

Abstract
Obesity promotes insulin resistance and chronic inflammation. Disrupting any of several distinct steps in lipid synthesis decreases adiposity, but it is unclear if this approach coordinately corrects the environment that propagates metabolic disease. We tested the hypothesis that inactivation of FAS in the hypothalamus prevents diet-induced obesity and systemic inflammation. Ten weeks of high-fat feeding to mice with inactivation of FAS (FASKO) limited to the hypothalamus and pancreatic β cells protected them from diet-induced obesity. Though high-fat fed FASKO mice had no β-cell phenotype, they were hypophagic and hypermetabolic, and they had increased insulin sensitivity at the liver but not the periphery as demonstrated by hyperinsulinemic-euglycemic clamps, and biochemically by increased phosphorylated Akt, glycogen synthase kinase-3beta, and FOXO1 compared with wild-type mice. High-fat fed FASKO mice had decreased excretion of urinary isoprostanes, suggesting less oxidative stress and blunted tumor necrosis factor alpha (TNFα) and interleukin-6 (IL-6) responses to endotoxin, suggesting less systemic inflammation. Pair-feeding studies demonstrated that these beneficial effects were dependent on central FAS disruption and not merely a consequence of decreased adiposity. Thus, inducing central FAS deficiency may be a valuable integrative strategy for treating several components of the metabolic syndrome, in part by correcting hepatic insulin resistance and suppressing inflammation.
Keywords: metabolic syndrome, insulin resistance, type 2 diabetes mellitus
Constant availability of food and sedentary living in contemporary wealthy cultures has dramatically limited the ability to utilize fat stores, resulting in an obesity epidemic. Worldwide, at least 1 in 10 adults is obese, and more than 25% are affected in many Western countries (1), leading to striking increases in type 2 diabetes and heart disease (2). Over a quarter of adults in the United States have the metabolic syndrome (3), a combination of central obesity, glucose intolerance/insulin resistance, dyslipidemia, and hypertension (4), that confers a 2- to 3-fold increase risk for cardiovascular morbidity and mortality (5).
Given the reluctance of most modern adults to eat less and exercise more, many groups have altered lipid metabolism in mice in hopes of establishing proof of principle for new obesity therapies. Mice are resistant to diet-induced obesity after genetic manipulations that decrease lipid synthesis (acetyl-CoA carboxylase-2, stearoyl-CoA desaturase-1, diacylglycerol acyltransferase-1) and adipogenesis [peroxisome-proliferator-activated receptor (Ppar)γ], increase mitochondrial gene expression (Pparδ), increase respiratory uncoupling (uncoupling protein-1, liver X receptor), or perturb intracellular signaling (Ikkβ, S6k1) (6–14). However, it is not clear that simply decreasing adiposity will universally improve the metabolic disease milieu. For instance, liver X receptor-deficient mice have increased metabolism, yet they are predisposed to the chronic inflammatory process of atherosclerosis (15). Stearoyl-CoA desaturase-1 null mice are also hypermetabolic and thin but are predisposed to acute colitis (16).
We recently identified hypothalamic expression of another key protein in lipid metabolism, as a mediator of energy homeostasis (17). FAS is ubiquitously expressed and catalyzes the first committed step in fatty acid biosynthesis (18). Pharmacologic inhibition of this enzyme with compounds such as C75 had previously implicated FAS in bioenergetics, but the lack of specificity of C75, notably its activation of the sympathetic nervous system (19), raised the possibility that mechanisms independent of FAS could be involved.
By mating FAS floxed mice (20) with RIPCre transgenic animals (known to express the Cre recombinase in pancreatic β cells and the hypothalamus), we generated animals with FAS deficiency limited to pancreatic β cells and the hypothalamus (FASKO). The lack of FAS, at least on a standard chow diet, did not affect β−cell function. However, hypothalamic FAS deficiency resulted in mice that were hypophagic, hypermetabolic, and lean on a standard chow diet (17). While these results provided unexpected insights into the role of hypothalamic FAS in feeding and behavior, they did not address effects of this manipulation on the risk for diet-induced obesity, insulin resistance, and chronic inflammation that are characteristic of human type 2 diabetes and metabolic syndrome. In addition, the high-fat feeding paradigm also allowed us to critically examine whether the beneficial metabolic effects in the FASKO mice are simply a consequence of decreased adiposity or due to a specific perturbation in central FAS signaling. Here we report the results of experiments testing the hypothesis that deficiency of hypothalamic FAS prevents diet-induced obesity and systemic inflammation, clinically relevant endpoints.
MATERIALS AND METHODS
Animals and feeding studies
Protocols were approved by the Washington University Animal Studies Committee. Generation of mice with FAS deletion in the hypothalamus and pancreatic β cells (FASKO), and wild-type (Cre negative with the FAS floxed allele) in a mixed (BL/6 and 129) background has been described (17). Starting at 6 weeks of age, FASKO and wild-type littermates were started on a Western-style diet (TD 88137, Harlan Teklad) [high-fat diet (HFD)] containing 21% (w/w) total lipid and 0.15% (w/w) total cholesterol for 10 weeks. Fatty acid composition of this diet is presented in supplementary Table I. Body weight was recorded weekly, and food intake [expressed as a function of lean body mass (g0.75)] was measured biweekly. On week 8, stools were collected to determine lipid content (21), and percentage of consumed lipid that was absorbed was calculated as (food intake × food lipid content) − (stool output × stool lipid content)/(food intake × food lipid content) × 100. After 10 weeks of high-fat feeding, another cohort of wild-type mice received an amount of food identical to that consumed by the FASKO mice (pair-fed) for an additional 15 days. These pair-fed wild-type mice were then subjected to the same experiments (see below) as freely fed wild-type and FASKO mice.
Analytical procedures
Body composition, indirect calorimetry, locomotor activity, serum glucose, insulin, nonesterified fatty acids, triglycerides, cholesterol, β-hydroxybutyrate, leptin, adiponectin, glucose, and insulin tolerance tests were performed as described (17, 20). Hepatic lipids were extracted in chloroform/methanol (2:1, v/v) followed by determination of triglyceride and cholesterol content (20). Serum lipoprotein profiles were analyzed by size exclusion chromatography (21).
Islet isolation and insulin secretion
Pancreatic islets isolated by collagenase digestion (22) were cultured with 3 mM and 16.7 mM glucose, or 10 mM arginine, and insulin secreted in the media was assayed (22). Insulin content of islets and whole pancreas was determined after acid-ethanol extraction (17).
