

Abstract
Plasma lipoprotein(a) [Lp(a)] concentration is related to risk of cardiovascular disease. The defining protein component of Lp(a) particles, apolipoprotein(a) [apo(a)], is encoded by the LPA gene. Apo(a) is extremely heterogeneous in size due to a common copy number variation, leading to a variable number of kringle-IV type 2 (KIV2)-like domains. Alleles with fewer KIV2 repeats, encoding smaller apo(a) isoforms, are associated with higher plasma Lp(a) concentrations. Two principal methods to detect variation in KIV2 repeat number are electrophoresis with immunoblotting to detect apo(a) protein isoforms or pulse-field electrophoresis of unamplified genomic DNA to detect the variation of the LPA gene. Both methods are technically challenging, laborious, and time consuming. Here, we report a rapid method to determine the number of KIV2 repeats in LPA from genomic DNA using quantitative real-time polymerase chain reaction (qPCR). With qPCR, we found KIV2 repeat number was correlated with both apo(a) isoform size as determined by immunoblotting (rs = 0.50, P < 1 × 10−6) and with plasma Lp(a) concentration (rs = 0.30, P < 1 × 10−6). The qPCR technique permits rapid evaluation of apo(a) size from genomic DNA, and thus would provide an adjunctive genomic variable, in addition to LPA single nucleotide polymorphisms, for evaluating the genetic determinants of plasma Lp(a) concentration in genetic epidemiology studies of cardiovascular disease outcomes.
Keywords: cardiovascular disease, apolipoprotein(a), quantitative real-time PCR, risk factors, genomics
Elevated plasma concentrations of lipoprotein(a) [Lp(a)] have been associated with risk of myocardial infarction and ischemic stroke (1–5). An interesting paradox is the apparent lack of association in African Americans (4), despite higher mean levels of Lp(a) than Caucasians (6). Myocardial infarction and stroke are the result of not only atherogenicity, or plaque formation, but also of thromboembolic events resulting from plaque rupture (7). Lp(a) is unique in its potential to be involved via either one or both of these pathways. Lp(a) could interfere with plasminogen activation (8) or platelet function (9), or it could contribute to inflammation (10) or endothelial dysfunction (11). Nevertheless, the lack of association in some populations has led to controversy regarding the use of Lp(a) as a marker of risk in common clinical practice.
Plasma levels of Lp(a) range over 1,000-fold between individuals, yet the plasma Lp(a) in a particular individual remains stable over a lifetime (12). Lp(a) is composed of an apolipoprotein(a) [apo(a)] molecule connected via a disulfide bond to the apolipoprotein B-100 of a proatherogenic LDL cholesterol particle (13). The interindividual variation in Lp(a) levels is 90% genetically determined by the LPA locus, a large gene found on chromosome 6 (132 kb in reference sequence NC_000006.10 build 36.3; 6q27; MIM: 152200) (14). Apo(a) is the large protein encoded by LPA, and is composed of a signal peptide region, many repeating kringle domains, and a protease domain (13). LPA contains 10 types of kringles that differ in sequence but are homologous with plasminogen kringle IV (KIV1–10), as well as a kringle homologous to plasminogen kringle V (KV) (Fig. 1) (13). Moreover, apo(a) has a variable number of repeats of one type of kringle domain, kringle-IV type 2 (KIV2), the result of genomic duplication and deletion of the two exons that encode for the KIV2 kringle (∼5 kb in size at the genomic DNA level) (13). The repeating KIV2 domains are an example of a common and functional copy number variation. The National Centre for Biotechnology Information reference sequence (reference#: NC_000006.10 build 36.3) contains 6 repeats of the kringle KIV2 domain, but the number of KIV2 repeats ranges from 5 to >50 in human populations (15). The genetically determined KIV2 repeat size affects the final size of the apo(a) protein, with larger isoforms being compromised with respect to protein folding, transport, and secretion. Null alleles have been identified, in which one allele has an exceedingly large number of KIV2 repeats and does not produce a secreted protein (16). Thus, the number of apo(a) KIV2 repeats is inversely proportional to plasma Lp(a) levels, determining between 20–40% of the variation in Lp(a) levels (17, 18). The biochemical effect of the kringle repeat number on the function of circulating Lp(a) is unknown.
Fig. 1.

