′-CTC AAC ACG GGA AAC CTC AC-3′; reverse primer, 5′-CGC TCC ACC AAC TAA GAA CG-3′; Universal ProbeLibrary probe no. 77. PCR was achieved with activation and denaturation steps for 2 min at 50°C and then for 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Baseline and threshold for Ct calculation were set automatically with the ABI Prism 7000 SDS software version 1.1, or set manually whenever necessary. Relative degrees of SphK-1 or SphK-2 mRNA expression were estimated by normalizing them to those of 18S in each set of RNA samples.
In some experiments, reverse transcribed templates were subjected to conventional semiquantitative PCR, essentially as described (20). Detailed protocols including the primer sequences, annealing temperature and PCR cycles in each assay condition are summarized in supplementary Table I. In preliminary experiments, we confirmed that the reaction could be quantified within the log phase of the amplification reaction with the number of amplification cycles in each primer pair.
Immunoblot analyses
Total cellular proteins were extracted using a buffer containing n-octyl β-glucopyranoside (described in Ref. 21). Protein determinations were made with Bicinchoninate Protein Assay Kit (Nacalai, Kyoto, Japan) with BSA as a standard. Equal amounts of cellular proteins were size-fractionated by gel electrophoresis and were transferred to nitrocellulose membranes (Amersham). Resulting membranes were then subjected to immunoblot analyses as described (22). Primary antibodies used were those raised against SphK-1 (PC727) (Calbiochem, San Diego, CA) and SphK-2 (sc-22704) (Santa Cruz Biotechnology, Santa Cruz, CA). Antibody to Rac1 (610650) (BD Transduction Laboratories, San Diego, CA) was used as a loading control (23).
Lipid extraction, OPA derivatization of S1P, and HPLC analysis
Lipid isolation and S1P measurement were carried out essentially as described (24). Briefly, lipids were extracted from cells cultured on a 100 mm culture dish by a previously described two-step lipid extraction method. An automated OPA derivatization procedure was performed using a Triathlon auto injector (Spark Holland, Emmen, The Netherlands) by adding 5 μL of a working derivatization solution to auto sampler vials that contain 45 μL of lipid samples. After incubation for 15 min at room temperature, 20 μL of resulting OPA-derivatized samples were injected into the HPLC system attached with a Triathlon Auto Sampler and a prepacked C18 reversed-phase column (TSKgel ODS-100Z, 4.6 × 150 mm, particle size 5 μm, Tosoh, Tokyo, Japan) at 35oC. The flow rate and a gradient program with acetonitrile and 0.07 M KH2PO4 as solvents were determined as described previously (24). The fluorescence intensity of the eluate was measured at 455 nm with excitation at 340 nm in a spectrofluorometer (470, Waters) equipped with a flow cell (16 μL) and a data processor (C-R6A, Shimazu, Kyoto, Japan).
We confirmed that increasing amounts of authentic S1P yields linearly increasing peak heights under these conditions between 0.2 to 200 pmole/injection (data not shown). Cellular S1P contents were normalized by the total cellular protein amount and were expressed as pmole/mg protein.
Treatment with DMS and DHS
DMS and DHS were dissolved into ethanol at a concentration of 50 mM and were stocked at −80°C under N2. Stock solution of DMS, DHS, or vehicle (ethanol) was added to the cultures every second day from the starting day of hormonal stimulation. Final concentration of ethanol was at a ratio of 1:10,000 (v/v).
Cell counting assay
Cells were trypsinized and subjected to cell number counting using a particle analyzer CDA-500 (Kobe, Japan) following supplier's instruction (20).
Transient transfection with siRNA
Gene silencing of mouse SphK-1 in 3T3-L1 cells was performed using sequence specific siRNA reagents (SI01431283, Qiagen). Twenty one nucleotide siRNA for mouse SphK-1 (CGAGCAGGUGACUAAUGAATT and UUCAUUAGUCACCUGCUCGTA) were designed that target 378 nucleotides downstream of the start codon. Control siRNA was designed by NIPPON EGT (Tokyo, Japan; ACCAUUACGGUCUGCUUCAdTdT and UGAAGCAGACCGUAAUGGUdTdT). One day after being split, cells were incubated with OPTI-MEM, which contained a complex of siRNA (20 nM) and Lipofectamine 2000 (0.5%, v/v) for 20 min. Thereafter, 4 vols of DMEM supplemented with D-glucose (1.0 g/L), but without FBS or penicillin/streptomycin, were added and incubation proceeded for 4 h. The medium was then replaced with Medium A. After cells had been incubated for another two days until confluence, adipogenesis was initiated by replacing Medium A with Medium B.
Oil Red O staining
The degrees of lipid accumulation were determined using Oil Red O solution essentially as described previously (25). Briefly, cells were washed with phosphate-buffered saline and were fixed with 10% formalin (pH 7.4 for 10 min). They were then stained with a solution comprising Oil Red O (1.8 mg/ml) and isopropanol (60% v/v) in water for 15min at room temperature, followed by washing twice with 60% isopropanol (v/v). They were finally scraped with 10% SDS (wt/vol) as dye extraction solution, and resulting Oil Red O staining was quantified by measuring the optical absorbance at 520 nm.
