

Abstract
Fibrous dysplasia (FD) of bone is an uncommon disease caused by sporadic, congenital mutations in the cAMP regulating protein, Gsα. It is an example of somatic mosaicism in which a wide spectrum of disease is possible. Widespread skeletal involvement is often associated with varying combinations of café-au-lait skin spots, and/or endocrine dysfunction (precocious puberty, renal phosphate wasting, hyperthyroidism, and/or growth hormone excess). Unrecognized and untreated endocrine dysfunction can exacerbate the skeletal disease. The diagnosis is usually established on clinical grounds on the basis of physical examination and typical radiographic appearance. Occasionally, gene testing of affected tissue may be helpful. The skeletal sites involved with disease are established at an early age, and the complications of fracture deformity are most pronounced in childhood. Bone pain in the absence of a fracture is more common in adults, but can also be present in children. Treatment with bisphosphonates is usually effective at relieving pain, but probably has no effect on the natural history of the disease. Scoliosis, which was previously thought to be an uncommon occurrence, has been shown to be common and progressive, and as such, warrants investigation and, when necessary, surgical treatment. The surgical management of FD remains challenging. Timing and technique remain controversial, but some consensus exists in that grafting materials (of any type) usually fail and should not be a central aspect of the surgical approach. Intramedullary devices are in general superior to side plates and screws. In extremely widespread disease with very early fracture and deformity, no surgical approach will affect final functional outcome. Efforts should be made for the initiation of international collaborative studies to better define optimal surgical approaches to the treatment of this challenging disease.
Keywords: Fracture, GNAS, Gs alpha, Pain, Outcomes, Scoliosis
Introduction
Fibrous dysplasia of bone (FD) is a sporadic, uncommon, fibroosseous skeletal disorder with a broad spectrum of severity. It was initially identified as a distinct entity in association with skin spots and hyperfunctioning endocrinopathies by Donovan McCune and Fuller Albright, and termed by Albright as osteitis fibrosa disseminata, reflecting the similarity of FD to the skeletal disease of hyperparathyroidism, osteitis fibrosa cystica [1, 2]. The disease was labeled polyostotic fibrous dysplasia (PFD) by Lichtenstein [3] in 1938, and it was Lichtenstein and Jaffe [4] who initially described the spectrum of the clinical, radiographic and histological findings. In addition, they introduced the idea that FD results from the “perverted activity of the specific bone-forming mesenchyme.” FD may exist as an isolated lesion involving a single skeletal site, monostotic fibrous dysplasia (MFD), or it may involve multiple bones, PFD. In addition, it may occur in association with distinct café-au-lait skin spots with a jagged (coast-of-Maine) border, and/or in association with a number of hyperfunctioning endocrinopathies (including precocious puberty, hyperthyroidism, growth hormone excess, rickets/osteomalacia, and others), in which case it is known as McCune–Albright syndrome (MAS) (reviewed in [5–7]). In addition, FD may present in association with intramuscular myxomas (usually in patients with long-standing disease and associated endocrine dysfunction), in which case it is referred to as Mazabraud’s syndrome [8, 9]. The precise prevalence of the various forms of FD are not known, but FD has been reported to account for approximately 5% of benign bone lesions [10], with the monostotic form reported to be eight to ten times more common than the polyostotic form [5]. The association of FD with the extraskeletal manifestations is even less common. Virtually any combination of bone disease with extraskeletal manifestations is possible: FD with skin lesions and no endocrine dysfunction, FD with endocrine dysfunction and no skin lesions, and classic skin and endocrine findings without FD [6].
Over the last 25 years, a large cohort of 150 patients with FD has been studied for a total of >500 patients-year of follow-up at the National Institutes of Health. This cohort has undergone extensive evaluation for both skeletal and extraskeletal manifestations of PFD; some of the clinical and basic science findings that pertain to orthopedics will be reviewed here. In addition, we present a suggested plan to evaluate a new patient from both an orthopedic and endocrine standpoint.
Pathophysiology
Bianco et al. [11, 12] were the first to demonstrate that FD is a disease of the bone marrow stromal cell (BMSC). The BMSCs form the structural framework upon which hematopoiesis occurs in the bone marrow, and a subset of BMSCs are multipotent stem cells capable of differentiating into multiple cells including osteoblasts, osteocytes, chondrocytes, bone marrow adipocytes, and probably other cells [13]. In FD, BMSCs begin to differentiate along the osteogenic lineage, but differentiation is arrested, and instead the cells proliferate, giving rise to the fibroosseous masses of tissue that make up FD.