Immunohistochemistry and morphometry
Adipose and liver tissues were processed as described (20). Adipose sections were stained with anti-F4/80 antibody (1:200, Abcam) and visualized using NovoRed (Vector) (23). Livers were stained with Oil Red O (Sigma) to visualize neutral lipids (20). Immunohistochemical and morphometric analyses for islet area, β cell, and non-β cell mass by point-counting morphometry utilized described methods (17).
Hyperinsulinemic-euglycemic clamps
Internal jugular catheters were placed (24), and after a 3-day recovery period, mice were subjected to an overnight fast. 3-[3H]glucose (PerkinElmer) was first infused (0.05 μCi/min) for 2 h to steady state. Human regular insulin was then bolused (50 μU/g) followed by a constant infusion at either 5 mU/kg/min (low-dose clamp) or 20 mU/kg/min (high-dose clamp) in separate cohorts of HFD fed animals. Infusion of 20% D-glucose was varied to maintain blood glucose at basal concentrations for at least 90 min. Rates of basal and clamped glucose production (Ra), and insulin-stimulated whole body glucose uptake (Rd) were determined as described (24).
Analysis of insulin signaling and immunoblotting
Acute insulin stimulation was performed by intraportal injections (0.1 or 10 U/kg body weight) of regular insulin into anesthetized mice. Ten min later, liver and gastrocnemius were clamp-frozen and homogenized in buffer containing protease and phosphatase inhibitors (20). Thirty μg of total protein was resolved by SDS-PAGE, electrotransferred onto PVDF membranes (Millipore), and immunoblotted with total and 473Ser-phosphoAkt (1:1,000, cell signaling), total and 9Ser-phospho glycogen synthase kinase-3beta (1:1.000, cell signaling), total and 256Ser-phosphoFOXO1 (1:1,000, cell signaling), TRB3 (1:2,500, Calbiochem), and actin (1:5,000, Sigma). Total and phospho-specific bands were detected by chemiluminescence (ECL kit, Amersham) (20).
Quantitative RT-PCR
RNA isolation, reverse transcription, and PCR (Applied BioSystems 7700) were performed using previously published primer-probe sequences (17, 20). Relative mRNA levels were calculated using the comparative CT and standard curve methods normalized to ribosomal protein L32, an invariant internal control.
Oxidative stress and inflammatory stimuli
Urine was collected for 24 h in the presence of the antioxidant butylated hydroxytoluene (0.005%), and 15-isoprostane F2t and creatinine were analyzed by enzyme-linked immunosorbent assay (Oxford Biochemicals) and colorimetric assay (Cayman Chemical), respectively. In a separate cohort of HFD fed wild-type and FASKO mice, lipopolysaccharide (7 mg/kg, serotype 0111:B4, Sigma) was injected intraperitoneally, and serum was obtained before injection and at 1, 3, and 6 h after injection. Tumor necrosis factor alpha (TNFα) and interleukin-6 (IL-6) were measured in serum by enzyme-linked immunosorbent assay (BD Biosciences).
Statistical analyses
Data are expressed as mean ± SEM. Statistical comparisons were performed using an unpaired, two-tailed Student's t-test or ANOVA (ANOVA). If the overall F was found to be significant (P < 0.05) for the latter, comparisons between means were made using appropriate posthoc tests.
RESULTS
FASKO mice resist HFD-induced weight gain and are hypermetabolic
Wild-type mice gained weight (35% and 23% increase from baseline in males and females, respectively) after 10 weeks of HFD (Fig. 1A, B). Weight gain was considerably less in FASKO mice (Fig. 1A, B). FASKO mice after HFD feeding had ∼30% lower adiposity and an ∼8% increase in lean mass (Fig. 1C). There was no genotype effect on nasoanal lengths (not shown).
Fig. 1.

Fatty acid synthase knockout (FASKO) mice resist high-fat diet (HFD)-induced weight gain. A, B: Total body weight (top panels) and body weight gained (bottom panels) during 10 weeks of high-fat feeding in male (n = 18) and female (n = 12) wild-type (WT) and FASKO mice. C: Body composition of male mice by dual energy X-ray absorptiometry after 10 weeks of HFD. Fat (top panel) and lean (bottom panel) mass are represented as percentage of total body mass. D: Representative hematoxylin and eosin-stained sections of perigonadal white adipose tissue (×20 magnification) after 10 weeks of HFD (top panel). Distribution curve of diameters (bottom panel) of 350 fat cells per mouse shows an equal preponderance of small (20–40 μm) and large (40–60 μm) adipocytes in both genotypes (n = 5 per group). All graphs represent mean ± SEM. * P < 0.05 compared with WT littermates.
Decreased adiposity in HFD fed FASKO animals was reflected by changes in white adipose tissue but not brown adipose tissue mass. Perigonadal fat pads weighed 3.17 ± 0.58 g in FASKO vs. 6.15 ± 1.12 g in wild-type, N = 10, P = 0.03). Intrascapular brown adipose tissue depots weighed 0.35 g in FASKO vs. 0.36 g in wild-type, P = NS. Adipocyte size was similar between genotypes (Fig. 1D).
FASKO mice ate 17–22% less than controls at baseline and at 4, 7, and 10 weeks of eating HFD (Fig. 2A), exhibiting normal fat absorption at the end of the dietary study (Fig. 2B). FASKO animals demonstrated a 14–19% increase in VO2 compared with their wild-type littermates at baseline and at 4, 7, and 10 weeks of eating HFD (Fig. 2C). The respiratory quotient was similar between genotypes at the end of the dietary study, suggesting that although FASKO mice had increased oxygen consumption, their substrate utilization was the same as wild-type (Fig. 2D). There were no genotype-specific differences in serum β-hydroxybutyrate (FASKO 193 ± 26 vs. wild-type 215 ± 57 μmol/L, N = 7, P = NS), heat production (Fig. 2E), body temperature (Fig. 2F), or in the expression of genes controlling fat oxidation in liver (Pparα, acyl-CoA oxidase, carnitine palmitoyl transferase-1, medium-chain acyl-CoA dehydrogenase), skeletal muscle (lipoprotein lipase), and brown adipose tissue (uncoupling protein-1) (Fig. 2G), all of which suggested that elevated metabolism in peripheral tissues is unlikely to contribute to the increased whole-body energy expenditure in HFD fed FASKO mice. In contrast, HFD fed FASKO animals had increased locomotor activity assessed using an infrared sensing technique to measure home-cage activity over 3 days (Fig. 2H). Together, these data suggest that enhanced oxygen consumption in FASKO animals on a HFD appears to be mostly due to increased physical activity, which in combination with hypophagia protects them from diet-induced obesity.
Fig. 2.