Adipose explants preparation
Thirteen-week-old C57BL/6 ob/ob male mice and WT C57BL/6 as littermate controls were purchased from Japan SLC (Shizuoka, Japan). All mice were housed one mouse per cage with temperature- and light-control (25°C and 12 h light/12 h dark cycle, respectively). Spleen and subcutaneous adipose tissue were isolated, immediately frozen in liquid nitrogen, and were stored at −80°C until RNA extraction. Tissue samples were grinded using a polytron homogenizer and subjected to column-based RNA isolation procedure as above.
LDH activity assay
LDH activity of culture medium was measured using a CytoTox-ONE Homogenous Membrane Integrity Assay kit (Promega), using DMEM and cell lysates extracted with a lysis buffer containing Triton-X (0.1% v/v) as negative and positive controls, respectively.
Approvals for usage of animal and recombinant DNA
The experimental protocol using genetically engineered as well as WT mice had been approved by the Institutional Animal Care and Use Committees at Kagawa University. The study procedures using recombinant DNA had been approved by the Recombinant DNA Usage Committees at Kagawa University.
Statistical analysis
Results are expressed as mean ± SEM of the pooled values derived from indicated number of independent experiments. Statistical significance was determined with ANOVA followed by Scheffe's F test among groups. In some experiments, unpaired Student's t-test was performed to analyze statistical significance between two. The threshold of significance was set at P = 0.05.
RESULTS
Adipogenesis promotes SphK expression and elevates S1P content in 3T3-L1 cells
In the present study, we utilized mouse 3T3-L1 preadipocytes as an in vitro model of adipogenesis, which differentiate into mature adipocytes within a few weeks when stimulated by a mixture of hormonal stimuli (26, 27) (see Fig. 4). We examined SphK mRNA expression levels in 3T3-L1 cells at various stages during differentiation initiated by the treatment with a mixture of insulin, dexamethasone, and IBMX. qRT-PCR analysis indicates that the expression levels of mRNA encoding both SphK-1 and SphK-2 normalized by those of 18S increased over time, and reached the maximum at day 5 by 37.6-folds (SphK-1) and 6.6-folds (SphK-2), respectively (Fig. 1A). In immunoblot analysis, we found that protein abundance of both SphK-1 and SphK-2 was augmented between 3 and 5 days of adipocyte differentiation (Fig. 1B). We then studied the S1P contents of 3T3-L1 cells during adipogenesis using a previously described HPLC detection system (24). Fig. 1C indicates that amounts of S1P in these cells significantly increased approximately by 7-folds compared with the basal state 5 days after hormonal stimulation. These results demonstrate that the expression levels of SphK-1 and SphK-2, two known isoforms of S1P-producing enzyme, significantly increase during differentiation processes of mouse 3T3-L1 preadipocytes at both levels of mRNA and protein, concomitantly with marked elevation of the intracellular contents of S1P.
Fig. 4.
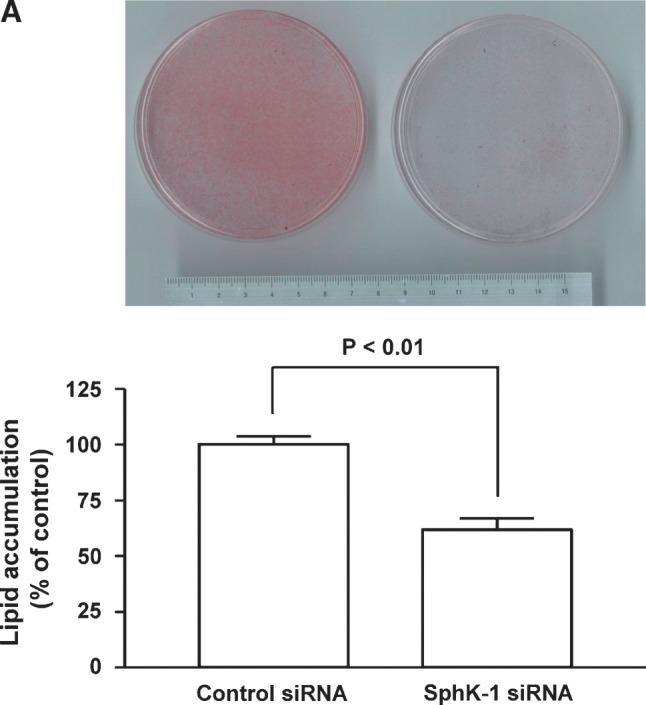

Effects of siRNA targeted to SphK-1 on adipogenesis of 3T3-L1 cells. Panel A shows the results of Oil Red O staining of 3T3-L1 cells. Cells had been transfected with control or siRNA targeted to SphK-1 40 h prior to hormonal stimulation. Upper half shows the macroscopic view of the cells on a 100 mm culture plates that had been transfected and were treated with hormonal stimuli for 8 days, followed by Oil Red O staining. Lower half is a graphic presentation of the quantification of Oil Red O staining that was determined by subjecting cell lysates to spectrophotometric analyses. Each data point represents mean ± SEM of pooled data derived from four independent experiments that yielded similar results. Panel B shows the results of semiquantitative RT-PCR assays. Forty h after transfection with siRNA, 3T3-L1 cells were subjected to differentiation regime, followed by RNA isolation at the time points as indicated. Steady-state mRNA abundance of adipocyte fatty acid binding protein-2 (aP2), peroxisome proliferator-activated receptor-γ (PPARγ), C/EBPα, and 18S were analyzed by semiquantitative RT-PCR and visualized on ethidium bromide-stained agarose gels. The experiment was repeated four times with identical results. Note that the degrees of adipocyte marker genes, aP2, PPARγ, and C/EBPα, are markedly lower in cells transfected with siRNA specific to SphK-1 compared with those with control oligonucleotides.
