

Abstract
TANK-binding kinase-1 (TBK1) and the inducible IκB kinase (IKK-i) have recently been shown to activate type I interferon (IFN) responses elicited by intracellular detection of RNA or DNA from infecting viruses. Detection of viral RNA is mediated by RIG-I or Mda5 pathways in which TBK1 and IKK-i have been demonstrated to play redundant roles in IFN activation. Here, we have examined if such redundancy occurs in the type I IFN response to DNA viral challenges by examining induction of IFNs and IFN-mediated signaling and gene programs in TBK1−/− macrophages. In contrast to the normal IFN responses in TBK1−/− macrophages infected with an RNA virus, IFN responses were severely abrogated during DNA virus infections in TBK1−/− macrophages. Since both TBK1 and IKK-i are expressed in macrophages, our studies suggest that TBK1 and IKK-i differ functionally in DNA virus–mediated IFN responses however are redundant in RNA virus-mediated IFN responses. Confirmatively, reconstitution of TBK1−/−IKK-i−/− fibroblasts revealed that TBK1 rescued IFN responses to transfected B-DNA to a much stronger degree than IKK-i. Finally, we demonstrate the requirement for the TBK1-IRF3 pathway in host defense against a DNA virus infection in vivo.
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
Keywords: Viral, Cytokines, Monocytes/Macrophages, Signal Transduction, Transgenic/Knockout Mice
Introduction
Type I interferons (IFNs)3 are cytokines that are rapidly produced in response to viral infections and act as critical mediators of host antiviral responses. IFNs activate numerous genes with such roles as growth inhibition and apoptosis. Additionally, IFNs activate the innate and adaptive immune systems through mechanisms such as promoting maturation of antigen presenting cells, and activating various activities of CD8+ T cells, B cells, and NK cells(1).
Type I IFNs include IFNβ and multiple IFNα species, which are transcriptionally controlled by the transcription factors interferon regulatory factors-3 (IRF3) and IRF7, in cooperation with NF-κB and AP-1(2). IRF3 is constitutively expressed, while IRF7 is expressed at low levels in most cell types but is upregulated in response to IFN signaling. In unstimulated cells, IRF3 and IRF7 reside in the cytoplasm, but in response to viral infection, they become activated by phosphorylation by virally activated kinases including TANK-binding kinase-1 (TBK1/T2K/NAK) or the inducible IκB Kinase (IKK-i/IKKε)(2-7). Phosphorylation allows the IRF to dimerize, translocate into the nucleus, and bind to interferon stimulatory response elements (ISRE) in the promoters of the IFNs and other target genes(8, 9). Both IRF3 and IRF7 have been shown to have essential roles in regulation of IFNβ, while IRF7 is the preferential regulator of the IFNα genes(10, 11). Once released from the cell, the IFNs bind to the type I IFN receptor (IFNAR), activating the JAK-STAT signaling pathway. This activates the IFN-stimulated gene factor-3 (ISGF3) transcription factor complex, consisting of IRF9, signal transducer and activator of transcription-1, (STAT1), and STAT2, which induces a large set of target genes important in antiviral responses, including IRF7 which acts to further enhance production of the IFNα family(12, 13). Thus, magnification of the anti-viral IFN response leads to high localized and systemic IFN levels for global protection from viral invasion.
In recent years, much progress has been made in understanding how the host detects viral infections and activates IFN production. Several members of the Toll-like receptor (TLR) family of transmembrane receptors were the first class of innate host pattern recognition receptors (PRR) found to recognize viral ligands and activate production of IFNs. TLR3 recognizes viral dsRNA or the dsRNA mimic polyinosinic acid:cytidylic acid (poly I:C)(14). TLR4 recognizes bacterial LPS and viral components such as the respiratory syncytial virus F protein and the murine mammary tumor virus and Moloney murine leukemia virus envelope proteins(15, 16). TLR3 and TLR4 are unique among TLRs in their ability to signal through the intracellular adaptor TIR domain-containing adapter inducing IFN-beta (TRIF) in order to activate IFN responses(17-22). TRIF activates IFN production through a signaling pathway dependent on TNF-receptor associated factor-3 (TRAF3), TBK1, and IRF3(6, 7, 21-26). In plasmacytoid dendritic cells (pDC) however, viral detection by TLR7 and TLR9 activates a unique pathway for IFNα production via the adaptor molecule MyD88(27, 28). The viral ligand for TLR7 is ssRNA species, while TLR9 recognizes CpG-DNA motifs present abundantly in bacterial and viral pathogen genomes(29-32). IRF7 is highly expressed in pDCs, and in response to TLR7 or TLR9 stimulation, is directly activated by the MyD88-dependent pathway which signals through TRAF3, TRAF6, Osteopontin, and the kinases IRAK1 and IKKα which have both been demonstrated to have the ability to phosphorylate IRF7(10, 25-28, 33-35).
Alternative mechanisms exist for infected host cells to detect cytoplasmic viral nucleic acids. The cytosolic RIG-I-like receptors (RLR) which include RIG-I (RIG-I, retinoic acid inducible gene-I ) and Mda5 (melanoma differentiation-associated gene-5), are RNA helicases which sense intracellular dsRNA of various RNA viruses(36-39). RLRs activate IFN responses through a complex involving IFNβ promoter stimulator-1 (IPS-1/MAVS/CARDIF/VISA) and TRAF3(40-47). Sendai virus infection has been shown to activate IFN production in a RIG-I dependent manner in macrophages and fibroblasts(38). Examination of the role for TBK1 and IKK-i in Sendai-virus mediated IFN activation revealed a cell-type dependent requirement for these kinases based on their basal expression levels(7). Fibroblasts express low basal levels of IKK-i, and were strongly dependent on TBK1 for IFN responses, although IKK-i played a role in late induction presumably following its transcriptional upregulation, as TBK1−/−IKK-i−/− fibroblasts were completely deficient in IFN responses(6, 7, 48). However, in macrophages, IKK-i is basally expressed much higher, and appears to play a primary role in RIG-I mediated IFN activation since a TBK1-deficiency did not produce any significant defects, while TBK1/IKK-i double knockout macrophages were completely defective in RIG-I-pathway induced IFN responses(7, 48).