A: Gene structure and linkage disequilibrium (LD) plot of LPA locus generated using HapMap CEPH (Centre d'Etude du Poymorphisme Humain) data. Red boxes indicate areas of complete LD, white boxes indicate areas of recombination, and blue boxes are uninformative areas. Exons are indicated by black boxes with every fifth exon labeled in top track. Physical position and exon numbers are taken from National Centre for Biotechnology Information reference sequence build 36.3. Kringles with sequence homology with the fourth plasminogen kringle (KIV) and fifth plasminogen kringle (KV) are indicated. Each kringle is encoded by two exons. B: Amino acid alignment showing sequence homology between kringle repeats.
Many studies have identified increased atherogenesis and coronary artery disease risk in individuals with fewer apo(a) KIV2 repeats (19–21). Regardless, many current studies attempting to find either genetic associations with Lp(a) levels or associations between Lp(a) levels and disease endpoints are reported without KIV2 repeat identification (22). The large size of the genomic repeat (∼5 kb) precludes repeat number identification using standard sequencing or genotyping techniques. Current methods used to identify the number of KIV2 repeats examine either the apo(a) protein size using electrophoresis with immunoblotting (15), or the number of tandem repeats using pulse-field electrophoresis of genomic DNA (23). Both of these techniques are time and labor intensive and require a high degree of technical skill. Quantitative real-time polymerase chain reaction (qPCR) has been used to verify the number of synthetic gene constructs successfully integrated into a plant genome after transformation (24). Similarly, qPCR has been used to verify copy number variations in humans (25). Thus, we hypothesized that qPCR could be used quickly and accurately to identify the number of KIV2 repeats in LPA from genomic DNA.
MATERIALS AND METHODS
Study subjects
Two hundred fifty-seven Alberta Hutterites were included in the study as previously described (18). The study was approved by ethical review panels of the Universities of Alberta and Toronto.
Biochemical analyses
Blood was collected after a 12- to 14-h fasting period, and plasma Lp(a) concentrations were determined using a sandwich enzyme-linked immunoadsorbent assay using monoclonal antibodies 3A5 and 5C4 as previously described (18). Apo(a) isoforms had been previously identified by resolving total plasma protein by 4% PAGE in the presence of SDS, followed by a sensitive chemiluminescent immunoblotting system (18). Alleles were then separated into 16 groups by apo(a) size (18). Genomic DNA was extracted from peripheral leukocytes using established protocols. A multiplexed qPCR was carried out using TaqMan® probes for LPA KIV2 and an endogenous single-copy control gene in the Applied Biosystems 7900HT Fast Real-Time PCR system. Custom TaqMan® gene expression LPA probes in exons 4 and 5 were designed using Applied Biosystems FileBuilder 3.1 (sequences are given in Table 1). TaqMan® RNase P (RNAP) control reagent was used as single-copy reference gene (Part Number 4316844). Reaction volumes contained: 5 uL of water, 1.25 uL of 20× TaqMan® primer/probe mix for LPA, 1.25 uL of 20× TaqMan® primer/probe mix for RNAP, 12.5 uL of 2× gene expression GX master mix (Applied Biosystems), and 4 uL of genomic DNA at a concentration of 5–7 ng/ul. Thermocycler conditions were as follows: 95°C hot-start for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C for 1 min.
TABLE 1.
Primer and probe sequences for Taqman reactions
| Target | Primer and Probe Sequences |
|---|---|
| LPA Exon 4 | Forward primer: GTCAGGTGGGAGTACTGCAA |
| Reverse primer: CGACGGCAGTCCCTTCTG | |
| Probe: CCTGACGCAATGCTCA | |
| LPA Exon 5 | Forward primer: GCACATACTCCACCACTGTCA |
| Reverse primer: GCGAGTGTGGTGTCATAGATGA | |
| Probe: CTTGGCAGGTTCTTCC |
Statistical methods
The total number of LPA KIV2 repeats as determined by immunoblotting was calculated by adding the predicted size of the two alleles if visualized, or doubling the allele count if only a single spot was observed. Cycle thresholds (CT) were identified using the relative quantification manager software (Applied Biosystems). The repeat number as determined by qPCR was calculated by determining the difference in CT between multiplexed target and control probes (ΔCT). The ΔCT was calculated for both the exon 4 (ΔCT4) and exon 5 (ΔCT5) probes for all individuals. The average difference between ΔCT4 and ΔCT5 (ΔΔCT) was calculated for all samples, and individuals whose ΔΔCT was greater than two standard deviations from the mean were excluded from the analysis. The average of ΔCT4 and ΔCT5 (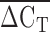 ) was then used for further analysis as the relative kringle repeat number. Finally, the nonparametric Spearman Rank Order Correlation (rs) was calculated in SAS (v9.1) to identify the correlation between ΔCT4 and ΔCT5, the correlation between the previously identified Lp(a) protein isoform size and relative KIV2 kringle repeat number, and the correlation between the plasma Lp(a) concentrations and relative KIV2 kringle repeat number.