Energy balance in response to HFD. A: Ad libitum daily food intake in WT and FASKO mice at the indicated times of the dietary study (n = 7-10). BW, body weight. B: Fat absorption, analyzed as described in Materials and Methods, at the end of 10 weeks of HFD (n = 7). C: Oxygen consumption (VO2) by indirect calorimetry integrated over a 24-h period at the indicated times of the dietary study (n = 12-15). D, E: Respiratory exchange ratio (D) and heat generation (E) by indirect calorimetry integrated over a 24-h period after 10 weeks of HFD (n = 12-15). F: Body temperature after 10 weeks of HFD (n = 11). G: Expression of the indicated genes involved in fatty acid oxidation was measured after 10 weeks of HFD in the liver, gastrocnemius (Sk.M), and brown adipose tissue by RT-PCR and normalized to L32 ribosomal mRNA levels in the same sample. Data represent three independent RT-PCR experiments for each gene with n = 4–5 mice per genotype. H: Locomotor activity after 10 weeks of HFD was quantified using infrared sensors over a 3-day period (n = 18). All graphs represent mean ± SEM. * P < 0.05 compared with WT littermates.
Effect of HFD on serum and tissue metabolites in FASKO mice
Serum chemistries and adipokines were unaffected by genotype at baseline (week 0, chow diet) (Fig. 3), consistent with our previous findings (17). High-fat feeding increased fasting glucose and insulin in both genotypes, but levels of both were lower at 7 and 10 weeks in FASKO mice (Fig. 3A, B). Nonesterified fatty acids levels (Fig. 3C) were decreased (perhaps reflecting suppression of peripheral lipolysis by insulin) in FASKO animals, suggesting the presence of enhanced insulin sensitivity in these mice with high-fat feeding. Fasting serum triglyceride levels increased over time in both genotypes, but were 28% lower at 10 weeks in FASKO animals compared with controls (Fig. 3D). Lower triglycerides were reflected by differences in VLDL in lipoprotein analyses (not shown). Serum cholesterol levels were unaffected by genotype (not shown). Consistent with increasing adiposity in wild-type mice, serum leptin levels rose ∼8-fold over the course of the dietary intervention, while levels were lower in obesity-resistant FASKO animals (Fig. 3E). FASKO mice also resisted the HFD-induced reduction in serum adiponectin (Fig. 3F).
Fig. 3.

Serum chemistries in response to Western diet. Serum was obtained from 5 h fasted animals at 6 weeks of age on a chow diet (week 0). Mice were then started on HFD, and serum from 5 h fasted mice was obtained at weeks 4, 7, and 10. Samples were assayed for glucose (A), insulin (B), nonesterified fatty acids (C), triglycerides (D), leptin (E), and adiponectin (F). Serum lipids were analyzed on the same day of sample collection, and hormones were measured in an aliquot of snap-frozen serum samples. Graphs represent mean ± SEM for 18–25 mice measured in five separate experiments over 24 months. * P < 0.05 compared with WT littermates.
Fatty acid profiling by mass spectrometry within the hypothalamus of HFD-fed animals showed no genotypic differences among several fatty acids, except for a significant reduction in palmitate (C16:0), the major product of the FAS reaction, in the FASKO mice (see supplementary Table II). These findings not only further validate the FASKO model, but also show that a diet rich in palmitate (see supplementary Table I) is unable to compensate for decreased de novo synthesis at this site.
Glucose homeostasis in HFD-fed FASKO mice
After 10 weeks of high-fat feeding, fed and 24 h-fasted serum glucose levels as well as serum insulin levels were lower in FASKO compared with control animals (Fig. 4A). There was no genotype effect on glucagon levels under these conditions (not shown). When given an acute bolus of D-glucose, HFD fed FASKO mice had lower glycemic excursions (Fig. 4B), indicating enhanced glucose tolerance.
Fig. 4.

Glucose homeostasis in high-fat fed FASKO mice. A: Serum glucose (top panel) and insulin (bottom panel) were determined in fed and fasted (24 h) mice after 10 weeks of HFD (n = 8–11). B, C: Mice were given a bolus i.p. injection of 2 g/kg glucose, and blood glucose (B) and insulin (C) were simultaneously measured at the indicated time points (n = 10). D: Serum insulin and glucose values at the 30 min time point during the GTT were used to obtain insulin-to-glucose ratio (n = 10). E: Insulin secretion in response to 3.3 mM or 16.7 mM glucose, and 3.3 mM glucose plus 10 mM arginine in islets isolated from 10-week HFD fed WT and FASKO animals (n = 7). F: Representative pancreatic sections (×10 magnification) immunostained with anti-insulin antibodies (top panel; arrows depict insulin-positive areas) and β cell mass (bottom panel) after 10 weeks of HFD feeding in WT and FASKO mice (n = 4). G: Insulin tolerance test (n = 8). Mice were given a bolus i.p. injection of 0.8 U/kg regular insulin, and blood glucose was subsequently measured at the indicated time points. H, I: Systemic glucose fluxes in hyperinsulinemic-euglycemic clamps following 10 weeks of HFD. Hepatic glucose output, represented as the rate of appearance (Ra) under basal and clamped (5 mU/kg/min insulin) conditions (H). Systemic insulin-dependent glucose uptake, represented as the rate of disappearance (Rd) (I) (n = 8 per group). Data are mean ± SEM. * P < 0.05 and ** P < 0.01, compared with WT littermates.
As FAS was also deleted in β cells (17), increased glucose tolerance in FASKO mice could be caused by enhanced β−cell function. However, two lines of evidence suggest that β-cell function is unaffected by FAS deletion. First, lower glucose levels in FASKO mice (Fig. 4B) were matched with lower insulin levels (Fig. 4C), yielding lower insulin-to-glucose ratios in FASKO mice (Fig. 4D). Second, ex vivo insulin secretory responses of islets to glucose and arginine were nearly identical between genotypes under basal (3.3 mM glucose) and stimulated (16.7 mM glucose) conditions (Fig. 4E). These data suggest that enhanced glucose tolerance in HFD fed FASKO animals is not due to insulin hypersecretion.
FASKO animals had lower β−cell mass (Fig. 4F) and a greater proportion of smaller-sized islets (19% in FASKO vs. 4% in WT had islet diameters of ∼75 μm or less), reflecting their insulin-sensitive metabolic and hormonal milieu (Fig. 3). Increased β-cell mass in WT mice (Fig. 4F) is an expected adaptive response to insulin resistance (25). FASKO mice were more sensitive to insulin than their wild-type littermates with insulin tolerance testing (Fig. 4G).