An additional pathway has been discovered for the detection of intracellular DNA, particularly B-form DNA, which relies on IRF3 to induce IFN responses(49-52). DNA-dependent activator of IFN-regulatory factors (DAI/ DLM-1/ZBP1) has been identified as an intracellular DNA receptor, although other unidentified DNA sensors likely exist as studies in DAI-knockout cells failed to identify a phenotype(49, 53, 54). Other signaling molecules with a potential role in this pathway include RIG-I and IPS-1 in humans but not mice (40, 47, 50, 55). Analysis of DNA transfection into fibroblasts again revealed a role for both TBK1 and IKK-i as TBK1−/−IKK-i−/− fibroblasts were completely unable to elicit IFN responses(50). However, examination of the specific roles for TBK1 versus IKK-i have not yet been done in cells such as macrophages which express high basal IKK-i levels. Thus whether TBK1 and IKK-i play redundant or complementary roles in IFN induction by intracellular DNA sensing pathways remains unknown.
In this study we utilized cells and mice lacking TBK1 to determine the relative contribution of TBK1 to IFN responses during DNA virus infections in vitro and in vivo. We demonstrate that unlike the RLR system, in which TBK1 and IKK-i appear to play overlapping roles in IFN induction, timely IFN responses to DNA virus infection or B-DNA transfection of macrophages were highly dependent on TBK1. Furthermore, when TBK1−/−IKK-i−/− fibroblasts were reconstituted with equivalent levels of TBK1 or IKK-i, TBK1 rescued IFN responses to transfected B-DNA to a much stronger degree than IKK-i, indicating a functional difference either in mechanism or efficiency of these kinases in IRF3 activation. RLR and intracellular DNA virus detection pathways converged at IRF3 as IRF3−/− macrophages were deficient in IFN responses to both RNA and DNA viruses. TBK1-dependent intracellular DNA virus detection pathways were dominant for macrophage activation of IFN responses, while the TLR9/MyD88-dependent, TBK1-independent DNA virus detection pathways were dominant in pDCs. Furthermore, we demonstrate that in vivo, following an intranasal infection with the murine gammaherpesvirus-68 (MHV-68), TBK1 and IRF3 are required for optimal anti-viral immunity and viral clearance. This study is the first to analyze the role of TBK1 in anti-viral responses in vivo.
Materials and Methods
Mice and Cells
TBK1−/−TNFR1−/− mice were a kind gift from Dr. W.C. Yeh (University of Toronto, Ontario, Canada). MyD88−/− and TLR9−/− mice were kindly provided by Dr. S. Akira (Osaka University, Osaka, Japan). IRF3−/− mice were a kind gift from Dr. T. Taniguchi (University of Tokyo, Tokyo, Japan). TRIFlps2/lps2 mice were kindly provided by Dr. B. Beutler (Scripps Research Institute, La Jolla, CA). All mice were maintained and bred under specific pathogen-free conditions in the University of California (Los Angeles, CA), Department of Laboratory Animal Medicine mouse facility, and experiments were conducted within the parameters of our approved protocol. Bone marrow derived macrophages (BMMs) were differentiated from marrow from 6−10 week-old mice as previously described(56). Briefly, bone marrow cells were harvested from mice and allowed to differentiate for 7−8 days in 2% L929-conditioned media before assays were done. Plasmacytoid dendritic cells (pDCs) were differentiated by culturing bone marrow cells with Flt3-ligand as previously described(25).
Viruses and Reagents
BMMs were stimulated with LPS (Sigma-Aldrich) at 10 ng/ml. B-DNA (poly dA-dT:dT-dA, Amersham Biosciences) was transfected into cells at 1ug/ml at a 1ug DNA:1ul Lipofectamine 2000 (Invitrogen) ratio. Flt3-ligand-derived dendritic cells were stimulated with CpG D19 (ggTGCATCGATGCAgggggG, where upper- and lower-case letters indicate bases with phosphodiester and phosphorothioate-modified backbones, respectively)(25).
Sendai virus (Z strain) was cultured in 10-day old chicken eggs. Herpes simplex virus-1 (HSV-1; strain 17) and murine gammaherpesvirus-68 (MHV-68; strain WUMS) were grown by culturing in vero cells. M3FL was produced using a two-step allelic exchange with MHV-68 BAC as the target(57-59). A cassette containing a viral M3 promoter-driven firefly luciferase was inserted between viral tRNA-1 and -2 (between genomic coordinate 746 and 747 of MHV-68 WUMS [U97553])(59, 60). The consequent mutation was confirmed by DNA sequencing, and the genomic integrity of mutated BAC MHV-68 was investigated by restriction enzyme digestion and southern blot analysis as previously described(57, 59). Cells were infected with viruses at a multiplicity of infection (MOI) of 1−3 for the time points indicated.
In vitro assays
Cell lysates were fractionated into nuclear and cytoplasmic fractions as described(18). Cell lysates run on SDS-PAGE gels were probed with antibodies against phosphorylated STAT1 (pY701) (Cell Signaling Technologies), STAT1, USF2, and HA (Santa Cruz Biotechnologies).
RNA was isolated using TRIzol (Invitrogen) and cDNA was synthesized using iScript (Bio-Rad) according to the manufacturer's instructions. Quantitative real time-PCR (Q-PCR) analysis was performed using the iCycler thermocycler (Bio-Rad) as previously described(18). Primer sequences for IFNβ, IFNα5, IP-10, MX1, ISG15, and β-actin have been described(7, 18). All gene expression data presented were normalized to β-actin levels for each sample and fold expression was determined as relative to media treated wild-type samples.
For ELISA assays, cells were cultured for 24 hours with indicated stimuli before supernatants were harvested. IFNβ levels were measured using a kit (Pestka Biomedical Laboratories). IFNα levels were measured using a rat-anti-mIFNα as a coating antibody and a rabbit-anti-mIFNα as a detection antibody (Pestka Biomedical Laboratories).