The molecular etiology of the disease, which leads to the arrest in differentiation that occurs in BMSCs, is activating mutations in the GNAS gene. GNAS codes for the alpha subunit of the signaling G protein, Gsα [14, 15]. Gsα is central in the cell signaling pathway that leads to the generation of the intracellular second messenger, cAMP, and activating mutations lead to ligand-independent cAMP/protein kinase A signaling. cAMP is involved in the signal transduction from multiple cell surface receptors, including parathyroid hormone (PTH), follicle stimulating hormone (FSH) and luteinizing hormone (LH), thyroid stimulating hormone (TSH), etc. All of the mutations in Gsα that have been identified in association with FD are at the 201Arg position. In >95% of the cases, arginine is replaced by either cysteine or histidine (R201C or R201H). These mutations result in inhibition of the intrinsic GTPase activity of Gsα protein and it is this aspect that leads to constitutive, ligand-independent generation of intracellular cAMP. In bone, it is as if the BMSCs harboring the Gsα mutation are under constant PTH stimulation, thus giving credence to Albright’s comparison of the similarity between osteitis fibrosa disseminata (what came to be called FD) and the bone disease of hyperparathyroidism, osteitis fibrosa cystica. The defect in Gsα also explains the molecular etiology of the associated extraskeletal manifestations of the café-au-lait spots (constitutive melanocyte simulating hormone signaling in skin melanocytes), precocious puberty (ovarian FSH signaling), hyperthyroidism (thyroid TSH signaling), etc.
Fibrous dysplasia is a genetic disease in which there has never been a well-documented case of vertical transmission from parent to offspring. This is a point worth making to parents and patients, as many have concerns that the disorder can be transmitted to the children of the patient or that subsequent children may also have the disease. It should also be emphasized that there is no evidence of the disease arising as a result of environmental (intrauterine) exposures, as this too is a major concern of parents. The observation that FD is not inherited, combined with the observation that the skin lesions in MAS follow the developmental lines of Blashko, and the fact that the bone disease often tends to involve one side of the skeleton, has led to the working, and generally accepted, hypothesis that FD is the result of somatic mutations that occur at some early stage of embryonic development. What follows from this hypothesis is that in patients with MAS, in whom tissues derived from all three germ layers are involved (bone/mesoderm, skin/ectoderm, and various endocrine tissues/endoderm), the mutation that gives rise to this phenotype must have occurred very early in development, at the inner cell mass stage [7] (Fig. 1). For disease that is isolated to the skeleton, but involves craniofacial and long bones, the mutation must have arisen later in development in the mesectoderm, as the craniofacial bones are of neuroectodermal origin, and long bones of mesodermal origin [7]. Thus, the phenotype and extent of skeletal involvement is dictated by the developmental stage and location in the developing organism where the mutation occurs (Fig. 1).
Fig. 1.
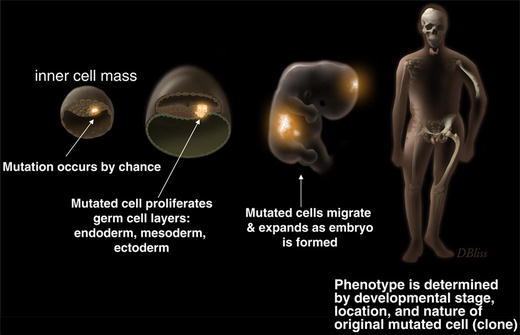
Development of fibrous dysplasia and McCune–Albright syndrome. The cartoon depicts the widely accepted hypothesis that FD/MAS is the result of somatic mutations that occur early in embryogenesis
Presentation and natural history
The result of a mutation involving a widely expressed, critical signaling protein that occurs sporadically at potentially any developmental stage is a disease of amazing complexity and variability. FD found in an adult with an isolated lesion in the fibula, discovered incidentally during a routine X-ray, is caused by the same molecular defect as the disease of a wheelchair-dependent, blind and deaf adolescent with total skeletal involvement, a history of innumerable fractures, skin macules, precocious puberty, hyperthyroidism, and acromegalic features [16].
Most children with FD present to the orthopedic surgeon do so because of a fracture through an FD lesion, a limp, or an incidentally discovered lesion when radiographs are obtained for another reason. The radiographic appearance of FD can appear markedly different depending on the age of the patient. The radiograph in the very young (<2 years of age) often lacks the classic “ground glass” appearance and has more of a streaked heterogeneous appearance, occasionally mistaken for enchondormatosis (Fig. 2a). With maturity, lesions become more lytic and develop a “ground glass” appearance (Fig. 2b), and with aging the lesions tend to become sclerotic at the edges of the lesion (Fig. 2c).
Fig. 2.
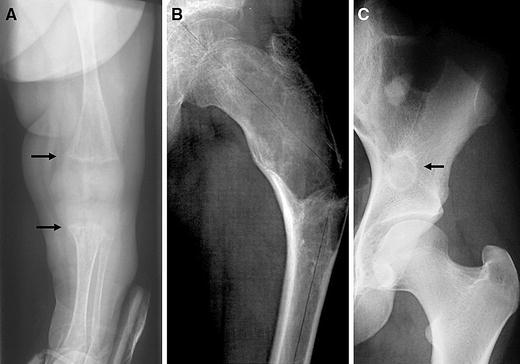
Radiographic appearance of FD. a In the very young, the lesions can have a streaky appearance, and be found in association with rickets (arrows). b The typical radiographic appearance of FD is described as “ground glass.” As seen in this radiograph of the proximal femur, which also demonstrates the pathognomonic, shepherd’s crook. c FD in older patients, or patients with relatively inactive disease will demonstrate a sclerotic rim around the FD (arrow)
When there is confusion about the diagnosis, a biopsy can be used to confirm the diagnosis. Histology usually shows what has classically been described as “Chinese characters” (Fig. 3a). The marrow cavity, which usually has a cellular fatty tissue, is replaced by fibroosseous tissue. With appropriate preparation, FD lesions also demonstrate a significant amount of osteoid (lesional osteomalacia), which may contribute to much of the clinical pathology observed (Fig. 3b). However, there are a number of histopathological subtleties that can be found in FD that either confuse or aid in the diagnosis. These subtleties and variants are beyond the scope of this discussion, but well-described elsewhere [17–19].