Fig. 1.



Abundance of sphingosine kinase (SphK)-1/SphK-2 mRNA/protein and sphingosine 1-phosphate (S1P) content of 3T3-L1 cells during adipogenesis. Shown are the results of experiments in which mouse 3T3-L1 cells underwent a treatment protocol with a hormonal stimulation to induce adipogenesis. Cells were treated with the adipogenic stimuli for the times indicated in each panel as described in detail under Materials and Methods. They were then harvested and subjected to either RNA isolation followed by real-time quantitative RT-PCR (qRT-PCR) (A), protein isolation followed by immunoblot analyses (B), or lipid isolation followed by HPLC analyses (C). In panel A, reverse-transcribed template cDNA samples were subjected to qRT-PCR using the primers specific to mouse SphK-1, SphK-2, as well as 18S. Each data point represents the fold increase in transcript expression levels of SphK-1 (closed circles) and SphK-2 (opened circles) relative to those of 18S, normalized to the values obtained from the cells before hormonal stimulation. n = 5. Panel B shows representative images of immunoblots probed for SphK-1, SphK-2, and Rac1 proteins, derived from five independent experiments that yielded equivalent results. Panel C shows the results of S1P measurement using HPLC. Each data point represents mean ± SEM of pooled data derived from three to eight independent experiments. * P < 0.01, ** P < 0.001 vs. values obtained at the time point 0 in each panel.
Pharmacological inhibition of SphK attenuates adipogenesis
After finding that hormonal stimulation elevates SphK expression and S1P contents in 3T3-L1 cells, we sought to explore functional association between SphK and adipogenesis. We first performed pharmacological experiments in which 3T3-L1 cells were treated with inhibitors of SphK, DMS, and DHS (28–31). When 3T3-L1 cells were treated by DMS or DHS (up to 5 μM from day 0 to day 8 of cell stimulation), the magnitudes of cellular lipid accumulation were markedly reduced (Fig. 2, Oil Red O staining). DMS (5 μM for 4 days) also led to significantly reduced expression level of aP2 mRNA by 47 ± 14% (conventional RT-PCR, P < 0.01 vs. vehicle, n = 3, supplementary Fig. I). Neither DMS nor DHS altered cell numbers, total protein recovery, and LDH release into culture medium of 3T3-L1 cells at 5 days and 7 days after hormonal stimulation, respectively (5 μM, supplementary Fig. II and data not shown). We also tested the effects of these SphK inhibitors in rat primary preadipocytes that undergo spontaneous adipogenesis at the day 7 of the culture (17, 18). Both DMS (5 μM) and DHS (2.5 μM) significantly attenuated the degrees of lipid accumulation compared with vehicle (supplementary Fig. III). Note that both inhibitors did not alter total amounts and release into culture medium of LDH of these cells at the day 7 (data not shown). Collectively these results demonstrate that pharmacological inhibition of SphK attenuates the degrees of adipogenesis.
Fig. 2.

Effect of N,N-dimethylsphingosine (DMS) and DL-threo-dihydrosphingosine (DHS), pharmacological SphK inhibitors, on adipogenesis. Confluent 3T3-L1 preadipocytes were treated with a hormonal stimulation to induce adipogenesis as above. DMS, DHS, or ethanol (vehicle) was also included throughout the treatment protocol using an adipogenic hormonal stimulation for 8 days, as described in detail under Materials and Methods. Cells were then subjected to Oil Red O staining followed by quantification using a spectrophotometer. The degree of Oil RedO staining in each treatment condition was normalized and expressed as a percentage of the values obtained in the absence of DMS/DHS in each culture. Closed (DMS) and open (DHS) circles represent means ± SEM of pooled data, derived from five independent experiments. * P < 0.05, ** P < 0.01 vs. values obtained with vehicle alone.