) was then used for further analysis as the relative kringle repeat number. Finally, the nonparametric Spearman Rank Order Correlation (rs) was calculated in SAS (v9.1) to identify the correlation between ΔCT4 and ΔCT5, the correlation between the previously identified Lp(a) protein isoform size and relative KIV2 kringle repeat number, and the correlation between the plasma Lp(a) concentrations and relative KIV2 kringle repeat number.
RESULTS
Lower ΔCT values were observed for LPA probes than RNAP probes, as one would expect given the greater amounts of LPA repeat DNA than the single-copy RNAP DNA. 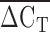 values ranged from 2.5 to 6.2 cycles faster for the LPA probe than the RNAP probe. The difference between ΔCT4 and ΔCT5 was greater than 2 standard deviations for eight individuals, who were excluded from subsequent analysis. A strong correlation was found between the probes targeted to exons four and five, which are located in the KIV2 repeat (rs = 0.85, P < 1 × 10−6; Fig. 2). A positive correlation between
values ranged from 2.5 to 6.2 cycles faster for the LPA probe than the RNAP probe. The difference between ΔCT4 and ΔCT5 was greater than 2 standard deviations for eight individuals, who were excluded from subsequent analysis. A strong correlation was found between the probes targeted to exons four and five, which are located in the KIV2 repeat (rs = 0.85, P < 1 × 10−6; Fig. 2). A positive correlation between 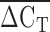 and Lp(a) isoform size identified by PAGE and immunoblotting was identified (rs = 0.50, P < 1 × 10−6; Fig. 3). An inverse relationship between
and Lp(a) isoform size identified by PAGE and immunoblotting was identified (rs = 0.50, P < 1 × 10−6; Fig. 3). An inverse relationship between 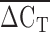 and the square root of plasma Lp(a) concentration was also observed (rs = 0.30, P < 1 × 10−6; Fig. 4). The correlation between the square root of plasma Lp(a) concentration and Lp(a) isoform size as identified by PAGE and immunoblotting (rs = 0.19, P = 0.0054) was weaker than the relationship observed using qPCR.
and the square root of plasma Lp(a) concentration was also observed (rs = 0.30, P < 1 × 10−6; Fig. 4). The correlation between the square root of plasma Lp(a) concentration and Lp(a) isoform size as identified by PAGE and immunoblotting (rs = 0.19, P = 0.0054) was weaker than the relationship observed using qPCR.
Fig. 2.

Correlation observed between the two probes found within the kringle-IV type 2 (KIV2) repeat: Δcycle thresholds (CT) CT4, the probe found in exon 4, and ΔCT5, the probe found in exon 5.
Fig. 3.

The relationship between apolipoprotein(a) [apo(a)] isoform as identified by PAGE and immunoblotting and 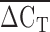 from quantitative real-time polymerase chain reaction (qPCR).
from quantitative real-time polymerase chain reaction (qPCR).
Fig. 4.

The relationship between plasma of lipoprotein(a) [Lp(a)] concentration and 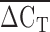 from qPCR.
from qPCR.
DISCUSSION
We demonstrate proof-of-concept that the relative number of KIV2 repeats can be identified by comparing the relative quantity of LPA target DNA compared with RNAP reference DNA using multiplexed real-time qPCR of genomic DNA. The 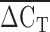 was correlated with Lp(a) isoform size as determined by PAGE and immunoblotting (rs = 0.50, P < 1 × 10−6). As well, the
was correlated with Lp(a) isoform size as determined by PAGE and immunoblotting (rs = 0.50, P < 1 × 10−6). As well, the 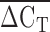 was correlated with plasma Lp(a) concentration in a sample of Alberta Hutterites (rs = 0.30, P < 1 × 10−6). The association between KIV2 repeat number using immunoblotting and Lp(a) concentration was similar to that reported in the literature (26). However, Lp(a) concentration was better correlated with the
was correlated with plasma Lp(a) concentration in a sample of Alberta Hutterites (rs = 0.30, P < 1 × 10−6). The association between KIV2 repeat number using immunoblotting and Lp(a) concentration was similar to that reported in the literature (26). However, Lp(a) concentration was better correlated with the 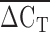 of qPCR than it was with immunoblotting.
of qPCR than it was with immunoblotting.