Fig. 3.
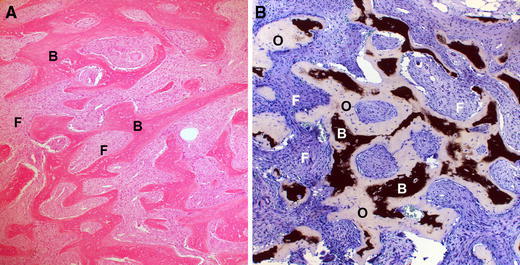
Histopathological appearance of FD. a H&E staining showing the classic “Chinese characters” made of trabeculae of bone (labeled B) with fibrous tissue interwoven between (labeled F). b Von Kossa stain for mineralization shows that the trabeculae of bone are composed of bone (black, labeled B) as well as osteoid (pale blue tissue labeled O). The fibrous tissue is labeled (F). The presence of the unmineralized bone confirms the osteomalacic nature of the lesions
Craniofacial Fibrous Dysplasia
Involvement of the craniofacial bones in FD is common. In fact, the skull base is the skeletal site most commonly involved with FD. As with the rest of the skeletal disease, craniofacial involvement can be trivial, asymptomatic and detected incidentally, or it can involve disfiguring involvement of the entire skull and be associated with vision and hearing loss. Skull disease in FD is sometimes associated with pain. The explanation for the pain is not clear, and there is no association between the extent or location of the disease and the likelihood of having pain. Patients with little skull disease may have debilitating pain, patients with extensive disease may be pain-free, and vice-versa. It has been our experience that unlike painful disease in the axial and appendicular skeleton, which usually responds to treatment with bisphosphonates, disease in the craniofacial region is less responsive. Pain can sometimes be debilitating and require treatment with relatively high doses of opiod analgesics. Such disease is best managed in conjunction with pain specialists.
There are no long-term studies of the natural history of craniofacial FD but, as can be seen in the appendicular skeleton, progression/expansion of craniofacial disease also has a tendency to slow or stop after skeletal maturation. There is one notable and important exception; that is when craniofacial disease is found in association with growth hormone excess, as seen in approximately 20% of the cases of MAS. Growth hormone excess is the single aspect of the disease that is associated with the craniofacial-specific morbidities of vision and hearing loss [20–22].
Fibrous dysplasia in the limbs and axial skeleton
Again, in fibrous dysplasia, there is a wide range of involvement. It can vary from an isolated single lesion in a single bone to involvement of the entire axial and appendicular skeleton. Limp, pain, or fracture through an FD lesion is the usual presentation. Lesions in the limbs also show great variability in involvement: they may be contained within a relatively normal cortex, or they can expand, deforming the bone and thinning the cortex, threatening structural stability of the bone. The radiographic appearance varies as well: from a classic ground glass appearance to that of a lytic or cystic appearance. The lesions may impact the skeleton minimally, or may cause tremendous deformity to the long bones. The spine, which was once not thought to be part of the constellation of FD, has recently been found to be involved frequently in the disorder and associated with scoliosis [23]. The wide variability in location of lesions, size of lesions and effect of the lesion on the bone create challenges for the orthopedist who would like to stabilize bones and prevent fracture and deformity. But such extreme variability makes individualized treatment a necessity. With aging, usually beyond skeletal maturity, FD lesions seem to be less aggressive. This is typically evidenced by the appearance of a sclerotic border around FD lesions. Clinically, the fracture rate improves over time [34]. However, in terms of the natural history of FD, more data needs to be collected to determine whether scoliosis progresses during adulthood, and whether there are changes in the angular deformities of the limbs over time [23, 49].
Fibrous dysplasia-associated malignancies
Malignant transformation of the benign neoplasm of FD is a relatively rare occurrence. The precise rate of malignant transformation is not known, but it is likely in the order of 1% or less. The most common presentation is the rapid expansion of a preexisting FD lesion in association with pain. Radiographically, there is disruption of the cortex in association with a soft tissue mass at the location of preexisting FD. Two large case series [23, 24] and two large reviews [25, 26] reported on approximately 123 cases of cancer, and found that approximately half of the cases arose in monostotic disease and half in polyostotic. Give that monostotic disease is more common, cancer is more likely to occur in polyostotic disease. In descending order of frequency, the craniofacial bones, femur, and tibia were the bones most likely to be involved. The most commonly seen histological types were osteosarcoma, fibrosarcoma, and chondrosarcoma, in descending order of frequency. There are reports suggesting that malignant transformation may be more common in Mazabraud’s syndrome (FD in association with intramuscular myxomas) [27, 28]. However, this may represent ascertainment bias, as the actual prevalence of intramuscular myxomas in association with FD may be more common than expected, as most of the intramuscular myxomas are asymptomatic and will go undetected.
Besides malignancies arising in bone, other malignancies may be found in association with FD, especially in the context of MAS. Both thyroid [29] and breast [30–32] cancer have been reported in association with MAS. Skeletal and extra-skeletal cancers may also be more common in those patients who also have growth hormone excess.