Hyperinsulinemic-euglycemic clamp experiments suggested that the liver was the major site affected in terms of glucose metabolism in mice with hypothalamic FAS inactivation. Hepatic glucose output was lower in HFD fed FASKO mice under basal and clamped (5 mU insulin/kg/min) conditions compared with littermate controls (Fig. 4H). No differences were seen between genotypes for insulin-dependent whole-body glucose disposal with either low-dose (5 mU/kg/min) (Fig. 4I), or high-dose (20 mU/kg/min) insulin (not shown). Consistent with the clamp data, mRNA levels for peroxisome-proliferator-activated receptor gamma coactivator-1α, Pepck, and glucose 6-phosphatase were decreased in FASKO compared with wild-type livers after 10 weeks of high-fat feeding (Fig. 5A). Glucokinase (GK) expression was increased in FASKO livers (Fig. 5A), reflecting enhanced hepatic insulin sensitivity.
Fig. 5.

Effect of hypothalamic FAS inactivation on liver and muscle insulin sensitivity. A: Expression of the indicated genes involved in glucose metabolism was measured in livers of wild-type and FASKO mice after 10 weeks of HFD by RT-PCR, and normalized to L32 ribosomal mRNA in the same sample. Each bar represents mean ± SEM of three independent RT-PCR experiments for each gene with n = 4 mice per genotype. * P < 0.05 vs. WT. B: Immunoblot analysis in liver of total and phosphorylated 473Ser-Akt, 9Ser- glycogen synthase kinase-3beta, 256Ser-Foxo1, TRB3, and actin following the dietary intervention in response to 0.1 U/kg intraportal bolus injection of Regular insulin. C: Densitometric quantification of all liver immunoblot analyses. Data represent mean ± SEM of five animals per group. * P < 0.05 vs. WT. D: Immunoblot analysis in gastrocnemius after 10 weeks of HFD of the above proteins following an intraportal bolus injection of 10 U/kg regular insulin. Data are representative of three independent experiments.
Key mediators of insulin signaling were assayed. Increased phosphorylation of Akt at serine 473, glycogen synthase kinase-3beta at serine 9, and Foxo1 at serine 256 were seen in FASKO compared with wild-type livers in response to 0.10 U/kg intraportal insulin (Fig. 5B, C). Protein levels for TRB3, an Akt inhibitor (26), were lower in FASKO livers (Fig. 5B, C). These differences were seen in the basal state as well as with acute insulin treatment (Fig. 5B). In contrast, these effects were not seen in gastrocnemius muscles even after 10 U/kg insulin (Fig. 5D), results consistent with the clamp data suggesting that peripheral sites of metabolism such as skeletal muscle are not involved in the insulin-sensitive phenotype of FASKO mice. In addition, there was neither a dietary nor genotype-specific effect on AMPK activity in the hypothalamus (see supplementary Fig. I).
HFD fed FASKO mice resist hepatic steatosis
FASKO livers weighed 27% less than controls with high-fat feeding (Fig. 6A). Decreased Oil Red O staining (reflecting decreased abundance of neutral lipids; Fig. 6B, bottom panel) in FASKO livers mirrored quantitative assessments of hepatic lipid content (Fig. 6C, D). Triglyceride content in gastrocnemius (Fig. 6E) was unaffected, consistent with the lack of effect on insulin signaling in this tissue (Fig. 5D). Expression of sterol-regulatory element binding protein-1c, an important regulator of hepatic lipogenesis (27), was decreased in FASKO livers (Fig. 6F). Gene expression of FAT/CD36, normally not abundant in liver (28), was higher in the livers of wild-type mice with increased triglyceride accumulation (Fig. 6F). Hepatic Pparγ expression, implicated in the development and maintenance of liver steatosis (29), was not affected by genotype under these conditions (not shown). Thus, the metabolic milieu in the FASKO animals favorably alters hepatic lipid metabolism to protect against diet-induced fatty liver.
Fig. 6.
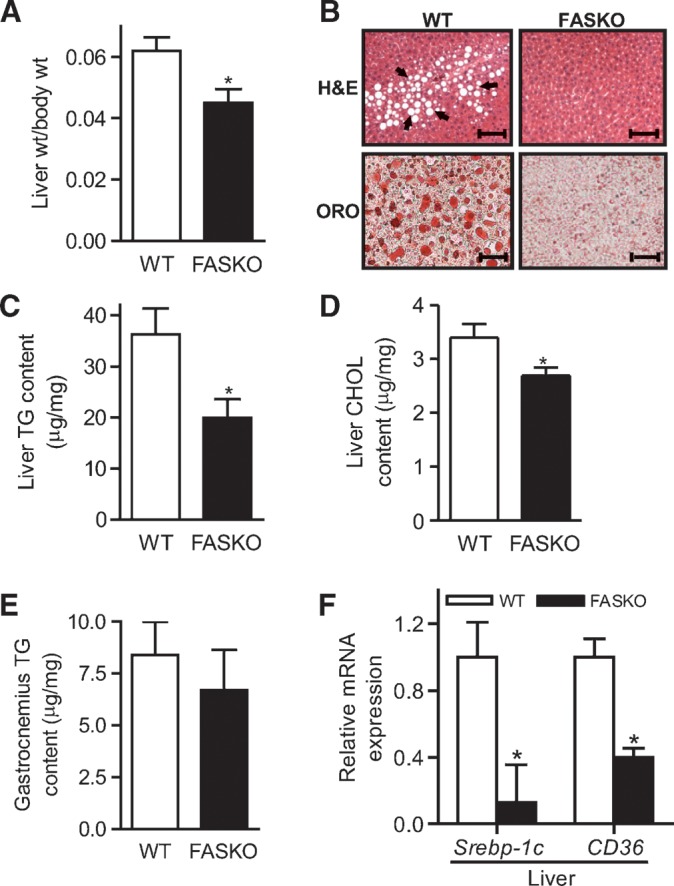
FASKO mice are resistant to HFD-induced fatty liver. A: Average weights of livers from WT and FASKO mice after 10 weeks of high-fat feeding (n = 18). B: Liver sections stained with H and E, and Oil Red O (ORO) at ×10 magnification. The prominent vacuolation seen in HFD-fed WT livers (arrows, top panel) stained positive for ORO (bottom panel). Sections are representative of 6–8 animals per group. C–E: Hepatic triglyceride (C) and cholesterol (D) content, and triglyceride content per unit mass in gastrocnemius (E) isolated from 10-week HFD-fed WT and FASKO mice (n = 7). F: Expression of hepatic sterol-regulatory element binding protein-1c and CD36 mRNA by RT-PCR normalized to L32 ribosomal mRNA after 10 weeks of HFD. Each bar represents mean ± SEM of three independent RT-PCR experiments for each gene with n = 4 mice per genotype. * P < 0.05 vs. WT.
