

Abstract
We previously reported that LDL modified by group V secretory phospholipase A2 (GV-LDL) promotes macrophage foam cell formation through a mechanism independent of scavenger receptors SR-A and CD36, and dependent on cellular proteoglycans. This study investigates the role of syndecans, a family of cell surface proteoglycans known to mediate endocytosis through macropinocytosis, in macrophage uptake of GV-LDL. LY 294002, a phosphatidylinositol 3-kinase inhibitor, significantly reduced internalization of 125I-labeled GV-LDL in J-774 macrophages, consistent with a macropinocytic uptake pathway. Using small, interfering RNA-directed gene silencing, we demonstrated a direct relationship between 125I-labeled GV-LDL binding and the level of syndecan-3 and syndecan-4 expression in J-774 cells. However, 125I-labeled GV-LDL uptake was significantly reduced only when syndecan-4 expression was suppressed. Peritoneal macrophages from syndecan-4-deficient mice exhibited markedly reduced uptake of fluorescently labeled GV-LDL compared with wild-type cells. Furthermore, cholesteryl ester accumulation induced by GV-LDL was dependent on syndecan-4 expression. Syndecan-4 expression and GV-LDL binding were significantly increased in J-774 cells treated with lipopolysaccharide, suggesting that GV-LDL uptake via this pathway may be enhanced during inflammation. Taken together, our data point to a novel role for syndecan-4 in mediating the uptake of GV-LDL, a process implicated in atherosclerotic lesion progression.
Keywords: atherosclerosis, foam cells, macropinocytosis, proteoglycan, lipoprotein modification
According to current paradigms, mechanisms that increase LDL1 influx and retention in the subendothelium and/or enhance LDL uptake by intimal macrophages are key initiating events in atherogenesis (1, 2). A number of processes that affect LDL composition and structure, such as oxidation or alterations in phospholipid content, have been shown to increase the binding affinity of LDL to extracellular matrix (ECM) proteoglycans, promote macrophage LDL uptake, or both (3–7). Accumulating evidence suggests that secretory phospholipase A2 (sPLA2) enzymes may promote atherosclerosis through modification of LDL. Of the 10 sPLA2 family members that have been described in mammals, group V (GV), GX, and very recently, GIII sPLA2 have been shown to be potent in hydrolyzing LDL in vitro (8–10). A role for GV sPLA2 in atherosclerotic lipid deposition in vivo has recently been substantiated by gain- and loss-of-function studies in LDL receptor-deficient mice (11). The ability of GV sPLA2 to hydrolyze LDL is significantly enhanced in the presence of arterial proteoglycans, and in turn, hydrolysis by GV sPLA2 increases the affinity of LDL for proteoglycans (12). This increased binding affinity of hydrolyzed LDL appears to be due to an alteration in the conformation of apolipoprotein B-100 (apoB-100) on the phospholipid-depleted LDL particle, which exposes cryptic proteoglycan binding sites (13). In addition to exhibiting enhanced binding to ECM proteoglycans, LDL particles modified by GV sPLA2 (GV-LDL) are avidly taken up by macrophages to form foam cells (14, 15). Interestingly, treatments that disrupt cellular proteoglycans significantly reduce macrophage uptake of GV-LDL (15). Thus, GV sPLA2 hydrolysis appears to bring about structural changes in LDL particles that promote interactions with both cellular and ECM proteoglycans. What remains to be determined is the precise molecular pathway by which hydrolyzed LDLs are taken up by macrophages, and their subsequent metabolic fate.
In the present study we investigated the cellular pathway involved in GV-LDL uptake by macrophages. Our results point to syndecans as major binding sites for GV-LDL, and indicate that syndecan-4 mediates GV-LDL uptake, possibly through macropinocytosis.
EXPERIMENTAL PROCEDURES
Isolation of mouse peritoneal macrophages
C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME). CD36−/− mice were kindly provided by R. L. Silverstein (Cleveland Clinic Foundation, Cleveland, OH) (16). The generation of mice with targeted deletion of syndecan-4 has already been described (17). Prior to collection of peritoneal macrophages, animals were injected intraperitoneally with a sterile solution (1 ml) of 1% Biogel 100 (Bio-Rad) in PBS. After 96 h, peritoneal macrophages were harvested by lavage with 5 ml of ice-cold PBS. All procedures were in accordance with the guidelines of the Veterans Affairs Institutional Animal Care and Use Committee.
Macrophage cell culture
Mouse peritoneal macrophages (MPMs) were seeded in 12-well dishes at a density of ∼2.0 × 106 cells/well in DMEM supplemented with 10% FBS, 100 U/ml penicillin/streptomycin, 2 mM l-glutamine, and 25 ng/ml macrophage colony-stimulating factor (Calbiochem) and allowed to attach for 4 h. Nonattached cells were then removed by washing the dishes with PBS, and then incubated overnight at 37°C prior to experiments. J-774 macrophage-like cells were obtained from the American Type Culture Collection. The cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin/streptomycin, and 2 mM l-glutamine. When indicated, cells were preincubated for 4 h with 1 U/ml heparinase III and 0.15 U/ml chondroitinase ABC cocktail (Sigma) in PBS containing 0.1 M sodium acetate and 0.1 mM calcium acetate (pH 7.0). Alternatively, cells were preincubated in culture medium containing 50 μM LY 294002 (LY 294) compound (Signagen, Gaithersburg, MD) for 1 h or for the indicated times with 100 ng/ml lipopolysaccharide (LPS) prior to binding and cell association assays. None of the treatments affected the cell viability.
RNA silencing with small interfering RNA
A set of predesigned, double-stranded synthetic oligonucleotides directed to mouse syndecan-3 with the sequences 5′-CCACUACUAGAAUCCUAAATT (sense), 5′-UUUAGGAUUCUAGUAGUGGTT (antisense) or syndecan-4: 5′-GGCACCUUAAUGCUGACUUTT (sense), 5′-AAGUCAGCAUUAAGGUGCCTG (antisense) were purchased from Ambion (Austin, Texas). For control, a validated non-targeting small interfering RNA (siRNA, Ambion) of equal concentration was used. The oligonucleotides (3 μg per 1 × 106 cells) were transfected into J-774 cells using Nucleofector Solution V (Amaxa, Gaithersburg, MD) and program X-001 of the nucleofector device (Amaxa) following the manufacturer's protocol. For silencing of both syndecan-3 and syndecan-4, 1.5 μg of each oligonucleotide per 1 × 106 cells was used. Transfections were carried out in bulk with each set of oligonucleotides, and cells were subsequently divided and plated in separate wells for analysis of RNA, protein, and ligand binding and uptake. Cells were collected 24 h after transfection for RNA preparation and 72 h after transfection for assessment of syndecan protein expression by flow cytometry, binding, or cell association assays.
Isolation, modification, and labeling of LDL
LDL (density 1.019–1.063) was isolated from the plasma of healthy volunteers by sequential ultracentrifugation and stored at 4°C under argon gas. For LDL hydrolysis, mouse GV sPLA2 was partially purified from conditioned media of COS-7 cells infected with a replication-defective adenoviral vector encoding this enzyme as previously described (14). Briefly, to prepare partially purified enzyme, media was sequentially chromatographed on Hi-Trap SP ion exchange and Hi-Trap heparin columns (Amersham Biosciences) as previously described for GIIa sPLA2 (18). Phospholipase activity of partially purified enzyme was determined using a colorimetirc assay with 1-palmitoyl-2-oleoyl-phosphatidyglycerol (POPG) as substrate (1 unit generates 1 nmol of FFA in 20 min using POPG as substrate). LDL particles (1 mg/ml) were incubated overnight in hydrolysis buffer (0.1 M HEPES, 0.1 M NaCl, 1 mM CaCl2, 10 mg/ml FA-free BSA, and 0.01% butylated hydroxytoluene) under argon gas at 37°C in the presence of 500 U/ml GV sPLA2. LDL hydrolysis was quantified by measuring the amount of FFAs released in the solution (14). As control, LDL was incubated overnight in hydrolysis buffer in the absence of the enzyme (“mock-LDL”). For the experiments described here, incubation with GV sPLA2 resulted in the hydrolysis of more than 70% of LDL phospholipid. As reported previously, there was no evidence that GV sPLA2 hydrolysis led to apoB degradation or alterations in the charge of LDL particles (15). To remove large aggregates, GV-LDL was filtered through a 0.1 μm filter (Whatman, Clifton, NJ) before experiments. To prepare oxidized LDL (ox-LDL), native LDL was dialyzed against 5 μM CuSO4 overnight at 4°C. Oxidation was stopped by adding EDTA at a final concentration of 1 mM. Ox-LDL was then dialyzed against 0.15 M saline and stored under argon gas. Relative electrophoretic mobility was assessed by separating the ligands for 90 min at 100 V on a 2% agarose gel (19). The relative mobility of ox-LDL compared with native LDL was ∼1.8. Thus, the relative mobility of the ox-LDL preparations corresponded to the “mildly” ox-LDL described by Kunjathoor et al. (20) (relative mobility ∼2.0), which has been shown to be preferentially internalized by macrophages via CD36. GV-LDL and ox-LDL were radiolabeled with 125I according to Bilheimer, Eisenberg, and Levy's (21) modification of the procedure described by Goldstein et al. Alternatively, native LDL and ox-LDL were labeled with Alexa-Fluor 568 and Alexa-Fluor 488, respectively (Molecular Probes, Eugene, OR), according to the manufacturer's instructions. Alexa-labeled native LDL was then hydrolyzed with GV sPLA2 as described previously.