It has long been recognized that Lp(a) concentrations are associated with the genetic variation in the LPA locus on chromosome 6 (27). However, as seen in Fig. 1, strong linkage disequilibrium exists throughout the kringle region of LPA. Thus, the reason a SNP in the LPA locus is associated with plasma Lp(a) concentration could be through linkage disequilibrium between the SNP and the number of KIV2 repeats. This hypothesis could be easily evaluated using our qPCR assay. The newest generation of microarray-based genotyping platforms (i.e., Affymetrix Human SNP Array 6.0 or Illumina Human 1M-duo beadchip) have been successful identifying copy number variations. However, computational techniques have been focused on identifying low-copy changes (0, 1, 2, 3, 4 copies) and delineation of large-copy changes as seen in the LPA locus (5–50 copies) have not been possible. Therefore, our qPCR method can be considered as an adjunctive technique for characterizing the genetic variation in the LPA locus.
One potential limitation of comparing CT values between different primer/probe sets is the possibility of variation in primer/probe set efficiency between individuals creates differences in CT, rather than relative differences in the quantity of starting target DNA. To mitigate the influence of primer/probe set efficiency, highly conserved exon sequence harboring no SNPs were selected for primer and probe sequences. Second, primer/probe sets were targeted to both of the exons within KIV2 and tested in independent reactions. Ideally, a perfect correlation would be observed between the two probes. In practice, a strong correlation was observed between the two probes (rs = 0.85, P < 1 × 10−6) and eight individuals were removed from the study due to disagreement between the probes (ΔΔCT > 2 standard deviations).
An initial concern was that qPCR would not be sufficiently sensitive to differentiate between alleles that vary by a single repeat (e.g., 9 versus 10 repeats). Upon examination of the data, no discrete incremental groupings were apparent. Therefore, we analyzed the 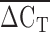 directly instead of attempting to group
directly instead of attempting to group 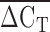 into copy number call bins. An important limitation of the qPCR technique is that it measures the total number of KIV2 repeats, not the number of KIV2 repeats in an allele specific fashion. An individual with 10 KIV2 repeats inherited from her mother and 20 KIV2 repeats from her father could possibly have a different biochemical phenotype than an individual who inherited 15 KIV2 repeats from both parents. Unfortunately, qPCR would not be able to differentiate between these two individuals. An individual who carries a null Lp(a) allele, would appear to be homozygous for the active allele using immunoblotting, but the qPCR would correctly identify the person as having a large number of KIV2 repeats. The individual in the lower right corner of Fig. 3 could be such an individual (
into copy number call bins. An important limitation of the qPCR technique is that it measures the total number of KIV2 repeats, not the number of KIV2 repeats in an allele specific fashion. An individual with 10 KIV2 repeats inherited from her mother and 20 KIV2 repeats from her father could possibly have a different biochemical phenotype than an individual who inherited 15 KIV2 repeats from both parents. Unfortunately, qPCR would not be able to differentiate between these two individuals. An individual who carries a null Lp(a) allele, would appear to be homozygous for the active allele using immunoblotting, but the qPCR would correctly identify the person as having a large number of KIV2 repeats. The individual in the lower right corner of Fig. 3 could be such an individual (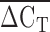 = 6.2, one spot of size 3 was visualized on the immunoblot).
= 6.2, one spot of size 3 was visualized on the immunoblot).
These limitations are balanced by the fact that real-time qPCR provides a fast and cost-effective method of identifying the relative total number of KIV2 repeats from genomic DNA samples that have been stored indefinitely. Thus, apo(a) KIV2 repeat size detected by qPCR is a potentially important new assay that could be used in adjunct to determination of plasma Lp(a) concentration. Future studies testing genetic association with either plasma Lp(a) concentration or cardiovascular disease endpoints might now include this rapid, cost-effective assessment of the apo(a) KIV2 repeat polymorphism in addition to the SNP arrays that have recently been used to study the locus at the genomic DNA level.