Gene silencing of SphK-1 by means of siRNA transfection
As a complementary approach of pharmacological experiments, we sought to perform genetic knockdown experiments of SphK isoform in these cells. We performed transient transfection of an siRNA targeted to mouse SphK-1 mRNA sequence (2). We first attempted to determine the effects of siRNA on endogenously expressed SphK-1 protein of day 5 after hormonal stimulation. As shown in Fig. 3, transfection with siRNA targeted to SphK-1 attenuated protein abundance of SphK-1, but not those of SphK-2 or Rac1, serving to establish the specificity of this siRNA to SphK-1. When cells underwent transfection with siRNA specific to SphK-1, the increases in S1P content 5 days after hormonal stimulation were attenuated by 61 ± 14% (P < 0.05, n = 3) compared with those transfected with control siRNA, indicating that this siRNA designed against SphK-1 is capable of effectively attenuating S1P producing activity of 3T3-L1 cells during adipogenesis. Together, these data indicate that transfection with siRNA targeted to mouse SphK-1 leads to specific knockdown of SphK-1 protein expression as well as marked attenuation of intracellular S1P content during adipogenesis.
Fig. 3.

Effects of small interfering RNA (siRNA) targeted to mouse SphK-1 on SphK-1 and SphK-2 protein expression in 3T3-L1 cells. The effects of siRNA targeted to mouse SphK-1 mRNA on protein expression levels of 3T3-L1 cells were examined in immunoblot analyses. 3T3-L1 cells were transiently transfected with siRNA specific to SphK-1 or control 40 h prior to hormonal stimulation. Five days after hormonal stimulation, cells were lysed and resulting lysates were subjected to immunoblot analyses using an antibody specific to SphK-1, SphK-2, and Rac1. Upper panels indicate representative images of immunoblots, derived from six independent experiments. The lower panel shows the results of densitometric analyses from pooled data. Protein abundance of SphK-1 as well as that of SphK-2 was normalized by that of Rac1 in control- and SphK-1-specific siRNA groups, respectively. The bars indicate percent changes obtained in cells treated with SphK-1-specific siRNA relative to those with control siRNA. ** P < 0.01 vs. values obtained with control siRNA.
siRNA against SphK-1 inhibits adipogenesis
We then explored whether or not silencing of SphK-1 gene alters the degrees of adipogenesis in 3T3-L1 cells. At day 8 of adipogenesis in cells transfected with control siRNA, numerous well-stained adipocytes became visible on Oil Red O staining that shows intracellular triacylglycerol as red color (Fig. 4A). However, in cells transfected with siRNA specific to SphK-1, the number of Oil Red O stain positive cells is dramatically less compared with those transfected with control siRNA at the identical period of differentiation (Fig. 4A). To further characterize the effects of siRNA specific to SphK-1 on adipogenesis, we examined adipocyte marker gene expression levels. PPARγ, aP2, and C/EBPα mRNA expression levels declined in the cells transfected with siRNA against SphK-1 compared with the control cells (Fig. 4B). These results demonstrate that gene silencing of SphK-1 by means of transient transfection with specific siRNA leads to marked attenuation of adipogenesis, at both levels of lipid accumulation and adipogenic marker gene expression, to a level comparable to those observed in cells that underwent pharmacological inhibition of SphK activity. siRNA directed to SphK-1, however, did not alter expression levels of Pref-1, an inhibitory gene of adipogenesis, or those of FAS, a lipogenic gene, compared with control siRNA (supplementary Fig. IV).
Elevated expression of SphK-1 transcripts in adipose tissue of obese animals
We sought to explore in vivo relevance of SphK expression in adipose tissue of living animals. We exploited ob/ob mice, which are mutant mice with impaired leptin functions and have been commonly used as an animal model of obesity (32). We isolated RNA samples from subcutaneous adipose tissue as well as those from a nonadipose tissue (spleen) from ob/ob and age- and sex-matched control WT mice (week 13, male). These animals weighed in average 59.7 ± 0.3 and 26.3 ± 0.5 g (n = 5), respectively. qRT-PCR analyses in mRNA samples derived from subcutaneous adipose tissue of ob/ob mice revealed that these animals exhibited higher levels of SphK-1 mRNA expression relative to 18S than did control animals. In contrast, the expression levels of the SphK-1 mRNA in spleen were similar in both WT and ob/ob animals (Fig. 5). Expression levels of SphK-2 transcripts were similar in both groups. Taken together, these data illustrate that abundance of SphK-1 mRNA is markedly higher in subcutaneous adipose tissue, but not in a nonadipose tissue (spleen), in 13-week-old ob/ob mice with obese phenotype compared with control animals.
Fig. 5.

Expression of SphK-1 and SphK-2 mRNA in adipose tissue of ob/ob mouse. Shown are the results of qRT-PCR analyses using RNA samples from obese mice or their littermates. RNA was extracted from subcutaneous adipose tissue and spleen of 13-week-old male mice of either ob/ob or WT genetic background. RNA samples were subjected to qRT-PCR assays as above using primer pairs directed to SphK-1, SphK-2, and 18S. Transcript abundance of SphK-1 as well as SphK-2 in each tissue sample was normalized to that of 18S. Values obtained from ob/ob mouse tissue was normalized to those obtained in WT samples, and are shown as relative abundance of SphK-1 (upper panel) and SphK-2 (lower panel). Each data point represents mean ± SEM of pooled data, derived from five independent animals in each genetic group.