Cell binding and cell association assays
For binding studies, cells were cooled to 4°C, and after washing with ice-cold PBS, were incubated with ice-cold buffer (50 mM Tris, 150 mM NaCl, 2 mg/ml FA-free BSA) containing the indicated concentrations of 125I-labeled ligands. After 2 h incubation at 4°C, medium was removed and cells were washed rapidly three times with washing buffer (50 mM Tris, 150 mM NaCl, 2 mg/ml FA-free BSA) followed by two washes with washing buffer without BSA. All washes were performed at 4°C with prechilled solutions. The cells were solubilized in 0.25 ml 0.1 N NaOH for 2 h on a rotary platform at room temperature. The total radioactivity was measured in a γ counter (Cobra II, Packard Instrument Co., Minneapolis, MN). For cell association assays, cells were incubated with the respective ligands for 6 h at 37°C and the total radioactivity was measured. To inhibit uptake via macropinocytosis, cells were preincubated with 50 μM LY 294 [a phosphatidylinositol 3 (PI3)-kinase inhibitor] for 1 h, and the respective ligands were added in the presence of the inhibitor. Dextran, known to be endocytosed via macropinocytosis (22), was labeled with 125I using the procedure described above and incubated with cells at a final concentration of 0.5 mg/ml.
Lipid extraction and cholesteryl ester measurements
After incubation with LDL ligands, cells were washed three times with washing buffer, followed by two washes with washing buffer without BSA. Lipids were extracted as described previously (23). Briefly, 1 ml hexane-isopropanol 3:2 (v/v) was added, and the cells were incubated at room temperature on a shaking platform for 1 h. Supernatants were dried in a Freezedry System (Freezone 4.5; Labconco, Kansas City, MO), and 1 ml 1% Triton X-100 in chloroform was added. Samples were dried again and dissolved in H2O. Aliquots were assayed for total and free cholesterol content using a colorimetric kit (Wako, Richmond, VA). Cholesteryl ester (CE) was calculated as the difference between the total and free cholesterol. After lipid extraction, cells were solubilized in 0.25 ml 0.1 N NaOH for 2 h on a shaking platform, and protein concentrations were determined using the BCA™ protein assay kit (Pierce, Rockford, IL).
Western blotting
Cellular lysates were prepared from J-774 cells after 4 h incubation with a heparinase III/chondroitinase ABC cocktail. Liver homogenates were prepared by sonication. Aliquots corresponding to 40 μg protein were separated on a 4–20% polyacrylamide gradient gel and immunoblotted with goat anti-mouse syndecan-3 (R and D Systems, Minneapolis, MN) or rabbit anti-mouse syndecan-4 (Santa Cruz, Santa Cruz, CA).
Confocal microscopy
J-774 cells were seeded on glass coverslips and grown until confluent. Cells were then preincubated at 37°C for 4 h in the presence or absence of 1 U/ml heparinase III and 0.15 U/ml chondroitinase ABC, followed by a 2 h incubation with 0.2 mg/ml Alexa-labeled ligands in culture medium and the enzyme cocktail. MPMs isolated from either C57BL/6, syndecan-4−/− or CD36−/− mice were incubated with 0.05 mg/ml Alexa-Fluor 488-labeled ox-LDL or Alexa-Fluor 568-labeled GV-LDL for 2 h. After incubations, cells were washed extensively and mounted on slides using Fluorescence Protecting Medium (Vectashield; Vector Laboratories, Burlingam, CA). Confocal microscopy was performed at the University of Kentucky Imaging Facility using a Leica laser scanning confocal microscope with argon (488 nm) and krypton (568 nm) lasers.
Indirect immunofluorescence
Cell surface syndecan expression in nonpermeabilized J-774 cells was assessed by confocal microscopy and by flow cytometry. J-774 cells were treated for 4 h with 1 U/ml heparinase III and 0.15 U/ml chondroitinase ABC and subjected to indirect immunofluorescence using goat anti-mouse syndecan-3 IgG (R and D Systems, Minneapolis, MN) or rat anti-mouse syndecan-4 IgG (BD Biosciences, Franklin Lakes, NJ). Alexa-Fluor 488-labeled rabbit anti-goat and goat anti-rat IgGs (Molecular Probes) were used as secondary antibodies at a dilution of 1:200. As control, cells were incubated in the absence of primary antibody. Confocal microscopy was carried out as described above. Alternatively, to quantify syndecan protein expression after the “silencing,” mean fluorescence intensity per cell was assessed by flow cytometry. Briefly, cells were incubated with heparinase III/chondroitinase ABC cocktail, followed by indirect immunofluorescence for syndecan-3 or syndecan-4 as described above, and harvested in 0.5 ml of PBS by gentle scraping.
Real-time RT-PCR
Total RNA was isolated from J-774 cells, MPMs, or liver homogenate using TriReagent (Molecular Research Center, Inc.). RNA (1–2 μg) was reverse transcribed into cDNA using a reverse transcription system (Promega). After 4-fold dilution, 5 μl was used as a template for real-time PCR. Amplification was done for 40 cycles using Power SYBR Green PCR Master Mix Kit (Applied Biosystems) and DNA Engine Optical 2 System (MJ Research, Inc.). Quantification was performed in duplicate using the standard curve method and normalizing to 18S. The primers used for various genes are as follows (gene, accession number: primers): mPerlecan, NM_008305: forward 5′-CTCAGTGTGACAAGTGTAAGCCTG-3′, reverse 5′-GGTTGTGGAAGGAAGAGAACGAC-3′; mSyndecan-1, NM_011519: forward 5′-ACTCTGACAACTTCTCTGGCTCTG-3′, reverse 5′-CTTCTTCTTCATCCGGTACAGCAT-3′; mSyndecan-2, NM_008304: forward 5′-CCCAAAGTGGAAACCATGAC-3′, reverse 5′-GATCACACCACCAGCAATGA-3′; mSyndecan-3, NM_011520: forward 5′-GATGATGAACTAGACGACCTCTAC-3′, reverse 5′-CTGTGTCCAAGGTAGCCACAGTAG-3′; mSyndecan-4, NM_011521: forward 5′-ATGGACCTGGAGCCAGCTCACCCCCAA-3′, reverse 5′-TCATGCGTAGAACTCATTGGTGGG-3′; m18S RNA, NR_003278: forward 5′-GCTGGAATTACCGCGGCT-3′, reverse 5′-CGGCTACCACATCCAAGGAA-3′.
Statistical analysis
Data are expressed as mean ± SE. Results were analyzed by Student's t-test or one-way ANOVA followed by Bonferroni's posttest. Values of P < 0.05 were considered statistically significant.