Impact of endocrine dysfunction on Fibrous Dysplasia
Endocrine dysfunction may impact on the disease in different ways, and it depends on the endocrine disease and the degree to which the endocrine dysfunction is controlled. Precocious puberty, together with bowing, can lead to extremely short stature (Fig. 4a). Yet, when growth hormone (GH) excess accompanies precocious puberty, stature can be normal, or even exceed the predicted height [20] (Fig. 4b). Renal phosphate wasting and hypophosphatemia, due to overproduction of the phosphate and vitamin D regulating hormone, FGF-23 [33], has been associated with earlier and more fractures [34], presumably by the direct effect of hypophosphatemia on inhibition of mineralization in FD, resulting in lesional osteomalacia [35, 36] (Fig. 3b). Thus, the management of FD should include an assessment of endocrine status for evidence of associated hyperfunctioning endocrinopathies, including renal phosphate wasting and hypophosphatemia [37].
Fig. 4.
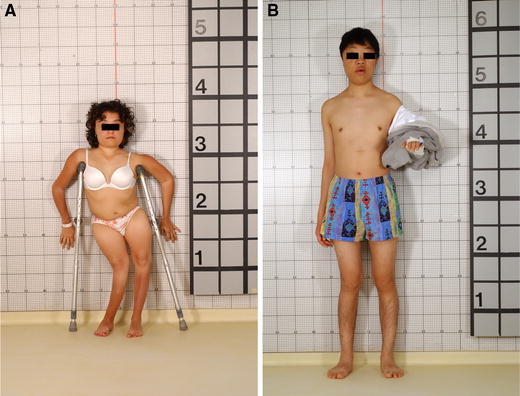
The varied phenotype of two patients with McCune–Albright syndrome. The patient depicted in a has had premature closure of the physes from precocious puberty, and that, together with lower extremity bowing (windswept deformity), has resulted in short stature. The patient in b has acromegaly in addition to precocious puberty, thus the presence of growth hormone “rescued” him from short stature
Evaluation and diagnosis
Imaging
Patients typically present with either a limp or a fracture, and a radiograph is usually the first imaging modality performed. The radiograph usually reveals a lytic, ground glass lesion consistent with FD. If there is doubt about the diagnosis, computed tomography (CT) is useful for demonstrating the medullary-based, fibroosseous nature of the FD, and magnetic resonance imaging (MRI) is useful for identifying/excluding fluid-filled non-FD cystic lesions. In order to determine the extent of disease, either a technetium bone scan or a skeletal survey should be performed. Bone scan is more sensitive in detecting skull, spine and rib lesions, as well as early, pre-clinical long bone lesions. By the age of 8–10 years, the vast majority of lesions (craniofacial and long bones) have already been established and are detectable by bone scan [38]. Thus, a bone scan performed early in the course of the disease is not only diagnostic, but can be used for prognosis.
Clinical examination of the patient for limb deformity, scoliosis, pelvic obliquity or facial asymmetry can help guide the choice of plain films. Non-contrast CT is the best imaging modality for craniofacial FD. CT with reformatting can also be useful in detecting subtle fractures within established FD lesions when radiographic findings are equivocal, but clinical suspicion for a fracture is high. (Figs. 5, 6) MRI can be helpful if there is a suspicion of an aneurysmal bone cyst, which can occur in association with long-standing FD (Fig. 7). Thus, the choice of imaging modalities must be tailored to the amount and location of involvement of the individual patient.
Fig. 5.
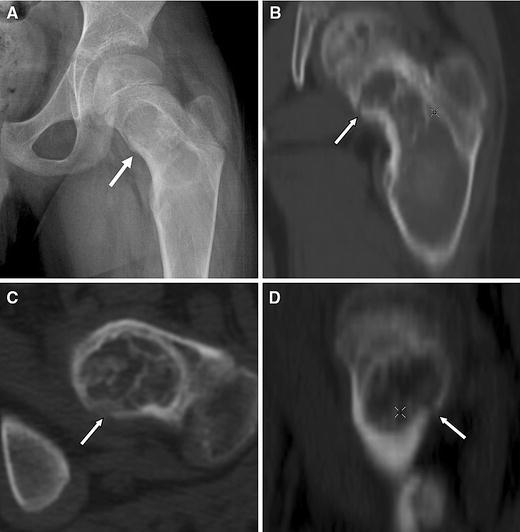
CT for detecting subtle fractures. a This radiograph was from a 9-year-old boy who complained of new-onset, focal pain in the groin. No discernable fracture is apparent. b–d Reformatted CT images of the lesion revealed a fracture in the medial proximal femur (arrows)
Fig. 6.
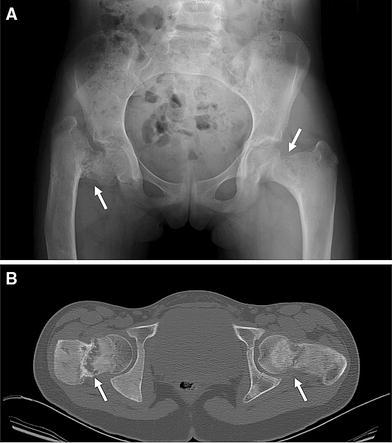
Use of CT imaging in FD. a This radiograph of a 10-year-old girl with FD revealed severe disease in both femurs, with a suggestion of a fracture. At the time of the radiograph, she was asymptomatic and ambulating without assistance. b CT at the level of the femora neck revealed the true extent of the disease, with almost complete disruption of the neck (arrows). The extent of the disease was not fully appreciated on the radiograph
Fig. 7.