Acknowledgments
Dr. Carl Breckenridge performed the apo(a) immunoblot assays. Dr. Hegele is a Career Investigator of the Heart and Stroke Foundation of Ontario and holds the Edith Schulich Vinet Canada Research Chair (Tier I) in Human Genetics and the Jacob J. Wolfe Distinguished Medical Research Chair.
Published, JLR Papers in Press, December 5, 2008.
Footnotes
This work was supported by a team grant from the CIHR (CTP-79853, MOP-37854), the Heart and Stroke Foundation of Ontario, and Genome Canada through the Ontario Genomics Institute. Mr. Lanktree is supported by the Canadian Institutes of Health Research MD/PhD Studentship Award.
References
- 1.Ridker P. M., C. H. Hennekens, and M. J. Stampfer. 1993. A prospective study of lipoprotein(a) and the risk of myocardial infarction. JAMA. 270 2195–2199. [PubMed] [Google Scholar]
- 2.Rhoads G. G., G. Dahlen, K. Berg, N. E. Morton, and A. L. Dannenberg. 1986. Lp(a) lipoprotein as a risk factor for myocardial infarction. JAMA. 256 2540–2544. [PubMed] [Google Scholar]
- 3.Ohira T., P. J. Schreiner, J. D. Morrisett, L. E. Chambless, W. D. Rosamond, and A. R. Folsom. 2006. Lipoprotein(a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 37 1407–1412. [DOI] [PubMed] [Google Scholar]
- 4.Moliterno D. J., E. V. Jokinen, A. R. Miserez, R. A. Lange, J. E. Willard, E. Boerwinkle, L. D. Hillis, and H. H. Hobbs. 1995. No association between plasma lipoprotein(a) concentrations and the presence or absence of coronary atherosclerosis in African-Americans. Arterioscler. Thromb. Vasc. Biol. 15 850–855. [DOI] [PubMed] [Google Scholar]
- 5.Rosengren A., L. Wilhelmsen, E. Eriksson, B. Risberg, and H. Wedel. 1990. Lipoprotein (a) and coronary heart disease: a prospective case-control study in a general population sample of middle aged men. BMJ. 301 1248–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorrentino M. J., C. Vielhauer, J. D. Eisenbart, G. M. Fless, A. M. Scanu, and T. Feldman. 1992. Plasma lipoprotein (a) protein concentration and coronary artery disease in black patients compared with white patients. Am. J. Med. 93 658–662. [DOI] [PubMed] [Google Scholar]
- 7.Hegele R. A. 1997. The genetic basis of atherosclerosis. Int. J. Clin. Lab. Res. 27 2–13. [DOI] [PubMed] [Google Scholar]
- 8.Lawn R. M., K. Schwartz, and L. Patthy. 1997. Convergent evolution of apolipoprotein(a) in primates and hedgehog. Proc. Natl. Acad. Sci. USA. 94 11992–11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez C., J. Rivera, S. Loyau, J. Corral, R. Gonzalez-Conejero, M. L. Lozano, V. Vicente, and E. Angles-Cano. 2001. Binding of recombinant apolipoprotein(a) to human platelets and effect on platelet aggregation. Thromb. Haemost. 85 686–693. [PubMed] [Google Scholar]
- 10.Syrovets T., J. Thillet, M. J. Chapman, and T. Simmet. 1997. Lipoprotein(a) is a potent chemoattractant for human peripheral monocytes. Blood. 90 2027–2036. [PubMed] [Google Scholar]
- 11.Schlaich M. P., S. John, M. R. Langenfeld, K. J. Lackner, G. Schmitz, and R. E. Schmieder. 1998. Does lipoprotein(a) impair endothelial function? J. Am. Coll. Cardiol. 31 359–365. [DOI] [PubMed] [Google Scholar]
- 12.Albers J. J., and W. R. Hazzard. 1974. Immunochemical quantification of human plasma Lp(a) lipoprotein. Lipids. 9 15–26. [DOI] [PubMed] [Google Scholar]
- 13.Berglund L., and R. Ramakrishnan. 2004. Lipoprotein(a): an elusive cardiovascular risk factor. Arterioscler. Thromb. Vasc. Biol. 24 2219–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boerwinkle E., C. C. Leffert, J. Lin, C. Lackner, G. Chiesa, and H. H. Hobbs. 1992. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J. Clin. Invest. 90 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraft H. G., S. Kochl, H. J. Menzel, C. Sandholzer, and G. Utermann. 1992. The apolipoprotein (a) gene: a transcribed hypervariable locus controlling plasma lipoprotein (a) concentration. Hum. Genet. 90 220–230. [DOI] [PubMed] [Google Scholar]