RESULTS
Macrophage uptake of GV-LDL depends on cellular proteoglycans
We reported previously (15) that uptake of GV-LDL, but not mock-LDL or ox-LDL, was reduced when MPMs were incubated with NaClO3 or heparin, two treatments that would be expected to disrupt the interaction of GV-LDL with cell surface proteoglycans (24, 25). To confirm a role for proteoglycans in macrophage uptake of GV-LDL, we investigated whether heparinase III + chondroitinase ABC alters the uptake of GV-LDL or ox-LDL by J-774 macrophage-like cells. J-774 cells were incubated for 4 h in the presence or absence of the enzyme cocktail and then for an additional 2 h with fluorescently labeled GV-LDL or ox-LDL (Fig. 1A). After the incubation, cells were fixed and subjected to confocal microscopy. Our results indicate that the uptake of GV-LDL, but not ox-LDL, was substantially reduced when glucosaminoglycan (GAG) side chains were cleaved from cell surface proteoglycans.
Fig. 1.
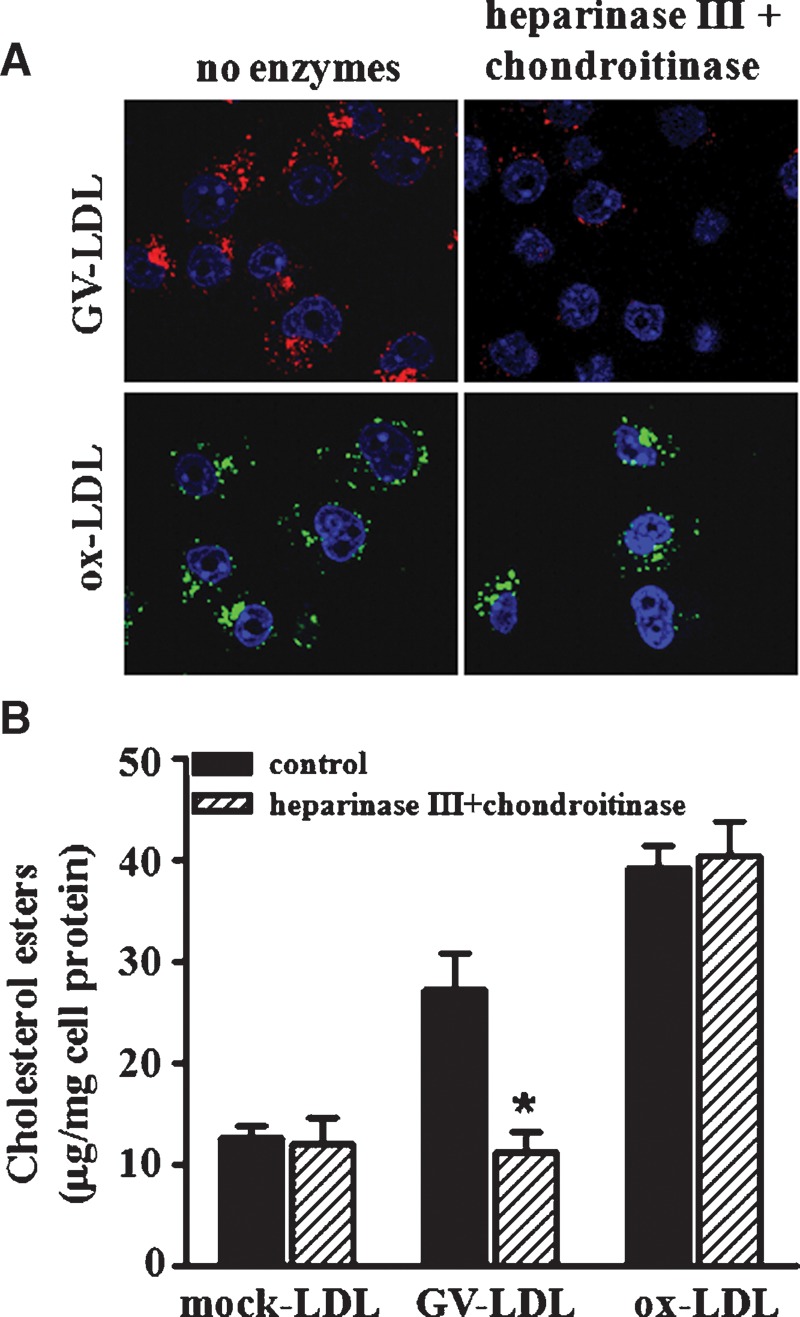
Macrophage uptake of group V sPLA2-modified LDL (GV-LDL) depends on cellular proteoglycans. J-774 cells were preincubated for 4 h in the presence or absence of 1 U/ml heparinase III + 0.15 U/ml chondroitinase ABC in PBS containing 0.1 M sodium acetate and 0.1 mM calcium acetate (pH 7.0). A: Cells were subsequently incubated for 2 h with complete media containing 0.05 mg/ml Alexa-568-labeled GV-LDL (red) or Alexa-488 ox-LDL (green), with or without the enzyme cocktail, and then processed for confocal microscopy. B: After the preincubation, cells were incubated for 24 h with 0.2 mg/ml mock-LDL, GV-LDL, or ox-LDL in culture media with the enzyme cocktail present, as indicated. Cellular lipids were extracted and cholesteryl esters were quantified. Values are presented as mean ± SE. * P < 0.05 compared with corresponding control cells.
As a more quantitative approach, we measured the effect of heparinase III/chondroitinase ABC treatment on intracellular CE accumulation in J-774 cells after 24 h incubations with 0.2 mg/ml mock-hydrolyzed LDL, GV-LDL, or ox-LDL (Fig. 1B). Similar to results reported previously for MPMs (15), hydrolysis of LDL by GV sPLA2 resulted in a significant 2.4-fold increase in CE accumulation in control J-774 cells compared with mock-hydrolyzed LDL. When cells were treated with the enzyme cocktail, CE accumulation induced by GV-LDL was significantly reduced, to a level comparable to mock-LDL. In contrast, treating cells with heparinase III/chondroitinase ABC had no effect on the amount of CE accumulation induced by ox-LDL. None of the treatments significantly changed cellular free cholesterol levels (data not shown). These data indicate that intact GAG side chains are necessary for macrophage uptake of GV-LDL, but not ox-LDL or mock-LDL.
Macrophage uptake of GV-LDL is disrupted by LY 294, an inhibitor of macropinocytosis
Our data indicate that GV-LDL is taken up by macrophages through a pathway that is independent of known receptors for modified LDL (15). Previous studies in human monocyte-derived macrophages identified a receptor-independent pathway leading to foam cell formation that involves macropinocytosis of native LDL, a process that is blocked by the PI3-kinase inhibitor LY 294 (26). Thus, it was of interest to determine whether LY 294 interferes with macrophage uptake of 125I-GV-LDL (Fig. 2A). 125I-labeled dextran was included in these experiments as a positive control (Fig. 2B). As expected, pretreatment with LY 294 resulted in significantly reduced (84.8 ± 8.9%) uptake of 125I-dextran. There was also a modest but significant 20.0 ± 6.7% decrease in the uptake of 125I-mock-LDL in LY 294-treated cells, consistent with a previous report that native LDL is partially taken up by macrophages through a macropinocytic pathway (26). Interestingly, the 2.3-fold increase in uptake of 125I-GV-LDL compared with 125I-mock-LDL was abolished when macrophages were treated with Ly 294 (Fig. 2A). In contrast, macrophage uptake of 125I-ox-LDL was not altered by Ly 294, providing additional evidence that macrophages internalize ox-LDL and GV-LDL through distinct pathways.
Fig. 2.

Macrophage uptake of GV-LDL is disrupted by LY 294, an inhibitor of macropinocytosis. Control J-774 cells and cells pretreated with 50 μM LY 294 for 1 h were incubated with 50 μg/ml 125I-mock LDL, 125I-labeled GV-LDL, or 125I-labeled ox-LDL (A), or 0.5 mg/ml 125I-labeled dextran (B), and the total cell-associated radioactivity was assessed after 4 h of incubation. Values are presented as mean ± SE.* P < 0.05 compared with control cells; ** P < 0.01.
GV-LDL binding to J-774 cells is syndecan-3 and syndecan-4 dependent
Syndecans comprise a family of cell surface heparan sulfate proteoglycans (HSPGs) that endocytose a variety of ligands, including lipoprotein lipase (LPL) enriched LDL (27). There is also evidence that syndecan-mediated endocytosis proceeds through macropinocytosis (22). Given our data that macrophage uptake of GV-LDL and not mock-LDL is reduced by agents known to disrupt cell surface HSPGs or macropinocytosis, we considered the possibility that syndecans play a role in the uptake of GV-LDL (15). Because expression of syndecans by J-774 cells had not been specifically addressed in the published literature, we performed RT-PCR, Western blotting, and indirect immunofluorscence to identify which members of the syndecan family are expressed by these cells. Liver was used as a positive control because this tissue has been shown to express all four syndecan isoforms (28, 29). Very low levels of syndecan-1 and -2 transcripts were detected in J-774 cells by RT-PCR, whereas syndecan-4 and syndecan-3 mRNAs were readily detected (Fig. 3A). Both syndecan-3 protein and syndecan-4 protein were detected by Western blotting (Fig. 3B), and indirect immunofluorescence staining confirmed cell surface expression of these two molecules (Fig. 3C).