For flow cytometry analysis, BMMs were cultured with media or the indicated virus or stimulus for 24 hours. Cells were stained with PE-labeled antibodies against CD86 or isotype control antibodies, and mean fluorescence intensity was determined using flow cytometry.
Vectors and Reconstituted Cell Lines
TBK1−/−IKK-i−/− Murine embryonic fibroblasts (MEF) were kindly provided by Dr. S. Akira. TBK1 and IKK-i were cloned into the pEBB eukaryotic expression vector containing a puromycin-resistant gene. Vectors were transfected into TBK1−/−IKK-i−/− MEFs and stably expressing cell lines were selected in the presence of 2.5 ug/ml puromycin.
In vivo mouse infections and imaging
Mice were first anaesthetized by intraperitoneal injection with 200 mg/kg ketamine, 4 mg/kg xylazine in PBS. 5,000 pfu of M3FL in 20ul of DMEM was administered into the right nostril. On days 5 and 7 following infection, mice were imaged using the in vivo imaging system (IVIS, Xenogen). Briefly, mice were anaesthetized by intraperitoneal injection with 200 mg/kg ketamine, 4 mg/kg xylazine in PBS, followed by intraperitoneal injection of 3mg D-luciferin/mouse prior to imaging. Grayscale photographs and color images of imaged mice were superimposed with LivingImage (Xenogen) and Igor (Wavemetrics) programs, similar to that previously described(61). The maximum photon flux value was measured for each mouse and expressed as photons/sec/cm2/steradian. On day 7 following imaging analysis, mice were euthanized and lungs were extracted and homogenized in PBS. Lung tissue homogenates were used to determine viral load by standard plaque assays or luciferase values were quantified on a luminometer using a luciferase assay system kit (Promega). Statistical analyses were done using the Mann Whitney U test.
Results
Activation of IFNs in response to DNA virus infections requires TBK1
The virus-induced type I IFN response involves activation of IRF3 and transcriptional up-regulation of a subset of primary response genes including IFNβ. Subsequently, autocrine and paracrine activity of type I IFNs activates the JAK–STAT pathway and up-regulates a subset of secondary response genes. To examine the role of TBK1 in virus-induced activation of the type I IFN response, induction of IFNs and IFN-regulated genes were compared in TBK1+/+ and TBK1−/− bone marrow derived macrophages (BMMs) at various time points after viral infection. Transcription of IFNβ, IFNα, and IFN-regulated genes such as IP-10 and MX1 in BMMs infected with the DNA viruses HSV-1 or MHV-68 required TBK1 (Figure 1A, 1D). This translated into a requirement for TBK1 for protein level expression of IFNβ and IFNα in response to DNA viruses (Figure 1B, 1E). Notably, infection with MHV-68 did not significantly upregulate IFNα expression as measured by either Q-PCR or ELISA (Figure 1D, 1E). However, induction of IFNs and IFN-regulated genes at both RNA and protein levels in BMMs infected with the RNA virus Sendai virus proceeded normally in the absence of TBK1 (Figure 1G, 1H), ((7, 48). IFNs have been shown to play critical roles in maturation of antigen presenting cells by upregulating expression of MHC and co-stimulatory molecules such as CD86(23). Thus, the mean fluorescence intensity of CD86 surface expression was examined in TBK1+/+ and TBK1−/− BMMs 24 hours after viral infections and compared to stimulation with media alone. TBK1 was required for upregulation of CD86 expression during infection with the DNA viruses HSV-1 and MHV-68 (Figure 1C, 1F). However, upregulation of surface expression of CD86 in response to Sendai virus did not require TBK1 (Figure 1I). Thus, in macrophages, TBK1 is required for timely induction of IFNs and IFN-mediated responses during DNA virus but not RNA virus infections.
Figure 1. TBK1 is required for IFN responses to DNA virus infections.
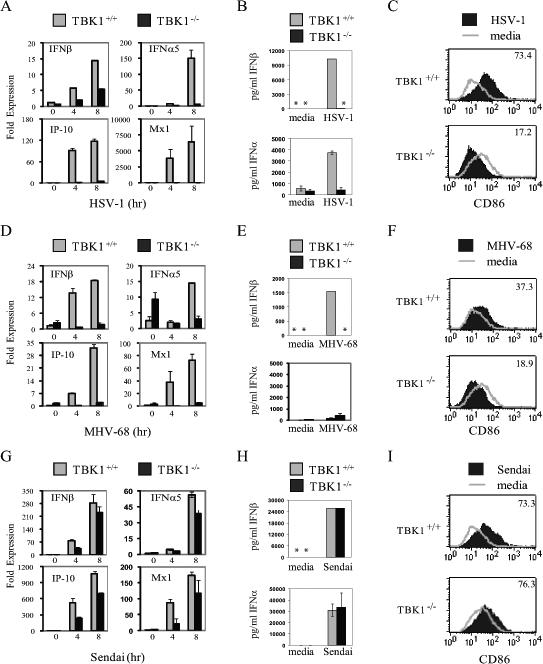
TBK1+/+TNFR1−/− and TBK1−/−TNFR1−/− BMMs were infected with the indicated virus at a multiplicity of infection (MOI) of 1−3. At the indicated times total RNA was extracted and expression of IFNβ, IFNα5, IP-10, and MX1 was analyzed by Q-PCR (A, D, and G) or IFNα and IFNβ levels in supernatants were quantitated by ELISA 24 hours after infection (B, E, and H). Asterisks (*) indicate IFN levels were undetectable by ELISA. 24 hours after infection or treatment with media alone CD86 expression was measured by flow cytometry and mean fluorescence intensity of the infected sample is indicated (C, F, and I). Results shown are representative of at least two independent experiments.