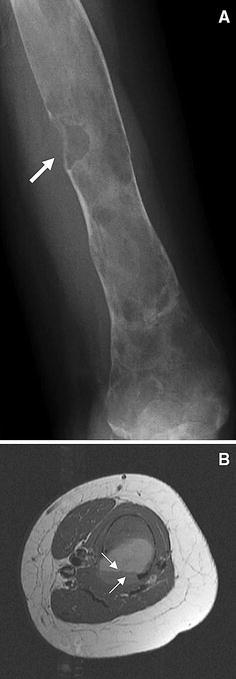
Aneurysmal bone cyst in FD. Aneurysmal bone cysts are uncommonly associated with fibrous dysplasia. a The radiograph demonstrated disruption of the cortex. b MRI imaging revealed a clear-cut fluid-fluid level (arrows), consistent with an aneurysmal bone cyst. These lesions can be aggressive, eroding bone as shown by the loss of the cortex in a and b, and bleed extensively. Adequate blood should be obtained if surgical intervention is planned
Biochemical and genetic testing
In FD, disease extent is best assessed by bone scan, but activity can also be assessed by measuring serum and urine markers of bone metabolism. This can be done with any number of assays that measure bone metabolism and include the bone formation markers alkaline phosphatase, bone-specific alkaline phosphatase, osteocalcin, etc., and serum and urine markers for bone resorption N-telopeptide of collagen, pyridinium cross links, and deoxypyridinoline cross links, etc. In general, all markers of bone metabolism are elevated in FD in parallel, relative to disease activity, and no specific assay is superior to another [39]. In fact, normal levels of the markers of bone metabolism can be used in support of excluding the diagnosis of FD in the case of what appears to be fairly extensive disease. However, in the case of low burden of disease, or in the case of older patients with “burnt out” disease, bone turnover markers may be only minimally elevated or normal. In consideration of cost, it may be most prudent to rely upon the least expensive of these markers, alkaline phosphatase, to estimate disease activity.
As part of the evaluation of patients with FD, other extraskeletal manifestations must be taken into consideration, and testing for these should be part of the routine evaluation. These laboratory tests are detailed in Table 1, and discussed in detail elsewhere [40].
Table 1.
Evaluation of extraskeletal disease associated with FD
| Extra-skeletal organ | Clinical manifestation | Testing | Comment |
|---|---|---|---|
| Gonad | |||
| Ovary Testicle |
Precocious puberty Precocious puberty, benign tumors |
Bone age, ultrasound, gonadotropin stimulation testing | Many men with FD will have benign testicular lesions only seen on ultrasound |
| Thyroid | Thyrotoxicosis | Thyroid function tests (TSH, T4, free T4, T3), ultrasound | Typically T3 toxicosis, can lead to intra-operative thyroid storm if undetected/treated |
| Pituitary | Growth hormone and prolactin excess | GH, IGF-1, prolactin, MRI | Untreated GH excess exacerbates craniofacial FD |
| Adrenal | Cushing syndrome | Serum and urine cortisol, dexamethasone suppression test, CT of adrenal glands | Only occurs in the neonatal period |
| Renal | Phosphate wasting, hypophosphatemia | Serum phosphate | Untreated hypophosphatemia may worsen FD |
| Parathyroid | Hyperparathyroidism | Serum PTH and calcium | The least frequent finding in FD/MAS, may or may not be part of the syndrome |
A more detailed explanation of testing and management of endocrine dysfunction can be found in Collins [40]
It is possible to perform genetic testing for mutations in GNAS, and this can occasionally be useful when there is uncertainty about the diagnosis. Several techniques to detect GNAS mutations have been described [14, 35, 41, 42]. However, there are a number of aspects related to genetic testing in this disease that must be considered prior to reliance upon the results to guide clinical decisions. First, one should remain cognizant that FD is a mosaic disease and for the most part only affected tissue will contain a significant number of cells harboring the mutation. Therefore, affected tissue must be used for DNA extraction and testing. Next, one must keep in mind that even affected tissue is an admixture of mutated and non-mutated cells [11], that bone is relatively acellular, and that the ability to detect mutated cells, which may be in low abundance, is dependent upon the sensitivity of the technique. The most sensitive technique for detecting low abundance mutated cells is that described by Karadag et al. [42]. While some report that the mutation can be detected in circulating white blood cells [43, 44], and this service is available from commercial laboratories, its utility remains to be established. In most cases, when the mutation can be detected in white cells, there is little question about the diagnosis on clinical grounds. However, in the rare cases of very extensive FD (panostotic), when there are no associated skin or endocrine lesions, FD may be difficult to distinguish from other skeletal dysplasias, such as severe osteogenesis imperfecta. In these cases, mutation testing may be particularly helpful. FD remains predominantly a diagnosis made on clinical grounds.