Less oxidative stress and inflammation in HFD fed FASKO mice
Nutrient excess, insulin resistance, hepatic steatosis, and other characteristics of the obese state contribute to the increased production of reactive oxygen species, which have been implicated in chronic inflammation and complications of diabetes (30). Urinary levels of a biomarker for oxidative stress, 15-isoprostane F2t, were lower in FASKO as compared with wild-type mice (Fig. 7A). Fewer mononuclear inflammatory cells, predominantly macrophages, were detected in FASKO as compared with wild-type adipose tissue (Fig. 7B). Mice were also challenged with a nonlethal dose of lipopolysaccharide, a potent endotoxin. As expected, endotoxinemia markedly increased cytokine levels in both genotypes. However, the peak levels for TNFα and IL-6 were substantially attenuated in FASKO as compared with wild-type serum (Fig. 7C, D).
Fig. 7.

Effect of 10 weeks of HFD on oxidative stress and systemic inflammation. A: Urinary 15-isoprostane F2t normalized to creatinine levels (n = 8–9 per genotype). Data represent mean values from two independent experiments each repeated twice. B: Representative perigonadal white adipose tissue sections (×40 magnification) immunostained with antibodies against the macrophage-specific antigen F4/80 (thick arrows), and counterstained with hematoxylin, demonstrating other mononuclear cells (thin arrows). F4/80-positive cells were more abundant and aggregated in the obese wild-type mice (top panel), whereas these cells were sparse and isolated in the leaner FASKO (bottom panel) mice. Sections are representative of n = 4 mice per genotype. C, D: Serum tumor necrosis factor alpha (TNFα) and interleukin-6 (IL-6) concentrations. After 10 weeks of HFD, animals were injected intraperitoneally with 7 mg/kg lipopolysaccharide, and serum TNFα (C) and IL-6 (D) concentrations were then measured at the indicated time points (n = 7 per group). A, C, D: Data represent mean ± SEM of two separate experiments over 18 months. * P < 0.05 vs. WT.
Pair-feeding studies
To determine whether changes in oxygen consumption, hepatic insulin signaling, and systemic inflammation are a consequence of the targeted disruption of hypothalamic FAS, we food restricted a separate cohort of wild-type mice by pair-feeding them to FASKO animals for 15 days following 10 wks of high-fat feeding. This intervention resulted in considerable weight loss leading to nearly identical body weight and adiposity in the pair-fed wild-type and freely- fed FASKO animals (Fig. 8A–C). The percentage of lean mass was unaffected (Fig. 8D). Remarkably, despite this reduction in adiposity, the pair-fed wild-type mice continued to show similar oxygen consumption as the ad libitum fed wild-type controls (Fig. 8E). The leaner pair-fed wild-type mice failed to increase hepatic 473Ser-Akt phosphorylation above that of obese freely fed wild-type controls (Fig. 8F), despite having similar fasting insulin levels as the FASKO mice (2.3 ± 0.6 ng/ml in ad libitum HFD-fed FASKO vs. 3.1 ± 0.8 ng/ml in pair-fed wild-type, n = 6, P = NS). This finding further confirms the increased hepatic insulin sensitivity in FASKO animals. Pair-feeding wild-type with FASKO animals also did not lower their peak serum TNFα concentration (at 1 h) in response to lipopolysaccharide injection, which remained comparable to freely fed obese wild-type control mice (Fig. 8G). Because, by design, adiposity in FASKO and pair-fed wild-type mice was the same, the higher oxygen consumption, increased Akt phosphorylation, and lower serum TNFα levels in response to endotoxin in the FASKO animals suggest that these effects are due to hypothalamic FAS inactivation, rather than a consequence of adiposity.
Fig. 8.

Pair-feeding studies in HFD fed WT and FASKO mice. A–E: Effect of pair-feeding WT mice to ad libitum fed FASKO animals on body weight gain (A), percent change in body weight compared with corresponding ad libitum fed controls (B), adiposity (C), percentage of lean mass (D), and oxygen consumption (by indirect calorimetry integrated over a 24 h period) (E). Results represent mean ± SEM of 7–10 animals per group. * P < 0.05 compared with WT control (Ctrl). F: Immunoblotting for 473Ser-phosphorylated and total Akt in the liver after pair-feeding in response to an intraportal bolus injection of 0.1 U/kg regular insulin (top panel), and corresponding densitometric quantification (bottom panel). Data represent mean ± SEM of four animals per group. * P < 0.05 vs. WT-Ctrl. # P < 0.05 vs. WT-pair-fed. G: Serum TNFα concentration at the indicated time points in control and pair-fed WT and FASKO mice in response to 7 mg/kg lipopolysaccharide injection. Results are mean ± SEM of 5–6 animals per group. * P < 0.05 vs. WT-Ctrl. # P < 0.05 vs. WT-PF. H: An integrative model for the regulatory role of brain FAS in energy homeostasis. Hypothalamic FAS deficiency impacts multiple organ systems culminating in resistance to diet-induced obesity (DIO) and increased insulin sensitivity.
DISCUSSION
Simply decreasing adiposity may not be sufficient for treating obesity-related disorders such as the metabolic syndrome and type 2 diabetes. There are examples of disruption of the expected inverse relationship between adiposity and aberrant metabolism. Despite decreased adiposity, inactivation of stearoyl-CoA desaturase-1 promotes the development of diabetes in leptin-deficient ob/ob mice (31). Conversely, despite increased adiposity, mice with inactivation of TLR-4 (32), as well as mice with overexpression of adiponectin (33), have enhanced insulin sensitivity. Novel therapies most likely to provide benefit will not only decrease adiposity but also ameliorate pathophysiologic conditions such as insulin resistance, fatty liver, and systemic inflammation. Here we demonstrate that central nervous system inhibition of de novo lipogenesis mediated by FAS protects mice from HFD-induced diabetes and results in enhanced hepatic insulin signaling, less fatty liver, decreased circulating triglycerides and free fatty acids, an improved adipokine profile, and evidence of decreased oxidative stress as well as systemic inflammation.
Our initial characterization of these mice on standard chow demonstrated decreased adiposity due to a combination of hypophagia and increased locomotor activity (17). The current results extend this work by showing that the decreased food intake and increased activity phenotype is preserved in the setting of high-fat feeding and protects mice from obesity. These findings, while logical, were not necessarily predictable. The product of the FAS reaction, mostly palmitate, appears to serve a signaling function that includes activation of Pparα signaling (20). Due to the fact that HFDs are enriched in palmitate (see supplementary Table I), it was possible that the provision of large amounts of this molecule could reverse the behavioral phenotype of these mice. This did not occur (17), indicating that either dietary palmitate cannot substitute for the product of the FAS reaction in brain or FAS is compartmentalized in the central nervous system to sites critical for energy homeostasis. Our pair-feeding studies support a potentially direct role for hypothalamic FAS in the regulation of bioenergetics (Fig. 8E).