Activation of the IFN program by B-DNA requires TBK1
B-DNA and DNA from various sources including DNA viruses have been shown to induce IFN responses when introduced into the cytoplasm of cells(50, 51). However, it is not yet known whether innate sensing of DNA viruses and B-DNA proceed through single or multiple DNA receptors and downstream pathways. To determine the role of TBK1 in type I IFN responses to B-DNA-transfection, induction of the IFN gene expression programs were compared in TBK1+/+ and TBK1−/− BMMs. We found that TBK1 was required for induction of IFNs and IFN-regulated genes at the RNA and protein levels following transfection with B-DNA (Figure 2A, 2C). TLR4 stimulation with LPS induces type I IFN responses(17, 18). TLR4-mediated activation of IFN gene expression programs were highly dependent on TBK1 (Figure 2B, 2D), (6, 7). TBK1 was also required for upregulation of surface expression of CD86 in response to B-DNA transfection (Figure 2E). Stimulation of TLR4 with LPS also required TBK1 for upregulation of CD86 expression (Figure 2F). Thus, similarly to that seen for DNA virus infections, TBK1 is required for induction of IFN responses to intracellular B-DNA, as well as TLR4 stimulation.
Figure 2. TBK1 is required for IFN responses to transfected B-DNA.
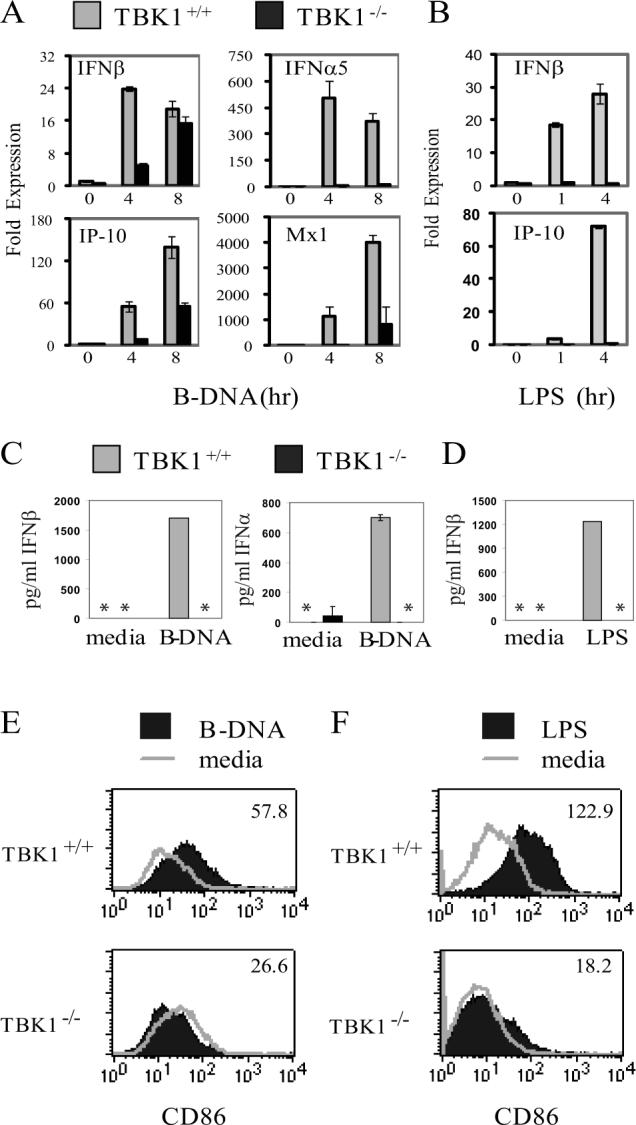
TBK1+/+TNFR1−/− and TBK1−/−TNFR1−/− BMMs were transfected with 1 ug/ml B-DNA (poly dA-dT:dT-dA) or stimulated with 10ng/ml LPS. At the indicated times total RNA was extracted and expression of IFNβ, IFNα5, IP-10, and MX1 was analyzed by Q-PCR (A and B) or IFNα and IFNβ levels in supernatants were quantitated by ELISA 24 hours after stimulation (C and D). Asterisks (*) indicate IFN levels were undetectable by ELISA. 24 hours after stimulation or treatment with media alone CD86 expression was measured by flow cytometry and mean fluorescence intensity of the stimulated sample is indicated (E and F). Results shown are representative of at least two independent experiments.
Activation of IFN-regulated signaling pathways by DNA viruses and B-DNA requires TBK1
Following production and release from the cell, IFNs bind and activate IFNAR. Activated JAK kinases phosphorylate STAT1, allowing formation of the ISGF3 transcription factor complex which regulates expression of IFN response genes. In the absence of TBK1, STAT1 phosphorylation was greatly abrogated in response to B-DNA (Figure 3A). Additionally, during infection with the DNA viruses HSV-1 and MHV-68, TBK1 was required for optimal STAT1 phosphorylation (Figure 3B, 3C). In contrast, Sendai virus-induced STAT1 phosphorylation did not require TBK1 (Figure 3D)(7). Thus, analysis of downstream signaling responses such as STAT1 phosphorylation further confirm the observation that TBK1 is required for timely induction of IFN responses during DNA transfection or infection with DNA viruses but not RNA viruses.
Figure 3. B-DNA and DNA virus activation of type I IFN mediated signal transduction require TBK1.
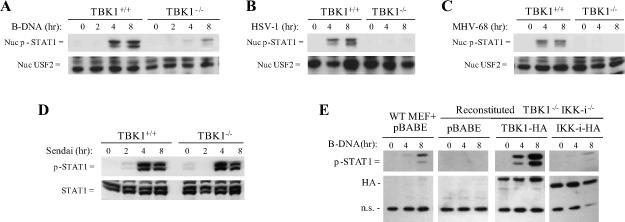
TBK1+/+TNFR1−/− and TBK1−/−TNFR1−/− BMMs were transfected with 1 ug/ml B-DNA (poly dA-dT:dT-dA) or infected with the indicated virus at a multiplicity of infection (MOI) of 1−3. At the indicated time points, cells were harvested, fractionated and nuclear fractions were probed for phospho-STAT1 and USF2 as a loading control for nuclear proteins (A, B, and C) or total cell extracts were probed for phospho-STAT1 and total STAT1(D). E, WT or TBK1−/−IKK-i−/− MEFs reconstituted with a pBABE vector or with HA-tagged TBK1 or IKK-i were transfected with 1 ug/ml B-DNA for the indicated time points. Total cell extracts were probed for phospho-STAT1 and HA. A non-specific (n.s.) HA band is shown as a loading control. Results shown are representative of at least two independent experiments.