Evaluation of a new patient with Polyostotic Fibrous Dysplasia
A clinical evaluation should include examination of gait and limbs, and of the spine for alignment. Limb length discrepancy is a common finding, either from bone deformity, pelvic obliquity or scoliosis, and should be assessed. Any areas of clinical deformity should be followed with plain films. The physical examination of a new patient should include a search for skin lesions suggestive of MAS (Fig. 8), which may not bear a relationship in location to the lesions in the skeleton.
Fig. 8.
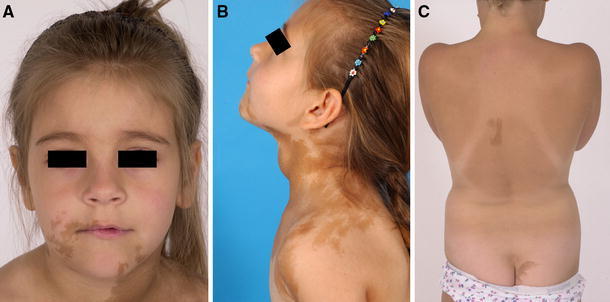
Café-au-lait spots found in MAS. a This girl demonstrates typical skin lesions on the face with jagged “coast-of-Maine” borders. Note that skin lesions are bilateral, but that the lesion on the neck tends to respect the midline. b The lateral view of the same patient seen in a demonstrates how the lesions can sometimes tend to follow the developmental lines of Blashko. Also of note is the obvious goiter in this girl with hyperthyroidism. c Skin lesions can often be subtle. These lesions demonstrate coast-of-Maine borders and respect the midline. Again, note that there are lesions on both the right and left. The location of the lesion on the buttocks is a typical location and appearance for a skin lesion seen in association with FD. This girl had FD and skin spots, but no endocrine dysfunction
A total body bone scan can help identify the location and extent of pathologic bone. The bone scan can be scored to generate a skeletal disease burden score that can then be used to predict future need for ambulation assistance and quality of life [45–47]. Early referral to a physiatrist and physical therapist for evaluation and treatment is an important part of the care. Laboratory testing to evaluate for the presence of endocrine dysfunction was discussed above (see Table 1). The presence of craniofacial FD is an indication for neuro-ophthalmologic and audiolgic testing. Clearly, treatment of patients with MAS and multiple organ involvement is best carried out by a team approach that includes assessment of skeletal and craniofacial involvement, endocrine evaluation, ophthalmologic assessment, hearing tests, and physiatry and physical therapy when needed.
Treatment
Orthopedic management
Fractures
In order to determine if new treatment modalities can decrease fractures in FD, the natural history of when fractures occur in childhood and adulthood must first be understood. In a study of 172 fractures in patients with an age range of 6–53, the peak fracture rate was between ages 5 and 10, and then decreased in adolescence (age 10–15), with even further decreases in number of fractures seen into adulthood [34]. The pattern of a peak in the fracture rate at age 5–10, followed by a decrease in the rate of fractures, was seen in the femur, tibia, humerus and forearm. The presence of renal phosphate wasting and hypophosphatemia was associated with more fractures that occurred at an earlier age. Thus, any patient who has multiple fractures, or whose first fracture occurs at a young age, should be evaluated for the presence of endocrinopathies, particularly renal phosphate wasting with resultant hypophosphatemia. Knowledge of the natural history of fractures in childhood makes it clear that if orthopedic procedures to stabilize the limbs, or medical therapies to strengthen the bone, are to have an effect on outcome, they must be introduced very early in childhood in order to have an impact on the peak fracture rate, which occurs from age 5 to 10. Likewise, once patients have reached adulthood the chance of fracture becomes much lower, so less aggressive management of lesions to prevent a fracture may be indicated (although attention to bone deformity per se may still need to be addressed). One of the limitations of the aforementioned study [34] is that few patients from age 60 to 90 were evaluated. Thus, the possibility of a second peak in the fracture incidence at the onset of senile osteoporosis cannot be ruled out.
There are no data to indicate when to operate prophylactically to prevent a fracture. Certainly patient complaints of pain are worrisome; although non-weight bearing bones, such as the skull and ribs, may also be painful, so pain per se does not equate to imminent fracture. Patient reports of increasing pain over time should prompt an evaluation. This could reveal a stress fracture, evidence of a fluid-filled cyst, or increasing deformity, worsening the mechanics of the bone. Radiographic evidence of a stress fracture is probably the best absolute indication for proceeding with elective stabilization. However, radiographs in complicated FD lesions can make the identification of stress fractures difficult. CT scans with reformatting can sometimes detect fractures that were not able to be identified on radiograph (Figs. 5, 6). Progression of pain (especially in association with a lesion that was previously pain-free), deformity, or loss of function, are also good indications.
Intramedullary rods are perhaps the most efficient devices at protecting bone [48–50]. However, there are limitations in using this device, particularly for correction of the proximal femur where a second generation IM rod is not yet readily available for use in children. Use of plate and screw fixation necessitates spanning the lesions and gaining screw purchase into normal bone to prevent eventual loss of fixation (Fig. 9). In children, the physis appears to limit the expansion of lesions in FD. We have rarely observed lesions in the femoral head. Thus, spanning the proximal femoral physis with a screw may be necessary for gaining adequate purchase into normal bone in the femoral head.
Fig. 9.