DISCUSSION
Present study provides several lines of evidence that two isoforms of SphK, S1P-producing enzymes, are expressed in adipocyte/adipose tissue, and that SphK may modulate functions of these cells. Both qRT-PCR and immunoblot assays reveal that abundance of SphK-1 as well as SphK-2 isoform increases over the period of adipogenesis in mouse 3T3-L1 cells at both levels of mRNA and protein (Fig. 1A, B). Furthermore, HPLC analyses show that intracellular S1P contents increase after hormonal stimulation (Fig. 1C). We therefore sought to explore functional relevance of SphK to adipogenesis. DMS and DHS have been characterized as specific SphK inhibitors and are widely used at the similar doses in various cell cultures (28, 29). During adipogenesis, they lead to attenuation of lipid droplet accumulation (Fig. 2) and that of aP2 gene expression (supplementary Fig. I). These agents suppress lipid accumulation after hormonal stimulation without altering the increases in cell numbers, total protein contents, and LDH release into culture medium over the course of differentiation (supplementary Fig. II and data not shown). We therefore interpret these data that they attenuate adipogenesis due to their pharmacological properties rather than just reflecting toxicity. Furthermore, DMS as well as DHS attenuates the magnitudes of lipid accumulation in rat preadipocytes (supplementary Fig. III). Thus, SphK system may play a significant role in promoting lipid droplet accumulation not only in the 3T3-L1 cell line but also in primary cultures of preadipocytes.
As a complementary approach of pharmacological experiments, we utilized a genetic loss-of-function approach of SphK protein in this model. We focused on SphK-1 isoform in the present study, and designed an siRNA targeted to mouse SphK-1 sequence. This siRNA is capable of significantly down-regulating SphK-1 protein abundance without affecting SphK-2 (Fig. 3). We chose to examine the effects of siRNA at day 5 of adipogenesis after hormonal stimulation, because basal SphK-1 protein abundance was so low that we could not rigorously quantify the effects of siRNA (data not shown). Note that the gene-silencing effects of siRNA could persist for several days, as we observed in the present study, at least in several other cell cultures (33, 34). Down-regulation of SphK-1 protein expression is associated with marked decreases in S1P contents, suggesting that SphK-1 occupies a significant portion of S1P-producing capacity of differentiating 3T3-L1 cells. Importantly, when hormonal stimulation is added to these cells whose SphK-1 expression has been silenced by siRNA, the magnitudes of lipid accumulation and adipogenic marker gene expression are markedly declined compared with control siRNA-treated cells (Fig. 4). Transfection procedure per se was without effect, because the degrees of Oil Red O staining were not influenced by control siRNA compared with untransfected cells (data not shown). Collectively, these results support a hypothesis that SphK-1, an isoform of S1P-producing enzyme, plays a key role in adipogenesis of 3T3-L1 cells.
SphK converts sphingosine and ATP to S1P and thereby influences intracellular contents of bioactive sphingolipids (35). Activation of this enzyme has emerged as an important point of signal transduction pathways that regulate a variety of physiological and pathophysiological processes, including cardiovascular development and vascular inflammation (36). Although SphK-1 and SphK-2 are widely expressed, these two isoforms exhibit distinct tissue distribution and developmental expression patterns with each other (3). Our results demonstrate that cultured adipocytes express both SphK isoforms in qRT-PCR assays and immunoblots (Fig. 1). While we focused on the SphK-1 isoform in our experiments with siRNA, more detailed quantitative characterizations of both SphK isoforms remain to be elucidated, in terms of S1P-producing capacity as well as functional regulation of these cells. Additionally it should be noted that elevation of intracellular S1P content seems to precede that of SphK mRNA/protein (Fig. 1). We raise several possibilities for this apparent discordance. First, SphK proteins may undergo various posttranslational regulations in terms of their enzymatic activity, notably at the levels of protein phosphorylation as well as of subcellular localization (as reviewed in Ref. 6). Second, it is also possible that expression level and/or catalytic activity of other S1P-related enzymes may change at the earlier time points of adipocyte differentiation of 3T3-L1 cells. Precise molecular mechanisms how intracellular S1P content gets elevated at the earlier stage of hormonal stimulation thus remain to be elucidated at this stage. S1P has been previously shown to modulate differentiation of several other cell types, including vascular endothelial cells (10), skeletal myoblasts (12), and osteoclasts (14). Our findings therefore suggest that adipocytes can be now added to this list of mammalian cell types, which differentiate under the influences of S1P-related molecules.
We also sought to explore in vivo relevance of SphK using adipose tissue samples derived from genetically engineered obese mice as a model. Mutant mice with leptin deficiency, ob/ob mice used in this study, become obese due to a dysregulation of feeding behavior characterized by hyperphagia and lower energy expenditure owing to the absence of leptin signaling (32). Our results indicate that SphK-1 mRNA expression is elevated in the adipose tissue derived from ob/ob mice compared with age- and sex-matched control animals (Fig. 5), while there was no change in SphK-1 mRNA expression in a nonadipose tissue (spleen) regardless of mice genotype. Under these conditions, we did not observe significant differences in SphK-2 mRNA abundance in both organs in either genotype. It should be noted that we were not able to effectively isolate sphingolipids and detect S1P using tissue samples derived from these mice by means of the lipid extraction procedure that we employed in cultured 3T3-L1 cells (data not shown). This is consistent with the notion that tissue S1P content is typically much lower compared with that in blood. However, the basal plasma levels of S1P are significantly higher in ob/ob mice compared with the lean controls (37). Thus, we suggest that SphK/S1P pathway may play roles in regulating (patho)physiological responses of obese animal organs in vivo as well.