TBK1 and IKK-i mediate distinct functions in IFN responses to DNA viruses
We have previously shown that reconstitution of TBK1−/− MEFs with either TBK1 or IKK-i rescues IFN production and downstream signaling responses during Sendai virus infection(7). To support our hypothesis that the role for IKK-i in IFN responses induced during DNA virus infections is different from the overlapping role it plays with TBK1 in the RIG-I signaling pathway, TBK1−/−IKK-i−/− fibroblasts were stably reconstituted with vectors expressing either HA-tagged TBK1 or IKK-i. When TBK1 and IKK-i were expressed at equivalent levels in TBK1−/−IKK-i−/− fibroblasts, TBK1 rescued STAT1 phosphorylation to a much stronger degree than IKK-i in response to DNA transfection (Figure 3E). Thus, there exists a functional difference either in mechanism or efficiency between TBK1 and IKK-i in activation of IFN responses by intracellular DNA detection pathways.
Activation of IFN programs in macrophages by DNA and RNA viruses is MyD88-independent but IRF3-dependent
Previous studies have shown that DNA and RNA viruses can be recognized by TLR9 or TLR7, respectively, which elicit IFNα production in pDCs via MyD88-dependent pathways. To address the role of TLR9 and TLR7 pathways in virus mediated IFN induction in macrophages, MyD88−/− BMMs were infected with MHV-68 or Sendai virus, and analyzed for IFN-responses. Induction of IFN gene programs and STAT1 phosphorylation did not require MyD88 in response to infection with either DNA or RNA viruses (Figure 4A-4D, S1A). Upregulation of CD86 surface expression in response to MHV-68 was also MyD88-independent (Figure S1B). Additionally, TLR9−/−, MyD88−/−, and TRIF−/− BMMs infected with HSV-1 all induced normal STAT1 phosphorylation (Figure S1C). Conversely, TBK1 was dispensable for IFNα production in response to DNA virus infections or stimulation with CpG in pDCs (Figure S1D). MyD88 however, was required for IFNα production in pDCs infected with DNA viruses (Figure S1E).
Figure 4. Macrophages use MyD88-independent IRF3-dependent pathways to recognize DNA and RNA virus infections.
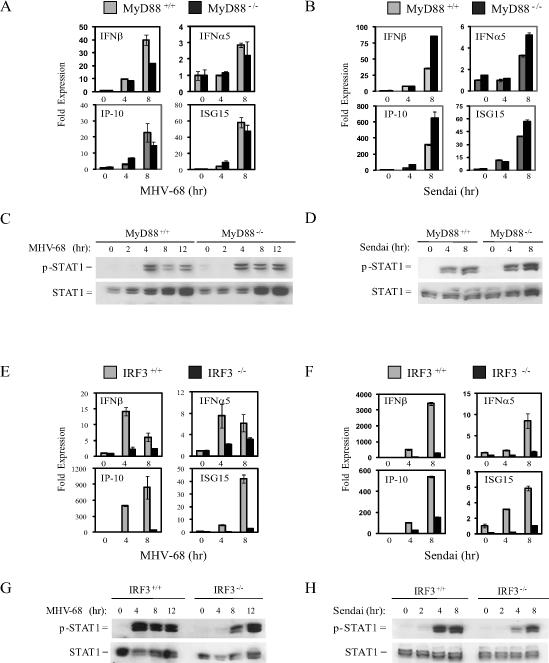
WT and MyD88−/− or IRF3−/− BMMs were infected with MHV-68 or Sendai virus at a multiplicity of infection (MOI) of 1−3. At the indicated time points total RNA was extracted and analyzed by Q-PCR for expression of IFNβ, IFNα5, IP-10, and ISG-15 (A, B, E, and F) or total cell extracts were probed for phospho-STAT1 and total STAT1 (C, D, G,and H).
Since TBK1 is differentially required for IFN responses to DNA and RNA viruses in macrophages, we examined the role for the downstream transcription factor IRF3 in BMM responses to MHV-68 and Sendai virus. IRF3−/− BMMs had significantly reduced expression of IFNs and IFN-regulated genes during infection with both MHV-68 and Sendai virus (Figure 4E, 4F). Furthermore, delayed STAT1 phosphorylation was observed in IRF3−/− BMMs during infection with either MHV-68 or Sendai virus (Figure 4G, 4H). Thus, despite the differential usage of TBK1, signals from intracellular sensors of both DNA and RNA viruses require IRF3 for optimal IFN induction in macrophages. Collectively, these data show that production of IFNs in response to DNA viruses utilizes a TBK-IRF3-dependent pathway in macrophages, but proceeds through the TLR9-MyD88 pathway in pDCs.
Resistance to pulmonary MHV-68 infection requires IRF3 and TBK1
MHV-68 has been used as a mouse model for Kaposi's sarcoma-associated herpes virus and Epstein-Barr virus infections. MHV-68 naturally infects the host most likely through the respiratory tract and establishes an acute infection in the lungs, followed by long term latency in splenic B cells, lung epithelial cells, DCs, and macrophages(62, 63). Although MHV-68 infection does not result in high systemic IFN levels, the IFN system is crucial for control of the viral infection as IFNAR−/− mice more susceptible to infection ((64, 65), Hwang S., Kim K.S., Flano E., Wu T.T., Tong L.M., Park A.N., Song M.J., Sanchez D.J., O'Connell R.M., Cheng G., Sun R. Conserved herpesviral kinase plays a critical role in viral persistence by inhibiting IRF-3 mediated type I interferon response. Cell Host & Microbe (in press)). Previous studies have indicated that pDCs are the major IFN producing cell type during in vivo infections with DNA viruses via the TLR9-MyD88 pathways(66, 67). However, we sought to determine the contribution of TBK1- IRF3-dependent pathways during DNA virus infections in vivo.