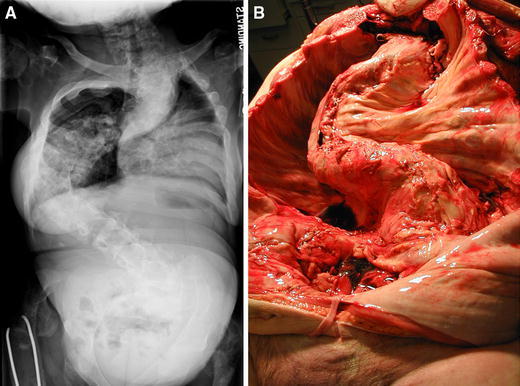
Scoliosis in association with FD. a The large magnitude curve in this 15-year-old girl, whose growth plates had been closed since the age of 10 due to precocious puberty, continued to progress after skeletal maturity. b A second patient who died from the complications of untreated scoliosis also developed this lesion after skeletal maturity. These dramatic cases emphasize the need to screen for, follow, and treat scoliosis in association with FD
Scoliosis
Scoliosis has been recently added to the constellation of orthopedic problems commonly found in association with PFD [23]. Previously, scoliosis and involvement of the spine with FD was thought to be an uncommon problem, mostly reported in case reports. Scoliosis can be difficult to access clinically in patients with lower extremity deformities causing limb length discrepancies or pelvic deformity causing pelvic obliquity. Thus, scoliosis may be attributed to secondary compensation and not correctly identified as a primary disorder. In a study of 62 patients with PFD, lesions in the spine were identified in 63% of the study group and scoliosis in 40–52% of the patients [23]. There was a statistically significant association between the lesions in the spine and the level at which the curve occurred, suggesting that the FD was causative. The location of café-au-lait lesions, and other areas of bone involvement (pelvis, ribs), did not correlate with scoliosis.
The care of scoliosis in children involves identification and aggressive monitoring. The role of bracing in adolescents with scoliosis and FD is unclear, and may need to be individualized to the patient. Certainly applying three-point fixation to ribs or pelvis that may have disease would be worrisome, while the patient with lesions only in the spine and not in the areas of contact of the brace would be more easily treated by bracing. Patients in our series with large curves have undergone successful posterior spinal fusions with no evidence of difficulty with fusion or loss of fixation in up to 22 years of follow-up. Because the etiology of the curve in FD is the deformity caused by bone that is more plastic in nature, it is unclear whether curve progression abates in adolescents or continues into adulthood. Our suspicion is that curves may continue to progress into adulthood, as we have identified patients who developed pulmonary symptoms after childhood and growth plate closure (Fig. 10). Identification of curves in children and adolescents requires a plan of monitoring during the adult years to avoid unchecked progression of scoliosis. The only endocrinopathy found to correlate with the presence of scoliosis was renal phosphate wasting, which suggests the osteomalacic bone with greater plasticity is part of the mechanism of the development of scoliosis.
Fig. 10.
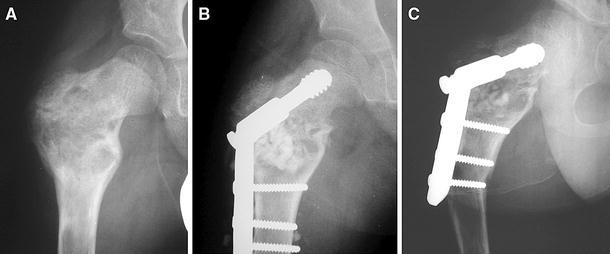
Difficulty in managing proximal femur disease. a A proximal femoral head stress fracture that required stabilization is shown. b A hip screw and side plate plus bone grafting was used to treat the fracture. c A year later, the bone graft has resorbed and the plate, which ended in bone involved with FD, is starting to cause bending at the end of the plate
Proximal femur
The pathognomonic lesion in FD is the shepherd’s crook deformity with bowing of the proximal femur and loss of the neck shaft angle (Fig. 2b). When surgery is indicated, the intervention should strive to restore the mechanical axes of the limb, thus reducing the deforming forces to the bones. Towards this end, medial displacement valgus osteotomy has been recommended [51]. Administration of the Pediatric Outcomes Data Collection Instrument (PODCI) functional outcomes questionnaire revealed that decreased neck shaft angle was also associated with decreased functional mobility for sports, while the amount of disease in the lower extremities was found to correlate with the transfer scale and the sports scale [47]. This raises the question as to whether or not procedures to maintain the femoral neck shaft angle can improve the functional outcome in children with large amounts of lower extremity disease. The answer to this will require a multicenter study, testing the ability of various procedures and the timing of the procedures to improve outcome.
The role of bone grafting
Enneking [52] popularized the use of cortical strut bone grafting in the treatment of FD in 1986. Later, cortical struts and subtotal resection of the lesions were proposed [53], and others have proposed the use of cryosurgery in association with grafting [54]. However, others have not been able to reproduce successful long-term results with these techniques, and have noted disappearance of bone graft observed over time and no decrease in the size of the lesion [49, 51, 55]. Since most published cohorts of surgically treated FD are small, it remains controversial as to whether bone grafting should be used and if so what kind of grafting materials should be employed. We have not observed good long-term retention of any graft material [34], and feel that there needs to be continued study, testing the utility of bone grafting in FD, ideally as part of a multicenter study.