The mechanisms whereby SphK promotes adipogenesis remain to be elucidated. S1P is capable of acting as an intracellular second messenger as well as an extracellular receptor ligand (4). We detected appreciable levels of mRNA expression that encode S1P1-3 receptor subtypes in mouse 3T3-L1 cells (conventional RT-PCR; data not shown). This is in agreement with an earlier report that rat adipocytes express several S1P receptors (38). Interestingly, in some cell types, lysophosphatidic acid, a lysophospholipid structurally related to S1P, could behave as a direct agonist of the PPARγ, thereby regulating genes whose expression are controlled by PPAR response elements (39, 40). Thus, SphK may possibly act on these and other molecules to modulate adipocyte functions.
Elevation of SphK abundance and S1P content in adipocytes and adipose tissue (Figs. 1 and 5) may provide implications for their roles not only in the processes of adipogenesis, but also in the (patho)physiology of mature adipocytes. For example, a recent report has shown that extracellularly added S1P is capable of suppressing insulin-induced ob gene expression and leptin production in primary culture of rat subcutaneous adipocytes (38). Like leptin, adipose tissue releases numerous bioactive molecules (termed “adipocytokines,” as reviewed in Ref. 41). One of such adipocytokines, tumor necrosis factor α, which exerts deleterious actions in remote cardiovascular organs (41), has been shown to activate SphK and increase S1P content in cultured human vascular endothelial cells (42). Thus, when taken together with these reports by others, our findings suggest that SphK/S1P system may exhibit complex patterns of cross-talk with other signaling machinery that are modulated by adipocytokines in adipose and in nonadipose tissue. Another implication of SphK induction during adipogenesis may derive from an interaction with angiogenesis, a physiological process involving the growth of new blood vessels from pre-existing ones. This is because enlargement of adipose tissue is largely dependent on the degrees of new blood vessel formation (43), and because S1P represents a strong stimulator of angiogenic responses (11, 44).
In conclusion, we demonstrated that expression levels of SphK is elevated in mouse 3T3-L1 cells during adipogenesis as well as in ob/ob mouse adipose tissue. Because inhibition of SphK leads to attenuated magnitudes of differentiation, our results suggest that this S1P-producing enzyme may play major roles to promote adipogenesis. We propose that a deeper understanding of SphK/S1P system in adipose tissue may lead to the identification of novel points of control in obesity-associated pathophysiological conditions, including cardiovascular and metabolic diseases.
Supplementary Material
Abbreviations
aP2, adipocyte fatty acid binding protein-2
DHS, DL-threo-dihydrosphingosine
DMS, N,N-dimethylsphingosine
IBMX, 3-isobutyl-1-methylxanthine
OPA o-phthalaldehyde
PPARγ, peroxisome proliferator-activated receptor-γ
qRT-PCR, real-time quantitative RT-PCR
siRNA, small interfering RNA
SphK, sphingosine kinase
S1P, sphingosine 1-phosphate
This study was supported in part by Grants-in-Aid from the Nankai Ikuei Foundation (to T.H., Kagawa, Japan), from the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to J.I., Tokyo, Japan), and from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to T.H., 19790650 and to J.I., 17790151).
Published, JLR Papers in Press, November 19, 2008.
Footnotes
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of one table and four figures.