The current data also show that the absence of FAS expression in pancreatic β cells does not affect β-cell function even under conditions of nutrient excess. In reductionist systems, de novo lipogenesis in islets may be important for normal function (34) but in intact mice, this process does not impact insulin secretion from islets isolated from mice following low-fat chow (17) or high-fat (Fig. 4) feeding.
Limitations to this work include the fact that these animals are in a mixed (BL/6 and 129) background and utilize the RIP-driven ectopic expression of the Cre recombinase, which has been shown to affect β-cell function (35). While genetic admixture and Cre could have confounding effects, controls were littermates of the FASKO mice and included Cre only in addition to floxed only mice. We also failed to observe any β−cell phenotype.
Beneficial effects at the liver were striking in high-fat fed FASKO mice. Hepatic glucose production was decreased (Fig. 4H) without an effect on peripheral glucose disposal (Fig. 4I), suggesting that much of the enhanced glucose tolerance in high-fat fed FASKO mice was due to effects at the liver. Consistent with this observation, insulin signaling was increased in liver but not muscle of FASKO as compared with control mice (Fig. 5). In FASKO as compared with control mice, the lipid content of liver but not muscle was decreased with high-fat feeding (Fig. 6). Expression of sterol-regulatory element binding protein-1c, an insulin responsive transcription factor important for lipogenesis, was substantially decreased in the FASKO mice (Fig. 6F), which likely contributes to their protection from fatty liver because their tissues were notable for the absence of up-regulated expression of genes involved in fat oxidation, uncoupled oxidative phosphorylation, or futile energy cycling (Fig. 2G). Consistent with our sterol-regulatory element binding protein-1c findings in liver, hepatic sterol-regulatory element binding protein expression is known to be increased in mouse models of obesity and diabetes (36). Additional mechanisms may also be involved in the protection from hepatic steatosis. CD36 expression was decreased in FASKO as compared with control livers (Fig. 6F), consistent with previous reports showing CD36 up-regulation in HFD-induced steatosis (29). FASKO mice are more active than controls (Fig. 2H), consistent with studies implicating increased physical activity in the protection against fatty liver in rodents (37).
Observations from the present study with high-fat feeding and our previous data in chow-fed FASKO mice (17) collectively support the notion of FAS functioning as a nutritional sensor within the hypothalamus. Inhibition of hypothalamic carnitine palmitoyl transferase-1 (38) and accumulation of malonyl-CoA (39) independently reduce food intake. Products of the FAS reaction serve as endogenous ligands to activate PPARα (17, 20). FASKO mice demonstrate reduced expression of carnitine palmitoyl transferase-1 and malonyl-CoA decarboxylase, targets of PPARα (17). Thus, manipulations of hypothalamic fatty acid oxidation (via inhibition of carnitine palmitoyl transferase-1) and synthesis (via accumulation of malonyl-CoA) that alter feeding behavior can be orchestrated through FAS.
Inactivation of hypothalamic FAS also affects glucose metabolism, providing another example of how FAS might serve as a nutritional sensor and contributing to a growing body of evidence identifying the brain-liver axis as a mediator of glucose homeostasis (24, 40, 41). Disrupting central nervous system liver neural signaling lowers blood glucose and blood pressure in the setting of glucocorticoid-induced insulin resistance (24). Central inhibition of lipid oxidation in the hypothalamus decreases hepatic glucose production in overfed rats (40). Central administration of lactate decreases glucose production in rats with diet-induced insulin resistance (41). Now we show that inactivation of lipogenesis in the hypothalamus (see supplementary Table II) reduces glucose production (Fig. 4H) and enhances hepatic insulin signaling in the setting of nutrient excess (Fig. 5B, C), independent of AMPK activation (see supplementary Fig. I) and adiposity (Fig. 8F). As glucose production by the liver is the main endogenous fuel source, hypothalamic FAS appears to regulate exogenous (via food intake) and endogenous (via glucose production) energy availability.
Systemic oxidative stress correlates with fat accumulation (42) and may promote insulin resistance (43, 44). Increasing oxidative metabolism increases production of reactive oxygen species, which may induce inflammation (30). Thus, while increasing fat oxidation might be considered beneficial by decreasing body weight (6–14), it may increase reactive oxygen species, and, depending on the tissue in which such species are generated, this effect could impair physiology. Global stearoyl-CoA desaturase-1 deficiency, which increases the rate of β-oxidation (11), increases proinflammatory cytokines and predisposes to acute colitis (16). Accelerating metabolism specifically in the vasculature increases the chronic inflammatory condition of atherosclerosis (45). In contrast, FASKO mice have less oxidant stress (Fig. 7A) and less systemic inflammation as manifested by decreased serum levels of TNFα and IL-6 (Fig. 7C, D), inflammatory markers elevated in insulin resistance and obesity (46, 47). At least some of this effect is mediated by macrophage accumulation in adipose tissue (23), and insulin resistance is ameliorated by inhibiting macrophage recruitment in animal models of obesity (48, 49). Consistent with these findings, macrophages were less apparent in adipose tissue of FASKO mice (Fig. 7B, bottom).
Although decreased inflammation in FASKO mice could reflect decreased adiposity, this effect appears to be due to a specific consequence of central FAS deficiency rather than body-fat content as suggested by pair-feeding studies (Fig. 8). Despite reducing adiposity of wild-type mice to the same level as the FASKO animals (Fig. 8C), the leaner pair-fed wild-type mice continued to manifest similar peak serum TNFα concentrations as the obese ad libitum HFD fed wild-type controls in response to endotoxin (Fig. 8G). These studies lend support to the notion that the brain can affect inflammation. Electrical stimulation of the vagus nerve in vivo during lethal endotoxinemia in rats attenuates TNFα levels and prevents shock (50). Perturbation of de novo lipogenesis in FASKO mice, which alters systemic energy balance, also appears to directly affect systemic inflammation.
In summary, our data suggest a critical role for hypothalamic FAS in the orchestration of bioenergetics, hepatic insulin signaling, and systemic inflammation resulting in obesity-resistance and enhanced insulin sensitivity (Fig. 8H). Medicinal chemists frequently pursue potential drug targets with a goal of excluding compounds from the central nervous system in hopes of diminishing behavioral side effects. The current data suggest that a strategy of targeting FAS at specific sites in the brain could coordinately improve multiple features of obesity-related disease.