Bioluminescence imaging was used to monitor an active infection of mice with an MHV-68 virus engineered to express luciferase under an M3 promoter (M3FL)(59). Mice were visualized at five and seven days post-infection and the maximum luminescence values were quantified for each mouse. IRF3−/− mice were more susceptible to M3FL infection than wild-type mice as evidenced by a statistically significant elevation of maximum luminescence values by approximately 1 log-fold in IRF3−/− mice on both days (Figure 5). Thus, IRF3 is required for optimal resistance to in vivo M3FL infections. Additionally, in TBK1+/+ mice, the infection remained confined to the nostril area with limited dissemination by day seven, while TBK1−/−mice showed elevated viral spread to the lungs and salivary glands over time (Figure 6A and 6B). A statistically significant elevation of maximum luminescence values by over 1 log-fold was seen on both days for TBK1−/− mice as compared to TBK1+/+ mice (Figure 6A and 6B). At day seven post-infection, whole lungs were extracted from infected mice, homogenized, and analyzed by luminometer and standard plaque assay for quantification of viral load. Both measurements confirmed that TBK1−/− mice had higher viral titers by approximately 1 log-fold in the lungs than TBK1+/+ mice (Figure 6C and 6D). Thus these studies show an indispensable role for the classical IRF3-TBK1 pathway in regulating in vivo immune resistance mechanisms during DNA virus infections.
Figure 5. IRF3 is required for in vivo resistance to DNA viruses.
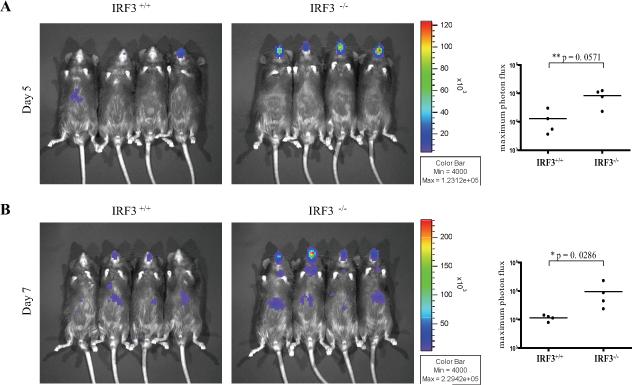
WT and IRF3−/− mice were infected intranasally with 5,000 pfu of M3FL. On days 5 (A) and 7 (B) following infection, mice were injected with D-luciferin and imaged. The maximum photon flux value (Log10 photons/sec/cm2/sr) was determined for each mouse.
Figure 6. TBK1 is required for in vivo resistance to DNA viruses.
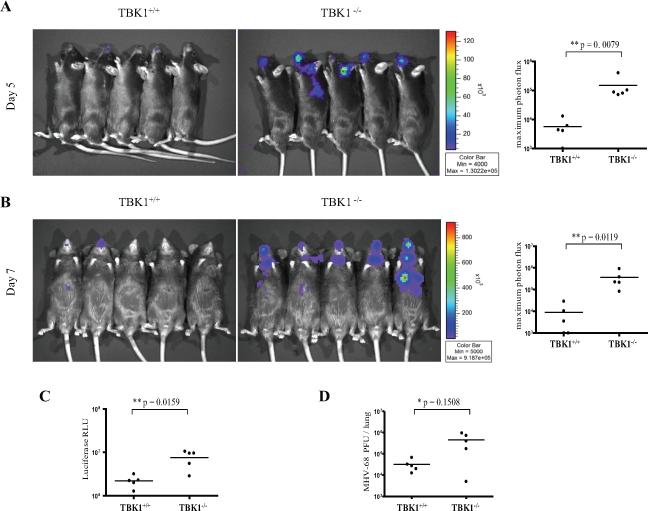
TBK1+/+TNFR1−/− and TBK1−/−TNFR1−/− mice were infected intranasally with 5,000 pfu of M3FL. On days 5 (A) and 7 (B) following infection, mice were injected with D-luciferin and imaged. The maximum photon flux value (Log10 photons/sec/cm2/sr) was determined for each mouse. Viral load in lung tissue was determined on day 7 by standard plaque assays (C) and luciferase values (D).
Discussion
Production of type I IFN is characteristic of virally infected cells and is vital to the host immune defense against viral infection. Multiple families of pattern recognition receptors including TLRs, RLRs, DAI, and other DNA sensors exist to recognize viral infections and activate IFN responses. As the pathways involved in type I IFN production are being carefully studied, the responsible signaling mediators during DNA virus infections in different cell types remains an open question. In this study, we identify a critical requirement for TBK1 in activation of the IFN response to DNA viruses in macrophages. Moreover, we demonstrate the in vivo requirement for the TBK1-IRF3 pathway in immune resistance to infection with the DNA virus MHV-68.
We and others have previously observed that the pathways leading to IFN activation during virus infections utilize different signaling molecules in various cell types. RIG-I mediates the activation of IFN responses to Sendai virus in conventional DCs and fibroblasts(38). In the RIG-I pathway, TBK1 and IKK-i appear to play redundant functions in IRF3 activation. During Sendai virus infection, fibroblasts which express low levels of IKK-i rely heavily on TBK1 for IRF3 activation(6, 7). However, macrophages express relatively high levels of IKK-i and thus activation of IRF3 can proceed normally in the absence of TBK1(7, 48). The roles for TBK1 and IKK-i in B-DNA induced IFN responses have been studied in fibroblasts and IKK-i has been theorized to play a redundant function with that of TBK1(50). However, the effect of a TBK1 deficiency during infection with DNA viruses had not yet been studied in macrophages or other cells highly expressing IKK-i.
In response to infection with the DNA viruses MHV-68 and HSV-1, BMMs derived from TBK1−/− mice were found to be defective in their ability to up-regulate production of IFNs, and activate IFN-regulated gene programs and downstream IFN-mediated signaling responses. Thus, although IKK-i is coexpressed in BMMs, TBK1 and IKK-i apparently play nonredundant roles in IFN induction in response to DNA viruses. Furthermore, TBK1−/−IKK-i−/− cells were reconstituted with equivalent levels of TBK1 or IKK-i in order to compare their relative contributions to the type I IFN response to DNA recognition. Reconstitution with TBK1 rescued the IFN response to a much stronger degree than reconstitution with IKK-i when cells were transfected with B-DNA. Thus, these studies indicate that although TBK1 and IKK-i both contribute to IFN production(50), they differ either in mechanism or efficiency in activation of IFN responses to intracellular DNA virus detection pathways.