Fig. 3.
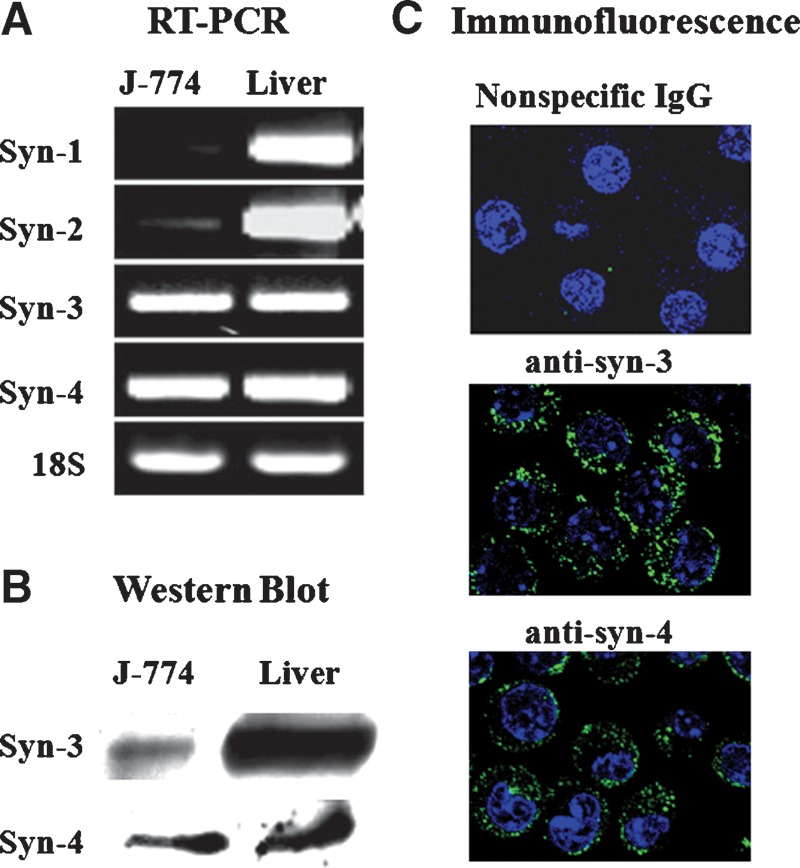
Syndecan-3 and -4 are expressed by J-774 macrophages. A: Expression of syndecan-1, -2, -3 and -4 mRNAs in J-774 cells and liver was assessed by RT-PCR using gene-specific primers. An equivalent amount of the RT reaction mix was separated on a 1.5% agarose gel. B: J-774 total cell lysate and liver homogenate (20 μg protein) were electrophoresed on a 4–20% polyacrylamide gradient gel and immunoblotted with goat anti-mouse syndecan-3 or rat anti-mouse syndecan-4 IgG. C: J-774 cells were incubated with 1 U/ml heparinase III + 0.15 U/ml chondroitinase ABC and then subjected to indirect immunofluorescence using goat anti-mouse syndecan-3 or rat anti-mouse syndecan-4 followed by confocal microscopy.
We employed a loss-of-function approach to investigate the role of syndecan-3 and syndecan-4 in macrophage binding and uptake of GV-LDL by J-774 cells. Specific siRNA-induced silencing resulted in ∼60–70% suppression of both syndecan-3 and syndecan-4 mRNA expression (Fig. 4A), which was accompanied by more modest 18.7 ± 3.6% and 22.3 ± 3.8% decreases in cell surface expression of syndecan-3 and syndecan-4 protein, respectively (Fig. 4B). Importantly, silencing of one molecule did not alter the expression of the other. In cells where both syndecans were suppressed by siRNA, protein expression of syndecan-3 was reduced 23.5 ± 5.9% and that of syndecan-4 26.5 ± 3.9% (Fig. 4B).
Fig. 4.

Macrophage syndecan-3 and syndecan-4 expression is suppressed by small, interfering RNAs (siRNAs). J-774 cells were transfected with siRNAs targeting syndecan-3 (Syn-3), syndecan-4, (Syn-4), both siRNAs together (Syn-3/4), or a validated nontargeting siRNA (Scr). A: After 24 h, total RNA was isolated and syndecan-3 and syndecan-4 mRNA expression was assessed by RT-PCR using gene-specific primers, and quantitative RT-PCR data were normalized to 18S. B: After 72 h, cells were incubated for 4 h with 1 U/ml heparinase + 0.15 U/ml chondroitinase ABC. Cells were then subjected to indirect immunofluorescence to measure syndecan-3 and syndecan-4 surface protein expression by flow cytometry. Data shown are relative mean fluorescent intensity per cell relative to cells transfected with control siRNA. Values are presented as mean ± SE. * P < 0.01.
To determine whether decreased cell surface expression of syndecan-3 or syndecan-4 results in a corresponding decrease in GV-LDL binding, we performed binding studies at 4°C using parallel dishes of J-774 cells (Fig. 5A). Compared with control cells, 125I-GV-LDL binding was significantly reduced by 15 ± 4.0% in macrophages with silenced expression of syndecan-3 and 20 ± 4.8% in cells with silenced expression of syndecan-4. For cells in which expression of both syndecans was suppressed, 125I-GV-LDL binding was reduced by 29 ± 5.5% (Fig. 5A). These results indicate a direct relationship between GV-LDL binding and the level of syndecan-3 and syndecan-4 expression, and possibly an additive effect of the two syndecans on binding. To provide additional evidence that syndecans mediate the interaction of GV-LDL with macrophages, we analyzed binding data from five separate experiments in which syndecan-3 and syndecan-4 were silenced to different extents. This analysis showed a significant correlation between the reduction in 125I-GV-LDL binding and the suppression of either syndecan-3 (r2 = 0.66, P < 0.05) or syndecan-4 (r2 = 0.76, P < 0.05) (data not shown). In contrast to GV-LDL, suppression of syndecan-3 or syndecan-4 had no effect on ox-LDL binding (not shown).
Fig. 5.

Macrophage GV-LDL binding and uptake is dependent on syndecan-4 expression. Separate dishes of transfected J-774 cells analyzed in Fig. 4 were used in 125I binding and uptake studies. A: Seventy-two hours after transfection, cells were cooled to 4°C and then incubated for 2 h at 4°C with 125I-labeled GV-LDL (50 μg/ml). Cells were washed, and cell-associated radioactivity was quantified. * P < 0.05 compared with control cells transfected with scrambled siRNA (Scr). B: Seventy-two hours after transfection, cells were incubated for 6 h at 37°C with 50 μg/ml 125I-labeled GV-LDL and washed, and total cell-associated radioactivity was quantified. Values are presented as mean ± SE. * P < 0.05 compared with control cells; ns, not significant.
Syndecan-4 mediates macrophage uptake of GV-LDL
In addition to GV-LDL binding, we also investigated the effect of syndecan-3 and syndecan-4 suppression on GV-LDL internalization by macrophages. Seventy-two hours after transfection with siRNAs, J-774 cells were incubated at 37°C for 6 h with 50 μg/ml 125I-labeled GV-LDL or ox-LDL, and the amount of total cell-associated radioactivity was quantified. Silencing of syndecan-3 or syndecan-4 did not significantly alter the amount of 125I-oxLDL cell association (not shown). In the case of 125I-GV-LDL, there was a significant 27.4 ± 5.1% decrease in cell association (Fig. 5B) in cells where cell surface syndecan-4 expression was reduced by 22.3 ± 3.8% (Fig. 4B). In contrast, GV-LDL uptake was not significantly reduced in cells transfected with syndecan-3 siRNA compared with control cells (Fig. 5B), despite a decrease of 18.7 ± 3.6% in syndecan-3 protein expression (see Fig. 4B). Although there was a trend for a larger decrease in GV-LDL uptake when both syndecan-3 and syndecan-4 were silenced compared with when syndecan-4 alone was silenced, this did not reach statistical significance (Fig. 5B), and probably reflects a more effective suppression of syndecan-4 protein expression (∼26.5% reduction) in the case of the doubly transfected cells (see Fig. 4B). Similarly to the 4°C binding studies, we analyzed cell association data from five separate experiments in which syndecan-3 and syndecan-4 were silenced to different extents. This analysis showed a significant correlation between the reduction in 125I-GV-LDL cell association and the suppression of syndecan-4 (r2 = 0.79, P < 0.05) but not syndecan-3 (r2 = 0.48) (data not shown). Moreover, we did not observe an effect on 125I-GV-LDL cell association even in an experiment in which syndecan-3 protein was suppressed ∼40% (data not shown).