Supplementary Material
Acknowledgments
We thank Dr. Barbara Mickelson at Harlan Teklad for her assistance with obtaining fatty acid composition of the high-fat diet. We are indebted to Dr. John Turk for his expert advice regarding lipid mass spectrometric analyses.
Abbreviations
HFD, high-fat diet
IL-6, interleukin-6
FASKO, fatty acid synthase knockout
Ppar, peroxisome-proliferator-activated receptor
TNFα, tumor necrosis factor alpha
Published, JLR Papers in Press, November 22, 2008.
Footnotes
This work was supported by the American Diabetes Association (Junior Faculty Award 1-07-JF-12 to MVC and a Mentor-Based Postdoctoral Fellowship Award), National Institutes of Health grants DK076729 and P50 HL083762, and the Clinical Nutrition Research Unit (DK56341) and Diabetes Research and Training Center (DK20579).
s⃞The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of two tables and one figure.
References
- 1.Ogden C. L., M. D. Carroll, L. R. Curtin, M. A. McDowell, C. J. Tabak, and K. M. Flegal. 2006. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 295 1549–1555. [DOI] [PubMed] [Google Scholar]
- 2.Li Z., S. Bowerman, and D. Heber. 2005. Health ramifications of the obesity epidemic. Surg. Clin. North Am. 85 681–701 (v.). [DOI] [PubMed] [Google Scholar]
- 3.Ford E. S., W. H. Giles, and A. H. Mokdad. 2004. Increasing prevalence of the metabolic syndrome among US adults. Diabetes Care. 27 2444–2449. [DOI] [PubMed] [Google Scholar]
- 4.Alberti K. G., P. Zimmet, and J. Shaw. 2006. Metabolic syndrome–a new world-wide definition. A consensus statement from the International Diabetes Federation. Diabet. Med. 23 469–480. [DOI] [PubMed] [Google Scholar]
- 5.Isomaa B., P. Almgren, T. Tuomi, B. Forsen, K. Lahti, M. Nissen, M. R. Taskinen, and L. Groop. 2001. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 24 683–689. [DOI] [PubMed] [Google Scholar]
- 6.Abu-Elheiga L., W. Oh, P. Kordari, and S. J. Wakil. 2003. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc. Natl. Acad. Sci. USA. 100 10207–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H. C., D. R. Jensen, H. M. Myers, R. H. Eckel, and R. V. Farese, Jr. 2003. Obesity resistance and enhanced glucose metabolism in mice transplanted with white adipose tissue lacking acyl CoA:diacylglycerol acyltransferase 1. J. Clin. Invest. 111 1715–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones J. R., C. Barrick, K. A. Kim, J. Lindner, B. Blondeau, Y. Fujimoto, M. Shiota, R. A. Kesterson, B. B. Kahn, and M. A. Magnuson. 2005. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. USA. 102 6207–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalaany N. Y., K. C. Gauthier, A. M. Zavacki, P. P. Mammen, T. Kitazume, J. A. Peterson, J. D. Horton, D. J. Garry, A. C. Bianco, and D. J. Mangelsdorf. 2005. LXRs regulate the balance between fat storage and oxidation. Cell Metab. 1 231–244. [DOI] [PubMed] [Google Scholar]
- 10.Li B., L. A. Nolte, J. S. Ju, D. H. Han, T. Coleman, J. O. Holloszy, and C. F. Semenkovich. 2000. Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat. Med. 6 1115–1120. [DOI] [PubMed] [Google Scholar]
- 11.Ntambi J. M., M. Miyazaki, J. P. Stoehr, H. Lan, C. M. Kendziorski, B. S. Yandell, Y. Song, P. Cohen, J. M. Friedman, and A. D. Attie. 2002. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA. 99 11482–11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Um S. H., F. Frigerio, M. Watanabe, F. Picard, M. Joaquin, M. Sticker, S. Fumagalli, P. R. Allegrini, S. C. Kozma, J. Auwerx, et al. 2004. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 431 200–205. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y. X., C. H. Lee, S. Tiep, R. T. Yu, J. Ham, H. Kang, and R. M. Evans. 2003. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 113 159–170. [DOI] [PubMed] [Google Scholar]
- 14.Yuan M., N. Konstantopoulos, J. Lee, L. Hansen, Z. W. Li, M. Karin, and S. E. Shoelson. 2001. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 293 1673–1677. [DOI] [PubMed] [Google Scholar]
- 15.Bradley M. N., C. Hong, M. Chen, S. B. Joseph, D. C. Wilpitz, X. Wang, A. J. Lusis, A. Collins, W. A. Hseuh, J. L. Collins, et al. 2007. Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. J. Clin. Invest. 117 2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C., Y. M. Shah, K. Morimura, K. W. Krausz, M. Miyazaki, T. A. Richardson, E. T. Morgan, J. M. Ntambi, J. R. Idle, and F. J. Gonzalez. 2008. Metabolomics reveals that hepatic stearoyl-CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab. 7 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakravarthy M. V., Y. Zhu, M. Lopez, L. Yin, D. F. Wozniak, T. Coleman, Z. Hu, M. Wolfgang, A. Vidal-Puig, M. D. Lane, et al. 2007. Brain fatty acid synthase activates PPARalpha to maintain energy homeostasis. J. Clin. Invest. 117 2539–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Semenkovich C. F. 1997. Regulation of fatty acid synthase (FAS). Prog. Lipid Res. 36 43–53. [DOI] [PubMed] [Google Scholar]
- 19.Cha S. H., Z. Hu, S. Chohnan, and M. D. Lane. 2005. Inhibition of hypothalamic fatty acid synthase triggers rapid activation of fatty acid oxidation in skeletal muscle. Proc. Natl. Acad. Sci. USA. 102 14557–14562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakravarthy M. V., Z. Pan, Y. Zhu, K. Tordjman, J. G. Schneider, T. Coleman, J. Turk, and C. F. Semenkovich. 2005. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 1 309–322. [DOI] [PubMed] [Google Scholar]
- 21.Gates A. C., C. Bernal-Mizrachi, S. L. Chinault, C. Feng, J. G. Schneider, T. Coleman, J. P. Malone, R. R. Townsend, M. V. Chakravarthy, and C. F. Semenkovich. 2007. Respiratory uncoupling in skeletal muscle delays death and diminishes age-related disease. Cell Metab. 6 497–505. [DOI] [PubMed] [Google Scholar]
- 22.Pappan K. L., Z. Pan, G. Kwon, C. A. Marshall, T. Coleman, I. J. Goldberg, M. L. McDaniel, and C. F. Semenkovich. 2005. Pancreatic beta-cell lipoprotein lipase independently regulates islet glucose metabolism and normal insulin secretion. J. Biol. Chem. 280 9023–9029. [DOI] [PubMed] [Google Scholar]