IRF3 has been shown to play a key role in the induction of IFN responses to RNA and DNA virus infections in this and other in vitro studies (Figures 4 & 5, (10, 11, 68-70)). However, the in vivo contribution of IRF3 during DNA virus infections appears to be dependent on route of infection as IRF3 was dispensable in induction of IFNs during i.v. HSV-1 infections but important in vaginal HSV2 infections(10, 71). Here we show that IRF3 was important for immunity to an intranasally introduced DNA virus, in which major IFN producers may be respiratory epithelial cells and alveolar macrophages. Thus, IRF3 is essential for DNA virus-mediated type I IFN responses in vitro and contributes to host immunity in vivo. Therefore, our studies are consistent with a model of DNA virus-induced IFN induction proceeding through a signaling pathway dependent on TBK1 and IRF3.
Previous studies have indicated that TLR9-independent detection of DNA viruses is due to intracellular recognition of viral DNA. Transfection of cells with various types of DNA including B-form DNA has been shown to elicit strong type I IFN responses(50, 51). However, whether synthetic B-DNA and viral DNAs are recognized by the same sensors remains to be determined. Our study found that TBK1 was required for the IFN response in BMMs when transfected with B-DNA. Thus, TBK1 is a convergence point for induction of IFNs following intracellular recognition of B-DNA and DNA virus infections.
In lieu of the many viral detection pathways identified, it is clear that different pathways are dominant in specific cell types and tissues. The TLR9/MyD88 pathway is activated during infection with DNA viruses in pDCs, resulting in production of high levels of IFNα via direct activation of IRF7(27, 28). We demonstrate that induction of the type I IFNs by DNA viruses in BMMs is independent of TLR9 and MyD88 signaling pathways, while in pDCs, IFN production requires MyD88 but not TBK1. Thus, the TBK1-IRF3 pathway clearly represents a DNA virus detection mechanism separate from the TLR9-MyD88 system.
Plasmacytoid dendritic cells (pDC) have been demonstrated to be primary in vivo producers of IFNα during infection with many viruses, via the TLR7/8 and TLR9 pathways. The role for different cell types and pathways in mediating in vivo anti-viral responses has recently been demonstrated for RNA viruses. pDCs were the strongest producers of IFNs during systemic Newcastle disease virus infection although macrophages and conventional DCs also contributed to IFN production(72). However, during pulmonary RNA virus infections, alveolar macrophages were the major producers of IFN via RLRs(72). The absence of either pDCs or the TLR9-MyD88 axis was detrimental to host control of DNA virus infections such as HSV-2 and murine cytomegalovirus in vivo(73, 74). However, the contribution of intracellular DNA virus detection pathways to host immunity has yet to be explored during in vivo infections.
Using a luciferase-expressing MHV-68 variant to monitor active infections of mice (M3FL), we find that mice lacking TBK1 or IRF3 were more susceptible to intranasal MHV-68 infection. Although we were unable to detect IFN production in these mice, this indicates that even low levels of IFN production through TBK1-IRF3 pathways makes important contributions to host immunity. Thus, while TLR9 detection of DNA viruses by pDCs is likely to additionally contribute to IFN production and subsequent immune defense, detection of infections through TBK1-dependent pathways by other cell types such as alveolar macrophages and respiratory epithelial cells is essential for optimal host control of viral infections.
Viruses are diverse and quickly evolving organisms and hosts have thus developed multiple mechanisms for detecting viral invasion and initiating IFN and immune responses. Although we have characterized the role for TBK1-pathways in response to DNA virus infections, future studies are needed to address the specific roles of TBK1 and IKK-i, as well as identify other signaling mediators and host receptors that contribute to DNA virus recognition. Since TBK1 is required for viral resistance in vivo, it would be interesting to determine the cell types that elicit IFN production via TBK1-pathways. Additionally, it would be interesting to determine the quality of IFN and immune responses initiated by TLR9-MyD88 versus TBK1-IRF3 pathways following virus infections by different routes. A clear understanding of these different pathways would contribute to better understanding how viruses are able to inhibit the IFN response and how we can in turn manipulate the host response to virus infections and other diseases in which IFNs contribute such as autoimmune disorders and cancer.
Supplementary Material
Footnotes
This work was supported in part by National Institutes of Health research grants RO1 CA87924, RO1 AI056154, and R37 AI47868. A. K. Miyahira was supported by the Howard Hughes Medical Institute Predoctoral Fellowship (Grant No. 59003787) and the Warsaw Dissertation Year Fellowship. A. Shahangian was supported by a UCLA Medical Scientist Training Program training grant. S. Hwang was supported by UCLA AIDS Institute and UCLA Center for AIDS Research (AI28697) and Universitywide AIDS Research Program Dissertation Award (D06-LA-4).
Abbreviations used in this paper: HSV, herpes simplex virus; IFN, interferon; IFNAR, type I interferon α/β receptor; IKK-i, inducible IκB kinase; IRF3, interferon regulatory factor-3; ISGF3, IFN-stimulated gene factor-3; ISRE, IFN-stimulated response elements; M3FL, MHV-68 virus expressing luciferase under an M3 promoter; Mda5, melanoma differentiation-associated gene-5; MHV-68, murine gammaherpesvirus-68; MyD88, myeloid differentiation factor-88; PAMP, pathogen-associated molecular patterns; pDC, plasmacytoid dendritic cell; PRR, pattern recognition receptor; RIG-I, retinoic acid inducible gene-I; STAT, signal transducer and activator of transcription; TBK1, TANK-binding kinase-1; TLR, Toll-like receptor; TRAF3, tumor necrosis factor receptor–associated factor-3; TRIF, Toll/IL-1 receptor domain-containing adaptor inducing IFNβ.