To more convincingly demonstrate that syndecan-4 mediates GV-LDL internalization, we compared the ability of MPMs isolated from wild-type (C57BL/6) and syndecan-4−/− mice to take up fluorescently labeled mock-LDL, GV-LDL, and ox-LDL. Results from semi-quantitative RT-PCR analyses indicated that MPMs express syndecan-3 and syndecan-4 mRNA at levels similar to J-774 cells (Fig. 6A). However, syndecan-1 and syndecan-2 were expressed at significantly higher levels in MPMs compared with J-774 cells. There was no evidence that deficiency in syndecan-4 leads to a compensatory upregulation of syndecans 1–3 in MPMs (Fig. 6A). Interestingly, in the case of syndecan-4−/− MPMs, internalization of GV-LDL was markedly reduced compared with C57BL/6 MPMs, whereas ox-LDL appeared to be readily taken up (Fig. 6B, middle panel). To further highlight the difference in uptake mechanisms for ox-LDL and GV-LDL, MPMs from CD36−/− mice were also incubated with the fluorescently labeled ligands (Fig. 6B, right panel). As reported previously, uptake of ox-LDL, but not GV-LDL, was dependent on CD36 expression (15, 20). Compared with GV-LDL and oxLDL, uptake of fluorescently labeled mock-LDL by C57BL/6 MPMs was negligible (data not shown).
Fig. 6.
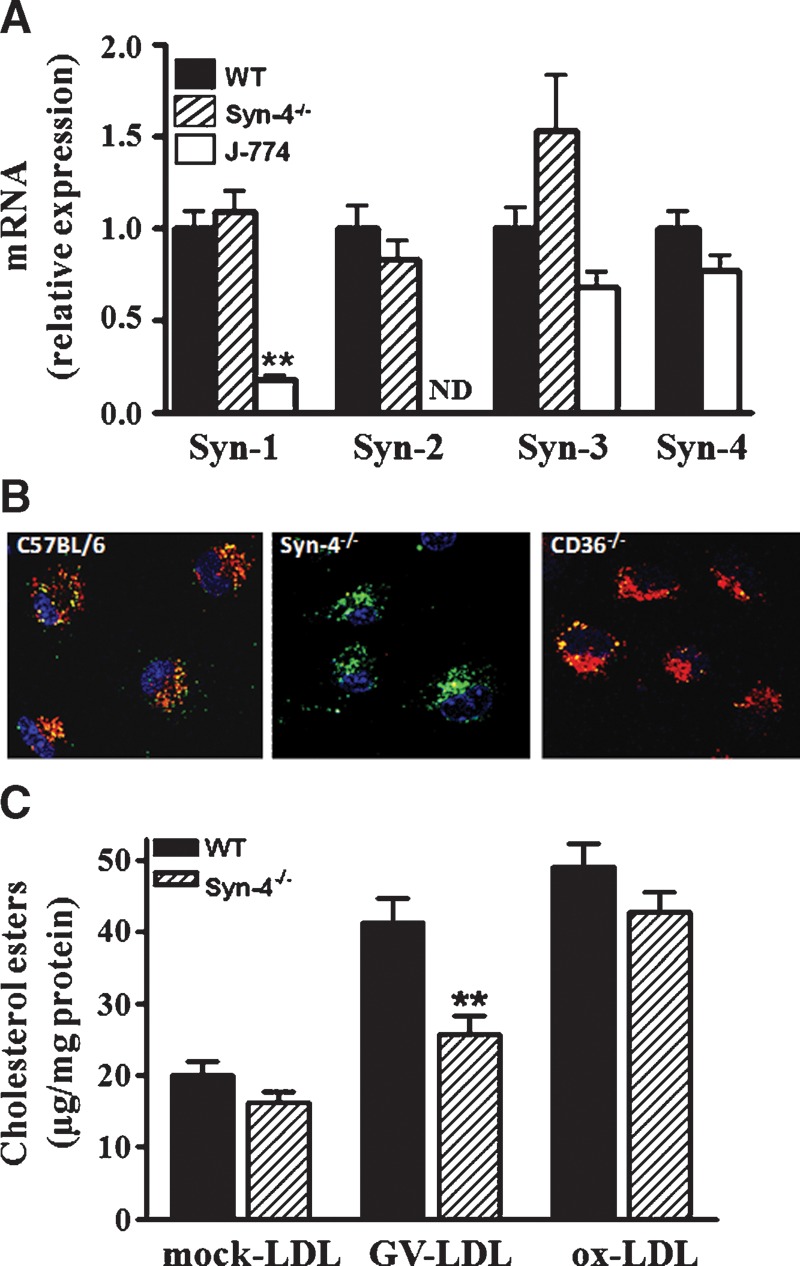
GV-LDL does not accumulate in macrophages from syndecan-4-deficient mice. A: Relative expression of syndecan 1–4 mRNA in macrophages from C57BL/6 or syndecan-4−/− mice was determined by real-time RT-PCR as described in Experimental Procedures. B: Peritoneal macrophages isolated from wild-type, syndecan-4−/−, or CD36−/− mice were coincubated with 50 μg/ml Alexa-Fluor 488-labeled ox-LDL (green) and 50 μg/ml Alexa-Fluor 568 GV-LDL (red) for 2 h. After the incubation, cells were subjected to confocal microscopy. C: Peritoneal macrophages isolated from wild-type or syndecan-4−/− mice were incubated with 0.2 mg/ml mock-LDL, GV-LDL, or ox-LDL for 48 h, and cell-associated cholesteryl ester was quantified. Values are presented as mean ± SE. ** P < 0.01 compared with values from wild-type mice.
The finding that targeted deletion of syndecan-4 leads to a defect in macrophage uptake of GV-LDL and not ox-LDL was confirmed by measuring CE accumulation in cells incubated with the ligands for 48 h (Fig. 6C). Similar to results for J-774 macrophages (Fig. 1B), C57BL/6 MPMs incubated with GV-LDL accumulated >2-fold more CE compared with MPMs incubated with mock-LDL (Fig. 6C). Notably, the bulk (more than 75%) of this increased CE accumulation was not observed when syndecan-4−/− MPMs were incubated with GV-LDL. Taken together, these findings support our previous conclusion that internalization of GV-LDL is not dependent on CD36 and that the uptake of ox-LDL and GV-LDL is mediated by different mechanisms. Our data also provide compelling evidence that GV-LDL uptake depends at least in part on syndecan-4 expression.
LPS increases syndecan expression and GV-LDL uptake by J-774 cells
According to previous reports, LPS upregulates syndecan-4 expression in mouse monocytes and endothelial cells (30). Consistent with this finding, we observed a ∼13-fold induction of syndecan-4 mRNA expression in J-774 cells treated with 100 ng/ml LPS (Fig. 7A). LPS treatment also resulted in a less dramatic ∼3-fold increase in syndecan-3 mRNA. These increases in mRNA expression were accompanied by ∼38% and ∼58% increases in cell surface expression of syndecan-3 and syndecan-4 protein, respectively (Fig. 7B). Interestingly, we also found a time-dependent increase in the binding of 125I-GV-LDL to cells treated with LPS compared with control cells (Fig. 7C) that was blocked by heparinase III/chondroitinase ABC cocktail (not shown). This finding provides the intriguing possibility that the interaction of GV-LDL with macrophages is enhanced during inflammation.
Fig. 7.