References
- 1.Futerman A. H., and Y. A. Hannun. 2004. The complex life of simple sphingolipids. EMBO Rep. 5 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kohama T., A. Olivera, L. Edsall, M. M. Nagiec, R. Dickson, and S. Spiegel. 1998. Molecular cloning and functional characterization of murine sphingosine kinase. J. Biol. Chem. 273 23722–23728. [DOI] [PubMed] [Google Scholar]
- 3.Liu H., M. Sugiura, V. E. Nava, L. C. Edsall, K. Kono, S. Poulton, S. Milstien, T. Kohama, and S. Spiegel. 2000. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 275 19513–19520. [DOI] [PubMed] [Google Scholar]
- 4.Hla T. 2004. Physiological and pathological actions of sphingosine 1-phosphate. Semin. Cell Dev. Biol. 15 513–520. [DOI] [PubMed] [Google Scholar]
- 5.Yatomi Y., R. J. Welch, and Y. Igarashi. 1997. Distribution of sphingosine 1-phosphate, a bioactive sphingolipid, in rat tissues. FEBS Lett. 404 173–174. [DOI] [PubMed] [Google Scholar]
- 6.Taha T. A., Y. A. Hannun, and L. M. Obeid. 2006. Sphingosine kinase: biochemical and cellular regulation and role in disease. J. Biochem. Mol. Biol. 39 113–131. [DOI] [PubMed] [Google Scholar]
- 7.Gardell S. E., A. E. Dubin, and J. Chun. 2006. Emerging medicinal roles for lysophospholipid signaling. Trends Mol. Med. 12 65–75. [DOI] [PubMed] [Google Scholar]
- 8.Hla T., and T. Maciag. 1990. An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to G-protein-coupled receptors. J. Biol. Chem. 265 9308–9313. [PubMed] [Google Scholar]
- 9.Chun J., E. J. Goetzl, T. Hla, Y. Igarashi, K. R. Lynch, W. Moolenaar, S. Pyne, and G. Tigyi. 2002. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 54 265–269. [DOI] [PubMed] [Google Scholar]
- 10.Lee M. J., S. Thangada, K. P. Claffey, N. Ancellin, C. H. Liu, M. Kluk, M. Volpi, R. I. Sha'afi, and T. Hla. 1999. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 99 301–312. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y., R. Wada, T. Yamashita, Y. Mi, C. X. Deng, J. P. Hobson, H. M. Rosenfeldt, V. E. Nava, S. S. Chae, M. J. Lee, et al. 2000. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Invest. 106 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meacci E., F. Cencetti, C. Donati, F. Nuti, M. Farnararo, T. Kohno, Y. Igarashi, and P. Bruni. 2003. Down-regulation of EDG5/S1P2 during myogenic differentiation results in the specific uncoupling of sphingosine 1-phosphate signalling to phospholipase D. Biochim. Biophys. Acta. 1633 133–142. [DOI] [PubMed] [Google Scholar]
- 13.Donati C., E. Meacci, F. Nuti, L. Becciolini, M. Farnararo, and P. Bruni. 2005. Sphingosine 1-phosphate regulates myogenic differentiation: a major role for S1P2 receptor. FASEB J. 19 449–451. [DOI] [PubMed] [Google Scholar]
- 14.Ryu J., H. J. Kim, E. J. Chang, H. Huang, Y. Banno, and H. H. Kim. 2006. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 25 5840–5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosen E. D., P. Sarraf, A. E. Troy, G. Bradwin, K. Moore, D. S. Milstone, B. M. Spiegelman, and R. M. Mortensen. 1999. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell. 4 611–617. [DOI] [PubMed] [Google Scholar]
- 16.Yves R., S. Aline, P. Laurence, R. Anne, C. Claudie, L. Fabrice, D. P. Elisabeth, P. Jean-Francois, W. Thierry, and J. Didier. 2004. Human adipocyte fatty acid-binding protein (aP2) gene promoter-driven reporter assay discriminates nonlipogenic peroxisome proliferator-activated receptor {gamma} ligands. J. Pharmacol. Exp. Ther. 311 467–475. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu K., M. Sakai, M. Ando, H. Chiji, T. Kawada, H. Mineo, and T. Taira. 2006. Newly developed primary culture of rat visceral adipocytes and their in vitro characteristics. Cell Biol. Int. 30 381–388. [DOI] [PubMed] [Google Scholar]
- 18.Mineo H., C. Oda, H. Chiji, T. Kawada, K. Shimizu, and T. Taira. 2007. Thiazolidinediones exhibit different effects on preadipocytes isolated from rat mesenteric fat tissue and cell line 3T3–L1 cells derived from mice. Cell Biol. Int. 31 703–710. [DOI] [PubMed] [Google Scholar]
- 19.Olsvik P. A., K. K. Lie, A. E. Jordal, T. O. Nilsen, and I. Hordvik. 2005. Evaluation of potential reference genes in real-time RT-PCR studies of Atlantic salmon. BMC Mol. Biol. 6 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Igarashi J., M. Miyoshi, T. Hashimoto, Y. Kubota, and H. Kosaka. 2007. Hydrogen peroxide induces S1P1 receptors and sensitizes vascular endothelial cells to sphingosine 1-phosphate, a platelet-derived lipid mediator. Am. J. Physiol. Cell Physiol. 292 C740–C748. [DOI] [PubMed] [Google Scholar]
- 21.Feron O., J. B. Michel, K. Sase, and T. Michel. 1998. Dynamic regulation of endothelial nitric oxide synthase: complementary roles of dual acylation and caveolin interactions. Biochemistry. 37 193–200. [DOI] [PubMed] [Google Scholar]