- 16.Gaw A., E. Boerwinkle, J. C. Cohen, and H. H. Hobbs. 1994. Comparative analysis of the apo(a) gene, apo(a) glycoprotein, and plasma concentrations of Lp(a) in three ethnic groups. Evidence for no common “null” allele at the apo(a) locus. J. Clin. Invest. 93 2526–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boerwinkle E., H. J. Menzel, H. G. Kraft, and G. Utermann. 1989. Genetics of the quantitative Lp(a) lipoprotein trait. III. Contribution of Lp(a) glycoprotein phenotypes to normal lipid variation. Hum. Genet. 82 73–78. [DOI] [PubMed] [Google Scholar]
- 18.Hegele R. A., W. C. Breckenridge, J. H. Brunt, and P. W. Connelly. 1997. Genetic variation in factor VII associated with variation in plasma lipoprotein(a) concentration. Arterioscler. Thromb. Vasc. Biol. 17 1701–1706. [DOI] [PubMed] [Google Scholar]
- 19.Paultre F., T. A. Pearson, H. F. Weil, C. H. Tuck, M. Myerson, J. Rubin, C. K. Francis, H. F. Marx, E. F. Philbin, R. G. Reed, et al. 2000. High levels of Lp(a) with a small apo(a) isoform are associated with coronary artery disease in African American and white men. Arterioscler. Thromb. Vasc. Biol. 20 2619–2624. [DOI] [PubMed] [Google Scholar]
- 20.Kraft H. G., A. Lingenhel, S. Kochl, F. Hoppichler, F. Kronenberg, A. Abe, V. Muhlberger, D. Schonitzer, and G. Utermann. 1996. Apolipoprotein(a) kringle IV repeat number predicts risk for coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 16 713–719. [DOI] [PubMed] [Google Scholar]
- 21.Sandholzer C., N. Saha, J. D. Kark, A. Rees, W. Jaross, H. Dieplinger, F. Hoppichler, E. Boerwinkle, and G. Utermann. 1992. Apo(a) isoforms predict risk for coronary heart disease. A study in six populations. Arterioscler. Thromb. 12 1214–1226. [DOI] [PubMed] [Google Scholar]
- 22.Scanu A. M. 2003. Lp(a) lipoprotein–coping with heterogeneity. N. Engl. J. Med. 349 2089–2090. [DOI] [PubMed] [Google Scholar]
- 23.Angles-Cano E., S. Loyau, G. Cardoso-Saldana, R. Couderc, and P. Gillery. 1999. A novel kringle-4 number-based recombinant apo[a] standard for human apo[a] phenotyping. J. Lipid Res. 40 354–359. [PubMed] [Google Scholar]
- 24.Bubner B., and I. T. Baldwin. 2004. Use of real-time PCR for determining copy number and zygosity in transgenic plants. Plant Cell Rep. 23 263–271. [DOI] [PubMed] [Google Scholar]
- 25.Wu Y. L., S. L. Savelli, Y. Yang, B. Zhou, B. H. Rovin, D. J. Birmingham, H. N. Nagaraja, L. A. Hebert, and C. Y. Yu. 2007. Sensitive and specific real-time polymerase chain reaction assays to accurately determine copy number variations (CNVs) of human complement C4A, C4B, C4-long, C4-short, and RCCX modules: elucidation of C4 CNVs in 50 consanguineous subjects with defined HLA genotypes. J. Immunol. 179 3012–3025. [DOI] [PubMed] [Google Scholar]
- 26.Gambhir J. K., H. Kaur, K. M. Prabhu, J. D. Morrisett, and D. S. Gambhir. 2008. Association between lipoprotein(a) levels, apo(a) isoforms and family history of premature CAD in young Asian Indians. Clin. Biochem. 41 453–458. [DOI] [PubMed] [Google Scholar]
- 27.Drayna D. T., R. A. Hegele, P. E. Hass, M. Emi, L. L. Wu, D. L. Eaton, R. M. Lawn, R. R. Williams, R. L. White, and J. M. Lalouel. 1988. Genetic linkage between lipoprotein(a) phenotype and a DNA polymorphism in the plasminogen gene. Genomics. 3 230–236. [DOI] [PubMed] [Google Scholar]