Lipopolysaccharide (LPS) increases syndecan expression and GV-LDL uptake by J-774 cells. A: Syndecan-3 and syndecan-4 mRNA expression in J-774 cells after 12 h incubation with 100 ng/ml LPS. The values shown are expressed as the fold increase compared with untreated J-774 cells, and have been normalized to 18S RNA. B: Cell surface syndecan-3 and syndecan-4 protein expression was quantified by flow cytometry as described in Experimental Procedures. The values shown are the percent increase in expression in J-774 cells after 12 h incubation with 100 ng/ml LPS compared with untreated J-774 cells. C: J-774 cells were preincubated with 0 ng/ml or 100 ng/ml LPS for the indicated time, cooled to 4°C, and incubated for 2 h with 50 μg/ml 125I-labeled GV-LDL. Cell-associated radioactivity was quantified. Values are presented as mean ± SE. * P < 0.05 compared with control cells.
DISCUSSION
In a recent study, we reported that deficiency of GV sPLA2 in bone marrow-derived cells significantly reduced atherosclerotic lesion area in LDL receptor-deficient mice, whereas retrovirus-mediated overexpression of GV sPLA2 in bone marrow-derived cells significantly increased lesion area (11). Accumulating evidence indicates that GV sPLA2 promotes atherosclerosis at least in part through its ability to modify LDL (12, 31, 32). In contrast to unmodified LDL, macrophages readily take up LDL hydrolyzed by GV sPLA2 to form foam cells in vitro (14, 15). Using MPMs deficient in CD36 and SR-A, we previously demonstrated that the binding, internalization, and intracellular accumulation of GV-LDL were independent of scavenger receptors known to internalize ox-LDL (15). On the other hand, treatments that disrupted cellular proteoglycans significantly reduced macrophage uptake of GV-LDL, but not native LDL or ox-LDL (15). Thus, macrophages appear to take up GV-LDL through a novel pathway that is distinct from ox-LDL uptake. Given its potentially important role in atherosclerotic lesion development, we set out to define in this study the cell surface proteoglycan(s) involved in macrophage uptake of GV-LDL.
In the current study, we demonstrate that treating J-774 cells with heparinase III/chondroitinase ABC significantly reduces uptake of GV-LDL, providing further evidence that GAG side chains present on the surface of macrophages are required for the internalization of this ligand. Accumulating evidence indicates that cell surface proteoglycans, in particular syndecans, directly mediate endocytic uptake of ligands, including lipoproteins (27, 33, 34). Thus, we initially considered each of the four members of the syndecan family as candidate molecules involved in GV-LDL uptake by J-774 macrophages. We determined that these cells express syndecan-3 and syndecan-4 mRNAs, whereas syndecan-1 and syndecan-2 were virtually undetectable. We confirmed by indirect immunofluorescence that syndecan-3 and syndecan-4 are expressed on the surface of J-774 cells.
To investigate the possible involvement of syndecan-3 and syndecan-4 in GV-LDL uptake by J-774 cells, we suppressed the expression of each of these molecules using specific siRNAs. The level of mRNA suppression for both molecules ranged between 60% and 80% in our different experiments. However, this decrease in mRNA was accompanied by only a modest decrease in cell surface protein expression, possibly due to a relatively long half-life for these HSPGs. Importantly, suppressing the expression of either molecule did not affect the expression of the other. Binding assays performed at 4°C using 125I-labeled ligands showed a direct correlation between syndecan-3 and syndecan-4 expression and GV-LDL binding. Moreover, we observed an additive effect on GV-LDL binding when both molecules were suppressed. Thus, our studies indicate that GV-LDL binds GAGs present on both syndecan-3 and syndecan-4 expressed on the surface of macrophages.
Our ability to only modestly suppress syndecan-3 and syndecan-4 protein expression complicated our ability to conclusively define the relative roles of these molecules in GV-LDL uptake in J-774 cells. Perhaps unexpectedly, in a number of siRNA studies, the cell association of GV-LDL appeared to be significantly reduced in J-774 cells only when syndecan-4 expression was suppressed. Because syndecan-3 and syndecan-4 suppression was somewhat variable from experiment to experiment, we analyzed binding and uptake data from five separate experiments in which syndecan-3 and syndecan-4 were silenced to different extents. This analysis showed a significant correlation between reduced GV-LDL binding and suppression of either syndecan, whereas GV-LDL cell association correlated only with syndecan-4. Indeed, GV-LDL uptake by J-774 cells was not significantly reduced even in an experiment in which syndecan-3 protein expression was reduced by 40%. We tentatively interpreted our findings to suggest that although both syndecans provide potential binding sites for GV-LDL, the actual uptake of this ligand is dependent on syndecan-4.
As another approach to investigating the role of syndecans in macrophage uptake of GV-LDL, we compared the interaction of this ligand with wild-type MPMs and MPMs that genetically lack syndecan-4. Interestingly, based on quantitative RT-PCR data, syndecan-1 and -2 appeared to be expressed at significantly higher levels in MPMs compared with J-774 cells. There was no evidence that deficiency of syndecan-4 in MPMs alters the expression of syndecans 1–3. Our results showed that the uptake of fluorescently labeled GV-LDL was substantially impaired in syndecan-4−/− MPMs. Furthermore, macrophage foam cell formation induced by GV-LDL was significantly reduced in syndecan-4−/− MPMs compared with wild-type, suggesting that syndecan-4 is the major uptake pathway for GV-LDL in these cells. When considering the results from the studies of MPMs, it is important to consider the effect of syndecan-4 deficiency on CE accumulation that occurs as a consequence of GV sPLA2 modification. Wild-type macrophages incubated with GV-LDL contained ∼2-fold more CE compared with cells incubated with mock-LDL. It is notable that the bulk of this increased CE accumulation (>75%) was not observed when syndecan-4−/− MPMs were incubated with GV-LDL. It is not clear whether the modest difference in CE accumulation in syndecan-4−/− MPMs incubated with GV-LDL compared with mock-LDL represents an alternative pathway for GV-LDL uptake, or imprecision in the methods employed in these studies. Thus, on the basis of current data, we cannot rule out the possibility that syndecans 1–3, or perhaps other cell surface HSPGs or pattern recognition receptors participate in the binding and internalization of GV-LDL by MPMs. One potential uptake mechanism could involve perlecan, which we found to be expressed by both macrophages and J-774 cells (data not shown). Perlecan is a secreted HSPG that may adhere to the cell surface through interactions with integrins (35, 36). According to a previous report, perlecans mediate internalization of LPL-enriched LDL through a unique endocytic pathway that involves β1 integrins and tyrosine kinases (36).
Previous investigations of syndecan-mediated LDL uptake have been performed in the presence of LPL, a heparin binding protein that serves as a bridge between the particle and cell surface HSPGs (27). In the case of GV-LDL, we envision that sPLA2 hydrolysis of phospholipids present on the shell of the LDL particle brings about alterations in the conformation of apoB to expose proteoglycan binding sites, as originally described for LDL hydrolyzed by GIIA sPLA2 by Flood et al. (13). Thus, hydrolysis by GV sPLA2 promotes the interaction of LDL with GAG side chains on the syndecan ectodomain, analogous to what has been suggested for ECM HSPGs. It seems likely that other members of the sPLA2 family shown to potently hydrolyze LDL, including GX and GIII sPLA2 (10, 37), also trigger alterations in the conformation of apoB, thereby promoting syndecan-4-mediated macrophage LDL uptake. Indeed, published reports substantiate the conclusion that LDLs modified by either GX or GIII sPLA2 induce macrophage foam cell formation in vitro (10, 37).