References
- 1.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I Interferons (alpha/beta) in Immunity and Autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 2.Malmgaard L. Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res. 2004;24:439–454. doi: 10.1089/1079990041689665. [DOI] [PubMed] [Google Scholar]
- 3.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 4.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 5.McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A. 2004;101:233–238. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med. 2004;199:1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 9.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 10.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 11.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 12.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- 13.Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. Embo J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 15.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 16.Rassa JC, Meyers JL, Zhang Y, Kudaravalli R, Ross SR. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc Natl Acad Sci U S A. 2002;99:2281–2286. doi: 10.1073/pnas.042355399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- 18.Doyle S, Vaidya S, O'Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 20.Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 23.Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han J, Beutler B. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol. 2003;4:1223–1229. doi: 10.1038/ni1010. [DOI] [PubMed] [Google Scholar]
- 24.Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, Honda K, Taniguchi T. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun. 2003;306:860–866. doi: 10.1016/s0006-291x(03)01049-0. [DOI] [PubMed] [Google Scholar]
- 25.Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 26.Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 27.Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, Takeuchi O, Akira S. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 28.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 31.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 32.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis ESC. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 33.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med. 2005;201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, Ohara O, Akira S, Kaisho T. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature. 2006;440:949–953. doi: 10.1038/nature04641. [DOI] [PubMed] [Google Scholar]
- 35.Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, Cantor H. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol. 2006;7:498–506. doi: 10.1038/ni1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 37.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 39.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr., Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 40.Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, Chen ZJ. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 41.Yoneyama M, Fujita T. RIG-I family RNA helicases: Cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 2007;18:545–551. doi: 10.1016/j.cytogfr.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 42.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 43.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 44.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 45.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 46.Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y, Cheng G. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. Embo J. 2006;25:3257–3263. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, Yamamoto M, Uematsu S, Ishii KJ, Takeuchi O, Akira S. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. 2006;203:1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsui K, Kumagai Y, Kato H, Sato S, Kawagoe T, Uematsu S, Takeuchi O, Akira S. Cutting edge: Role of TANK-binding kinase 1 and inducible IkappaB kinase in IFN responses against viruses in innate immune cells. J Immunol. 2006;177:5785–5789. doi: 10.4049/jimmunol.177.9.5785. [DOI] [PubMed] [Google Scholar]
- 49.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 50.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, Sato S, Yamamoto M, Uematsu S, Kawai T, Takeuchi O, Akira S. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 51.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 52.Hochrein H, Schlatter B, O'Keeffe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Suter M, Wagner H. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A. 2004;101:11416–11421. doi: 10.1073/pnas.0403555101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, Akira S. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- 54.Wang Z, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, Tamura T, Takaoka A, Nishikura K, Taniguchi T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci U S A. 2008;105:5477–5482. doi: 10.1073/pnas.0801295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng G, Zhong J, Chung J, Chisari FV. Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc Natl Acad Sci U S A. 2007;104:9035–9040. doi: 10.1073/pnas.0703285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature. 2002;416:190–194. doi: 10.1038/416190a. [DOI] [PubMed] [Google Scholar]
- 57.Song MJ, Hwang S, Wong WH, Wu TT, Lee S, Liao HI, Sun R. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc Natl Acad Sci U S A. 2005;102:3805–3810. doi: 10.1073/pnas.0404521102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith GA, Enquist LW. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J Virol. 1999;73:6405–6414. doi: 10.1128/jvi.73.8.6405-6414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hwang S, Wu T, Tong LM, Martinez-Guzman D, Colantonio AD, Uittenbogaart CH, Sun R. Persistent Gamma-Herpesvirus Replication and Its Dynamic Interaction with Host In Vivo. J Virol. 2008;82:12498–509. doi: 10.1128/JVI.01152-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinez-Guzman D, Rickabaugh T, Wu TT, Brown H, Cole S, Song MJ, Tong L, Sun R. Transcription program of murine gammaherpesvirus 68. J Virol. 2003;77:10488–10503. doi: 10.1128/JVI.77.19.10488-10503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luker GD, Bardill JP, Prior JL, Pica CM, Piwnica-Worms D, Leib DA. Noninvasive bioluminescence imaging of herpes simplex virus type 1 infection and therapy in living mice. J Virol. 2002;76:12149–12161. doi: 10.1128/JVI.76.23.12149-12161.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nash AA, Dutia BM, Stewart JP, Davison AJ. Natural history of murine gamma-herpesvirus infection. Philos Trans R Soc Lond B Biol Sci. 2001;356:569–579. doi: 10.1098/rstb.2000.0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stewart JP, Usherwood EJ, Ross A, Dyson H, Nash T. Lung epithelial cells are a major site of murine gammaherpesvirus persistence. J Exp Med. 1998;187:1941–1951. doi: 10.1084/jem.187.12.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dutia BM, Allen DJ, Dyson H, Nash AA. Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology. 1999;261:173–179. doi: 10.1006/viro.1999.9834. [DOI] [PubMed] [Google Scholar]
- 65.Barton ES, Lutzke ML, Rochford R, Virgin H. W. t. Alpha/beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. J Virol. 2005;79:14149–14160. doi: 10.1128/JVI.79.22.14149-14160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 67.Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. 2005;175:6723–6732. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- 68.Paladino P, Cummings DT, Noyce RS, Mossman KL. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J Immunol. 2006;177:8008–8016. doi: 10.4049/jimmunol.177.11.8008. [DOI] [PubMed] [Google Scholar]
- 69.Nociari M, Ocheretina O, Schoggins JW, Falck-Pedersen E. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J Virol. 2007;81:4145–4157. doi: 10.1128/JVI.02685-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Collins SE, Noyce RS, Mossman KL. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol. 2004;78:1706–1717. doi: 10.1128/JVI.78.4.1706-1717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gill N, Deacon PM, Lichty B, Mossman KL, Ashkar AA. Induction of innate immunity against herpes simplex virus type 2 infection via local delivery of Toll-like receptor ligands correlates with beta interferon production. J Virol. 2006;80:9943–9950. doi: 10.1128/JVI.01036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S. Alveolar Macrophages Are the Primary Interferon-alpha Producer in Pulmonary Infection with RNA Viruses. Immunity. 2007;27:240–252. doi: 10.1016/j.immuni.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 73.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198:513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, Alexopoulou L, Flavell RA, Beutler B. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.