- 22.Igarashi J., P. A. Erwin, A. P. Dantas, H. Chen, and T. Michel. 2003. VEGF induces S1P1 receptors in endothelial cells: implications for cross-talk between sphingolipid and growth factor receptors. Proc. Natl. Acad. Sci. USA. 100 10664–10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J., S. M. DeYoung, M. Zhang, M. Zhang, A. Cheng, and A. R. Saltiel. 2005. Changes in integrin expression during adipocyte differentiation. Cell Metab. 2 165–177. [DOI] [PubMed] [Google Scholar]
- 24.Ruwisch L., M. Schafer-Korting, and B. Kleuser. 2001. An improved high-performance liquid chromatographic method for the determination of sphingosine-1-phosphate in complex biological materials. Naunyn Schmiedebergs Arch. Pharmacol. 363 358–363. [DOI] [PubMed] [Google Scholar]
- 25.Lagathu C., C. L. Yvan, J. P. Bastard, M. Maachi, B. A. Quignard, J. Capeau, and M. Caron. 2006. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia. 49 2162–2173. [DOI] [PubMed] [Google Scholar]
- 26.Green H., and M. Meuth. 1974. An established pre-adipose cell line and its differentiation in culture. Cell. 3 127–133. [DOI] [PubMed] [Google Scholar]
- 27.Mandrup S., and M. D. Lane. 1997. Regulating adipogenesis. J. Biol. Chem. 272 5367–5370. [DOI] [PubMed] [Google Scholar]
- 28.Yatomi Y., F. Ruan, T. Megidish, T. Toyokuni, S. Hakomori, and Y. Igarashi. 1996. N,N-dimethylsphingosine inhibition of sphingosine kinase and sphingosine 1-phosphate activity in human platelets. Biochemistry. 35 626–633. [DOI] [PubMed] [Google Scholar]
- 29.Edsall L. C., J. R. Van Brocklyn, O. Cuvillier, B. Kleuser, and S. Spiegel. 1998. N,N-Dimethylsphingosine is a potent competitive inhibitor of sphingosine kinase but not of protein kinase C: modulation of cellular levels of sphingosine 1-phosphate and ceramide. Biochemistry. 37 12892–12898. [DOI] [PubMed] [Google Scholar]
- 30.Yang L., Y. Yatomi, K. Satoh, Y. Igarashi, and Y. Ozaki. 1999. Sphingosine 1-phosphate formation and intracellular Ca2+ mobilization in human platelets: evaluation with sphingosine kinase inhibitors. J Biochem (Tokyo). 126 84–89. [DOI] [PubMed] [Google Scholar]
- 31.Choi O. H., J. H. Kim, and J. P. Kinet. 1996. Calcium mobilization via sphingosine kinase in signalling by the Fc epsilon RI antigen receptor. Nature. 380 634–636. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y., R. Proenca, M. Maffei, M. Barone, L. Leopold, and J. M. Friedman. 1994. Positional cloning of the mouse obese gene and its human homologue. Nature. 372 425–432. [DOI] [PubMed] [Google Scholar]
- 33.Chiu Y. L., and T. M. Rana. 2003. siRNA function in RNAi: a chemical modification analysis. RNA. 9 1034–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartlett D. W., and M. E. Davis. 2006. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 34 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tani M., M. Ito, and Y. Igarashi. 2006. Ceramide/sphingosine/sphingosine 1-phosphate metabolism on the cell surface and in the extracellular space. Cell. Signal. 19 229–237. [DOI] [PubMed] [Google Scholar]
- 36.Hla T., M. J. Lee, N. Ancellin, J. H. Paik, and M. J. Kluk. 2001. Lysophospholipids–receptor revelations. Science. 294 1875–1878. [DOI] [PubMed] [Google Scholar]
- 37.Samad F., K. D. Hester, G. Yang, Y. A. Hannun, and J. Bielawski. 2006. Altered adipose and plasma sphingolipid metabolism in obesity: a potential mechanism for cardiovascular and metabolic risk. Diabetes. 55 2579–2587. [DOI] [PubMed] [Google Scholar]
- 38.Jun D. J., J. H. Lee, B. H. Choi, T. K. Koh, D. C. Ha, M. W. Jeong, and K. T. Kim. 2006. Sphingosine-1-phosphate modulates both lipolysis and leptin production in differentiated rat white adipocytes. Endocrinology. 147 5835–5844. [DOI] [PubMed] [Google Scholar]
- 39.McIntyre T. M., A. V. Pontsler, A. R. Silva, A. St Hilaire, Y. Xu, J. C. Hinshaw, G. A. Zimmerman, K. Hama, J. Aoki, H. Arai, et al. 2003. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc. Natl. Acad. Sci. USA. 100 131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang C., D. L. Baker, S. Yasuda, N. Makarova, L. Balazs, L. R. Johnson, G. K. Marathe, T. M. McIntyre, Y. Xu, G. D. Prestwich, et al. 2004. Lysophosphatidic acid induces neointima formation through PPARgamma activation. J. Exp. Med. 199 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuzawa Y. 2005. White adipose tissue and cardiovascular disease. Best Pract. Res. Clin. Endocrinol. Metab. 19 637–647. [DOI] [PubMed] [Google Scholar]
- 42.Xia P., L. Wang, J. R. Gamble, and M. A. Vadas. 1999. Activation of sphingosine kinase by tumor necrosis factor-alpha inhibits apoptosis in human endothelial cells. J. Biol. Chem. 274 34499–34505. [DOI] [PubMed] [Google Scholar]
- 43.Duan H. F., C. T. Wu, Y. Lu, H. Wang, H. J. Liu, Q. W. Zhang, X. X. Jia, Z. Z. Lu, and L. S. Wang. 2004. Sphingosine kinase activation regulates hepatocyte growth factor induced migration of endothelial cells. Exp. Cell Res. 298 593–601. [DOI] [PubMed] [Google Scholar]
- 44.Allende M. L., T. Yamashita, and R. L. Proia. 2003. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 102 3665–3667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.