Using an overexpression approach in transfected Chinese hamster ovary cells, Fuki et al. (27) demonstrated that syndecan-1, syndecan-2, and syndecan-4 all promote LPL-dependent LDL binding, internalization, and degradation. The role of syndecan-3 in lipoprotein uptake was not investigated. By studying chimeric receptor constructs in which the proteoglycan ectodomain of syndecan-1 was replaced with the ectodomain of the human IgG Fc receptor Ia, the authors established that syndecans directly mediate efficient internalization and lysosomal delivery of surface-bound ligand, rather than acting as coreceptors (27). This process was shown to be triggered by the clustering of the transmembrane and cytoplasmic domains of the core syndecan protein followed by internalization via a nonclathrin-coated pit pathway (27, 33). Similarly, syndecan-1 has been shown to directly mediate internalization of apoE-VLDL in transfected human fibroblasts through a clathrin-independent pathway that requires lipid rafts (38). Syndecan-4-mediated endocytosis is effected through a CDC42-dependent macropinocytic pathway that requires activation of Rac1 and lipid rafts (22). Our data that GV-LDL uptake is blocked by LY 294, an inhibitor of PI3 kinase, is consistent with a macropinocytic uptake pathway for this ligand. Inhibition of PI3 kinase has been shown to block macropinocytosis, but not micropinocytosis, by interfering with the closure of membrane ruffles to form macropinosomes (26, 39). The finding that PI3 kinase activity is not required for endocytosis of ox-LDL provides further evidence that different pathways are involved in the uptake of ox-LDL and GV-LDL. In time-course studies, we previously showed that endocytosis of GV-LDL by C57BL/6 MPMs is markedly delayed compared with oxLDL, and once taken up, a considerable portion of GV-LDL and ox-LDL do not initially colocalize (15). However, in the current study, colocalization of GV-LDL and ox-LDL was evident in C57BL/6 MPMs after longer incubation periods (6 h). This colocalization of GV-LDL and ox-LDL after longer incubations is consistent with our previous report that GV-LDL is eventually degraded by macrophages, albeit at a slower rate compared with ox-LDL (15). Our data are also consistent with previous findings that syndecan-mediated internalization is kinetically distinct from receptor-mediated endocytosis (27).
An important aspect of the atherosclerotic process is a chronic inflammatory environment localized in the vessel wall. Interestingly, we found a significant increase in syndecan-4 mRNA and protein expression in J-774 cells treated with LPS, consistent with previous reports (30). This increased syndecan expression was accompanied by a time-dependent increase in GV-LDL binding, suggesting the possibility that macrophage foam cell formation induced by GV-LDL may be enhanced during inflammation. It is also possible that other pathophysiological conditions known to affect HSPG levels and/or sulfation of GAG chains, such as insulin resistance and diabetes (40, 41), may alter the interaction of macrophages with GV-LDL. A potential role for syndecan-4 in atherogenic processes is substantiated by several studies showing expression of this molecule by macrophages and in the vasculature, and evidence that this molecule is strongly upregulated under acute injury and chronic pro-atherogenic conditions (42–44).
In conclusion, our data point to a physiologic modification of LDL that triggers an increased uptake by macrophages through a syndecan-4-dependent pathway. Upregulation of this pathway may be a pathological consequence of inflammation. These in vitro studies identify a novel mechanism leading to foam cell formation distinct from the classical understanding that macrophage scavenger receptors are essential for initiating a key step in atherogenesis.
Abbreviations
apoB-100, apolipoprotein B-100
CE, cholesteryl ester
ECM, extracellular matrix
GAG, glucosaminoglycan
GV, group V
HSPG, heparan sulfate proteoglycan
LPS, lipopolysaccharide
MPM, mouse peritoneal macrophage
ox-LDL, oxidized LDL
PI, phosphatidylinositol
sPLA2, secretory phospholipase A2
Published, JLR Papers in Press, December 3, 2008.
Footnotes
This work was funded by National Institutes of Health Grant RO1HL-071098 to N.R.W. The authors thank Kathy Forrest and William Bailey for excellent technical assistance.
References
- 1.Williams K. J., and I. Tabas. 1995. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 15 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tabas I., K. J. Williams, and J. Boren. 2007. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 116 1832–1844. [DOI] [PubMed] [Google Scholar]
- 3.Steinberg D., S. Parthasarathy, T. E. Carew, J. C. Khoo, and J. L. Witztum. 1989. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 320 915–924. [DOI] [PubMed] [Google Scholar]
- 4.Stocker R., and J. F. Keaney, Jr. 2004. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 84 1381–1478. [DOI] [PubMed] [Google Scholar]
- 5.Xu X. X., and I. Tabas. 1991. Sphingomyelinase enhances low density lipoprotein uptake and ability to induce cholesteryl ester accumulation in macrophages. J. Biol. Chem. 266 24849–24858. [PubMed] [Google Scholar]
- 6.Hakala J. K., K. Oorni, M. O. Pentikainen, E. Hurt-Camejo, and P. T. Kovanen. 2001. Lipolysis of LDL by human secretory phospholipase A(2) induces particle fusion and enhances the retention of LDL to human aortic proteoglycans. Arterioscler. Thromb. Vasc. Biol. 21 1053–1058. [DOI] [PubMed] [Google Scholar]
- 7.Oorni K., P. Posio, M. Ala-Korpela, M. Jauhiainen, and P. T. Kovanen. 2005. Sphingomyelinase induces aggregation and fusion of small very low-density lipoprotein and intermediate-density lipoprotein particles and increases their retention to human arterial proteoglycans. Arterioscler. Thromb. Vasc. Biol. 25 1678–1683. [DOI] [PubMed] [Google Scholar]
- 8.Gesquiere L., W. Cho, and P. V. Subbaiah. 2002. Role of group IIa and group V secretory phospholipases A(2) in the metabolism of lipoproteins. Substrate specificities of the enzymes and the regulation of their activities by sphingomyelin. Biochemistry. 41 4911–4920. [DOI] [PubMed] [Google Scholar]
- 9.Pruzanski W., L. Lambeau, M. Lazdunsky, W. Cho, J. Kopilov, and A. Kuksis. 2005. Differential hydrolysis of molecular species of lipoprotein phosphatidylcholine by groups IIA, V and X secretory phospholipases A2. Biochim. Biophys. Acta. 1736 38–50. [DOI] [PubMed] [Google Scholar]
- 10.Sato H., R. Kato, Y. Isogai, G-i. Saka, M. Ohtsuki, Y. Taketomi, K. Yamamoto, K. Tsutsumi, J. Yamada, S. Masuda, et al. 2008. Analyses of group III secreted phospholipase A2 transgenic mice reveals potential participation of this enzyme in plasma lipoprotein modification, macrophage foam cell formation, and atherosclerosis. J. Biol. Chem. 283 33483–33497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bostrom M. A., B. B. Boyanovsky, C. T. Jordan, M. P. Wadsworth, D. J. Taatjes, F. C. de Beer, and N. R. Webb. 2007. Group V secretory phospholipase A2 promotes atherosclerosis: evidence from genetically altered mice. Arterioscler. Thromb. Vasc. Biol. 27 600–606. [DOI] [PubMed] [Google Scholar]
- 12.Rosengren B., H. Peilot, M. Umaerus, A-C. Jonsson-Rylander, L. Mattsson-Hulten, C. Hallberg, P. Cronet, M. Rodriguez-Lee, and E. Hurt-Camejo. 2006. Secretory phospholipase A2 group V: lesion distribution, activation by arterial proteoglycans, and induction in aorta by a Western diet. Arterioscler. Thromb. Vasc. Biol. 26 1579–1585. [DOI] [PubMed] [Google Scholar]
- 13.Flood C., M. Gustafsson, R. E. Pitas, L. Arnaboldi, R. L. Walzem, and J. Boren. 2004. Molecular mechanism for changes in proteoglycan binding on compositional changes of the core and the surface of low-density lipoprotein-containing human apolipoprotein B100. Arterioscler. Thromb. Vasc. Biol. 24 564–570. [DOI] [PubMed] [Google Scholar]