- 23.Weisberg S. P., D. McCann, M. Desai, M. Rosenbaum, R. L. Leibel, and A. W. Ferrante, Jr. 2003. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernal-Mizrachi C., L. Xiaozhong, L. Yin, R. H. Knutsen, M. J. Howard, J. J. Arends, P. Desantis, T. Coleman, and C. F. Semenkovich. 2007. An afferent vagal nerve pathway links hepatic PPARalpha activation to glucocorticoid-induced insulin resistance and hypertension. Cell Metab. 5 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weir G. C., and S. Bonner-Weir. 2004. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 53 (Suppl 3): S16–S21. [DOI] [PubMed] [Google Scholar]
- 26.Du K., S. Herzig, R. N. Kulkarni, and M. Montminy. 2003. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 300 1574–1577. [DOI] [PubMed] [Google Scholar]
- 27.Shimomura I., Y. Bashmakov, S. Ikemoto, J. D. Horton, M. S. Brown, and J. L. Goldstein. 1999. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA. 96 13656–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abumrad N. A., M. R. el-Maghrabi, E. Z. Amri, E. Lopez, and P. A. Grimaldi. 1993. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J. Biol. Chem. 268 17665–17668. [PubMed] [Google Scholar]
- 29.Inoue M., T. Ohtake, W. Motomura, N. Takahashi, Y. Hosoki, S. Miyoshi, Y. Suzuki, H. Saito, Y. Kohgo, and T. Okumura. 2005. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem. Biophys. Res. Commun. 336 215–222. [DOI] [PubMed] [Google Scholar]
- 30.Hotamisligil G. S. 2006. Inflammation and metabolic disorders. Nature. 444 860–867. [DOI] [PubMed] [Google Scholar]
- 31.Flowers J. B., M. E. Rabaglia, K. L. Schueler, M. T. Flowers, H. Lan, M. P. Keller, J. M. Ntambi, and A. D. Attie. 2007. Loss of stearoyl-CoA desaturase-1 improves insulin sensitivity in lean mice but worsens diabetes in leptin-deficient obese mice. Diabetes. 56 1228–1239. [DOI] [PubMed] [Google Scholar]
- 32.Shi H., M. V. Kokoeva, K. Inouye, I. Tzameli, H. Yin, and J. S. Flier. 2006. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 116 3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J. Y., E. van de Wall, M. Laplante, A. Azzara, M. E. Trujillo, S. M. Hofmann, T. Schraw, J. L. Durand, H. Li, G. Li, et al. 2007. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Invest. 117 2621–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brun T., E. Roche, F. Assimacopoulos-Jeannet, B. E. Corkey, K. H. Kim, and M. Prentki. 1996. Evidence for an anaplerotic/malonyl-CoA pathway in pancreatic beta-cell nutrient signaling. Diabetes. 45 190–198. [DOI] [PubMed] [Google Scholar]
- 35.Lee J. Y., M. Ristow, X. Lin, M. F. White, M. A. Magnuson, and L. Hennighausen. 2006. RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J. Biol. Chem. 281 2649–2653. [DOI] [PubMed] [Google Scholar]
- 36.Shimomura I., Y. Bashmakov, and J. D. Horton. 1999. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 274 30028–30032. [DOI] [PubMed] [Google Scholar]
- 37.Rector R. S., J. P. Thyfault, R. T. Morris, M. J. Laye, S. J. Borengasser, F. W. Booth, and J. A. Ibdah. 2008. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am. J. Physiol. Gastrointest. Liver Physiol. 294 G619–G626. [DOI] [PubMed] [Google Scholar]
- 38.Obici S., Z. Feng, A. Arduini, R. Conti, and L. Rossetti. 2003. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat. Med. 9 756–761. [DOI] [PubMed] [Google Scholar]
- 39.Hu Z., Y. Dai, M. Prentki, S. Chohnan, and M. D. Lane. 2005. A role for hypothalamic malonyl-CoA in the control of food intake. J. Biol. Chem. 280 39681–39683. [DOI] [PubMed] [Google Scholar]
- 40.Pocai A., T. K. Lam, S. Obici, R. Gutierrez-Juarez, E. D. Muse, A. Arduini, and L. Rossetti. 2006. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J. Clin. Invest. 116 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chari M., C. K. Lam, P. Y. Wang, and T. K. Lam. 2008. Activation of central lactate metabolism lowers glucose production in uncontrolled diabetes and diet-induced insulin resistance. Diabetes. 57 836–840. [DOI] [PubMed] [Google Scholar]
- 42.Rosen P., P. P. Nawroth, G. King, W. Moller, H. J. Tritschler, and L. Packer. 2001. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab. Res. Rev. 17 189–212. [DOI] [PubMed] [Google Scholar]
- 43.Xu H., G. T. Barnes, Q. Yang, G. Tan, D. Yang, C. J. Chou, J. Sole, A. Nichols, J. S. Ross, L. A. Tartaglia, et al. 2003. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Houstis N., E. D. Rosen, and E. S. Lander. 2006. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 440 944–948. [DOI] [PubMed] [Google Scholar]
- 45.Bernal-Mizrachi C., A. C. Gates, S. Weng, T. Imamura, R. H. Knutsen, P. DeSantis, T. Coleman, R. R. Townsend, L. J. Muglia, and C. F. Semenkovich. 2005. Vascular respiratory uncoupling increases blood pressure and atherosclerosis. Nature. 435 502–506. [DOI] [PubMed] [Google Scholar]
- 46.Hotamisligil G. S., N. S. Shargill, and B. M. Spiegelman. 1993. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 259 87–91. [DOI] [PubMed] [Google Scholar]
- 47.Petersen E. W., A. L. Carey, M. Sacchetti, G. R. Steinberg, S. L. Macaulay, M. A. Febbraio, and B. K. Pedersen. 2005. Acute IL-6 treatment increases fatty acid turnover in elderly humans in vivo and in tissue culture in vitro. Am. J. Physiol. Endocrinol. Metab. 288 E155–E162. [DOI] [PubMed] [Google Scholar]
- 48.Kanda H., S. Tateya, Y. Tamori, K. Kotani, K. Hiasa, R. Kitazawa, S. Kitazawa, H. Miyachi, S. Maeda, K. Egashira, et al. 2006. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest. 116 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lumeng C. N., J. L. Bodzin, and A. R. Saltiel. 2007. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borovikova L. V., S. Ivanova, M. Zhang, H. Yang, G. I. Botchkina, L. R. Watkins, H. Wang, N. Abumrad, J. W. Eaton, and K. J. Tracey. 2000. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 405 458–462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.