- 14.Wooton-Kee C. R., B. B. Boyanovsky, M. S. Nasser, W. J. de Villiers, and N. R. Webb. 2004. Group V sPLA2 hydrolysis of low-density lipoprotein results in spontaneous particle aggregation and promotes macrophage foam cell formation. Arterioscler. Thromb. Vasc. Biol. 24 762–767. [DOI] [PubMed] [Google Scholar]
- 15.Boyanovsky B. B., D. R. van der Westhuyzen, and N. R. Webb. 2005. Group V secretory phospholipase A2-modified low density lipoprotein promotes foam cell formation by a SR-A- and CD36-independent process that involves cellular proteoglycans. J. Biol. Chem. 280 32746–32752. [DOI] [PubMed] [Google Scholar]
- 16.Febbraio M., N. A. Abumrad, D. P. Hajjar, K. Sharma, W. Cheng, S. F. A. Pearce, and R. L. Silverstein. 1999. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J. Biol. Chem. 274 19055–19062. [DOI] [PubMed] [Google Scholar]
- 17.Ishiguro K., K. Kadomatsu, T. Kojima, H. Muramatsu, S. Tsuzuki, E. Nakamura, K. Kusugami, H. Saito, and T. Muramatsu. 2000. Syndecan-4 deficiency impairs focal adhesion formation only under restricted conditions. J. Biol. Chem. 275 5249–5252. [DOI] [PubMed] [Google Scholar]
- 18.Sartipy P., B. Johansen, G. Camejo, B. Rosengren, G. Bondjers, and E. Hurt-Camejo. 1996. Binding of human phospholipase A2 type II to proteoglycans. Differential effect of glycosaminoglycans on enzyme activity. J. Biol. Chem. 271 26307–26314. [DOI] [PubMed] [Google Scholar]
- 19.Noble R. P. 1968. Electrophoretic separation of plasma lipoproteins in agarose gel. J. Lipid Res. 9 693–700. [PubMed] [Google Scholar]
- 20.Kunjathoor V. V., M. Febbraio, E. A. Podrez, K. J. Moore, L. Andersson, S. Koehn, J. S. Rhee, R. Silverstein, H. F. Hoff, and M. W. Freeman. 2002. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 277 49982–49988. [DOI] [PubMed] [Google Scholar]
- 21.Bilheimer D. W., S. Eisenberg, and R. I. Levy. 1972. The metabolism of very low density lipoprotein proteins. I. Preliminary in vitro and in vivo observations. Biochim. Biophys. Acta. 260 212–221. [DOI] [PubMed] [Google Scholar]
- 22.Tkachenko E., E. Lutgens, R-V. Stan, and M. Simons. 2004. Fibroblast growth factor 2 endocytosis in endothelial cells proceed via syndecan-4-dependent activation of Rac1 and a Cdc42-dependent macropinocytic pathway. J. Cell. Sci. 117 3189–3199. [DOI] [PubMed] [Google Scholar]
- 23.Carr T. P., C. J. Andresen, and L. L. Rudel. 1993. Enzymatic determination of triglyceride, free cholesterol, and total cholesterol in tissue lipid extracts. Clin. Biochem. 26 39–42. [DOI] [PubMed] [Google Scholar]
- 24.Williams K. J., G. M. Fless, K. A. Petrie, M. L. Snyder, R. W. Brocia, and T. L. Swenson. 1992. Mechanisms by which lipoprotein lipase alters cellular metabolism of lipoprotein(a), low density lipoprotein, and nascent lipoproteins. Roles for low density lipoprotein receptors and heparan sulfate proteoglycans. J. Biol. Chem. 267 13284–13292. [PubMed] [Google Scholar]
- 25.Hoogewerf A. J., L. A. Cisar, D. C. Evans, and A. Bensadoun. 1991. Effect of chlorate on the sulfation of lipoprotein lipase and heparan sulfate proteoglycans. Sulfation of heparan sulfate proteoglycans affects lipoprotein lipase degradation. J. Biol. Chem. 266 16564–16571. [PubMed] [Google Scholar]
- 26.Kruth H. S., N. L. Jones, W. Huang, B. Zhao, I. Ishii, J. Chang, C. A. Combs, D. Malide, and W-Y. Zhang. 2005. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J. Biol. Chem. 280 2352–2360. [DOI] [PubMed] [Google Scholar]
- 27.Fuki I. V., K. M. Kuhn, I. R. Lomazov, V. L. Rothman, G. P. Tuszynski, R. V. Iozzo, T. L. Swenson, E. A. Fisher, and K. J. Williams. 1997. The syndecan family of proteoglycans. Novel receptors mediating internalization of atherogenic lipoproteins in vitro. J. Clin. Invest. 100 1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernfield M., R. Kokenyesi, M. Kato, M. T. Hinkes, J. Spring, R. L. Gallo, and E. J. Lose. 1992. Biology of the syndecans: a family of transmembrane heparan sulfate proteoglycans. Annu. Rev. Cell Biol. 8 365–393. [DOI] [PubMed] [Google Scholar]
- 29.Carey D. J., D. M. Evans, R. C. Stahl, V. K. Asundi, K. J. Conner, P. Garbes, and G. Cizmeci-Smith. 1992. Molecular cloning and characterization of N-syndecan, a novel transmembrane heparan sulfate proteoglycan. J. Cell Biol. 117 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishiguro K., K. Kadomatsu, T. Kojima, H. Muramatsu, M. Iwase, Y. Yoshikai, M. Yanada, K. Yamamoto, T. Matsushita, M. Nishimura, et al. 2001. Syndecan-4 deficiency leads to high mortality of lipopolysaccharide-injected mice. J. Biol. Chem. 276 47483–47488. [DOI] [PubMed] [Google Scholar]
- 31.Murakami M., and I. Kudo. 2003. New phospholipase A(2) isozymes with a potential role in atherosclerosis. Curr. Opin. Lipidol. 14 431–436. [DOI] [PubMed] [Google Scholar]
- 32.Webb N. R. 2005. Secretory phospholipase A2 enzymes in atherogenesis. Curr. Opin. Lipidol. 16 341–344. [DOI] [PubMed] [Google Scholar]
- 33.Fuki I. V., M. E. Meyer, and K. J. Williams. 2000. Transmembrane and cytoplasmic domains of syndecan mediate a multi-step endocytic pathway involving detergent-insoluble membrane rafts. Biochem. J. 351 607–612. [PMC free article] [PubMed] [Google Scholar]
- 34.Tkachenko E., J. M. Rhodes, and M. Simons. 2005. Syndecans: new kids on the signaling block. Circ. Res. 96 488–500. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi K., J. A. Madri, and P. D. Yurchenco. 1992. Endothelial cells interact with the core protein of basement membrane perlecan through beta 1 and beta 3 integrins: an adhesion modulated by glycosaminoglycan. J. Cell Biol. 119 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuki I. V., R. V. Iozzo, and K. J. Williams. 2000. Perlecan heparan sulfate proteoglycan: a novel receptor that mediates a distinct pathway for ligand catabolism. J. Biol. Chem. 275 25742–25750. [DOI] [PubMed] [Google Scholar]
- 37.Hanasaki K., K. Yamada, S. Yamamoto, Y. Ishimoto, A. Saiga, T. Ono, M. Ikeda, M. Notoya, S. Kamitani, and H. Arita. 2002. Potent modification of low density lipoprotein by group X secretory phospholipase A2 is linked to macrophage foam cell formation. J. Biol. Chem. 277 29116–29124. [DOI] [PubMed] [Google Scholar]
- 38.Wilsie L. C., A. M. Gonzales, and R. A. Orlando. 2006. Syndecan-1 mediates internalization of apoE-VLDL through a low density lipoprotein receptor-related protein (LRP)-independent, non-clathrin-mediated pathway. Lipids Health Dis. 5 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Araki N., M. T. Johnson, and J. A. Swanson. 1996. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 135 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kjellen L., D. Bielefeld, and M. Hook. 1983. Reduced sulfation of liver heparan sulfate in experimentally diabetic rats. Diabetes. 32 337–342. [DOI] [PubMed] [Google Scholar]
- 41.Ebara T., K. Conde, Y. Kako, Y. Liu, Y. Xu, R. Ramakrishnan, I. J. Goldberg, and N. S. Shachter. 2000. Delayed catabolism of apoB-48 lipoproteins due to decreased heparan sulfate proteoglycan production in diabetic mice. J. Clin. Invest. 105 1807–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houston M., M. A. Julien, S. Parthasarathy, and E. L. Chaikof. 2005. Oxidized linoleic acid regulates expression and shedding of syndecan-4. Am. J. Physiol. Cell Physiol. 288 C458–C466. [DOI] [PubMed] [Google Scholar]
- 43.Smith S., S. Mountcastle, A. Burridge, T. F. Dodson, A. A. Salam, K. Kasirajan, R. Milner, R. Veeraswamy, and E. L. Chaikof. 2008. A single-institution experience with the aneurx stent graft for endovascular repair of abdominal aortic aneurysm. Ann. Vasc. Surg. 22 221–226. [DOI] [PubMed] [Google Scholar]
- 44.Li L., T. L. Couse, H. DeLeon, C. P. Xu, J. N. Wilcox, and E. L. Chaikof. 2002. Regulation of syndecan-4 expression with mechanical stress during the development of angioplasty-induced intimal thickening. J. Vasc. Surg. 36 361–370. [DOI] [PubMed] [Google Scholar]