Surgical indications
Absolute surgical indications for the treatment of FD are lacking. An individual surgeon’s experience is limited by both the rarity and spectrum of severity found in this disorder. In our cohort, we found that in many patients who failed surgical management it was most often due to loss of fixation, with cut-out of metal devices. We suggest prior to undertaking surgical intervention that consideration of endocrinopathies be entertained, particularly as hyperparathyroidism or phosphate wasting can further weaken bone that is already osteomalacic [36]. Selection of the proper surgical candidate and orthopedic device is vital to a good outcome. Caution should be used in the panostotic form of the disorder where there is no normal bone with which fixation can gain purchase. There are patients in whom no amount of surgical intervention will prevent progression to the need for assisted ambulation.
Medical management
Early non-surgical attempts of the treatment of FD included glucocorticoids, calcitonin, and external beam radiation [56–58]. None of these treatments were effective, and radiation predisposes FD to malignant transformation [24]. The only medications that have shown any efficacy in the treatment of FD are the bisphosphonates. While FD is a disease of the osteoblast, bisphosphonates, which inhibit osteoclasts, were advocated for two reasons. First, it was felt that lesion expansion was mediated by osteoclastic resorption of adjacent normal bone, and that bisphosphonates would inhibit this, and thus stop lesion expansion. Second, FD is a “high turnover” bone disease, sometimes with dramatic elevations in markers of bone turnover and occasionally evidence of increased numbers of osteoclasts in FD lesions [59]. Bisphosphonate use was postulated to inhibit these processes. The first report of the use of bisphosphonates in FD was by Liens et al. [60], wherein they treated nine patients with high dose, intravenous pamidronate. The authors subsequently increased the size of the group, and reported on long-term outcomes [61, 62]. Others have used similar high dose, intravenous regimens and reported similar results [63–66]. The universal findings in all of these studies are that high-dose, intravenous pamidronate decreases pain and markers of bone metabolism. Some report improvement in the radiographic appearance, bone density, and fracture recurrence or rate. Oral bisphosphonates and other intravenous bisphosphonates have also been used with similar findings [67, 68]. The most rigorously conducted study to date using high dose, intravenous pamidronate (1–1.5 mg/kg per day on three consecutive days, given every 4 months) was by Plotkin et al. [69]. This study entailed pre- and post-treatment biopsies of affected and unaffected bone and longitudinal radiographs of sentinel FD lesions. They found pain and markers of bone metabolism were decreased, but there was no improvement in the radiographic appearance or the propensity to fracture. This work was also important and reassuring in showing that there was no effect on longitudinal growth of the children, nor was there evidence of a significant deleterious effect of bisphosphonates on the unaffected, normal bone in children with FD treated with high dose, intravenous pamidronate. The benefits and risks of this class of drugs, the indications for treatment, and the most effective treatment regimen (depending on the indication) remain to be defined. Ongoing, placebo-controlled trials of bisphosphonates in patients with FD in the US and Europe [70] may help to define the role of these drugs. At this time, the only clear indication for the use of bisphosphonates in FD is for pain relief. When these drugs are used, the minimum dose needed to relieve pain should be used. This is especially true given the recently-described association between the use of high dose bisphosphonates and the development of osteonecrosis of the jaw [71].
Future treatment and research
Stem cells
Stem cell treatment for FD is attractive. After all, FD is a disease of the bone marrow stromal cell (mesenchymal stem cell) [11, 12, 72]. As such, as a stem cell disease, it may be a candidate for mesenchymal stem cell tissue engineering [13, 73]. However, there are a number of significant barriers to overcome before this approach is practical. FD is a somatic disease, so treatment would need to be targeted to affected parts of the skeleton. Restoration of functionality to established lesions would involve the recreation of the fine bone architecture and use of carriers, scaffolds and lattices that are not yet available in the clinics. However, given the broad interest in tissue engineering techniques, and recent significant gains in the preclinical [74] and clinical use of bone marrow stromal cells [75], this approach may be within our reach in the not so distant future.
Medical treatment
In theory, any new medication in use or in development with bone specificity is potentially of use in the treatment of FD. Drugs currently in use, other then other bisphosphonates, include those approved for the treatment of osteoporosis: raloxifene and parathyroid hormone. The use of either of these in the treatment of FD is intuitively unappealing. Raloxifene is an estrogen receptor modulator and in children this would act as an anti-estrogen in bone, thus having potentially deleterious effects on unaffected bone and likely to have no effect on FD. Parathyroid hormone activates the Gsα-cAMP signaling pathway, which is already activated in FD and therefore of obvious concern. There are new drugs in the development pipeline with “anti-resorptive” action including denosumab, a monoclonal antibody-derived drug that inhibits RANK ligand, and balicatib, an inhibitor of the enzyme central in osteoclast action, cathepsin K. The mechanism of action of these drugs is similar to that of the bisphosphonates, in that they inhibit osteoclastic bone resorption, and advantages over the bisphosphonates may not be significant. A new anabolic drug, a neutralizing antibody to sclerostin, the protein involved in the pathophysiology of van Buchem’s disease, is in development. It may be of benefit in FD.
Fibrous dysplasia is a disease that is ideally positioned for molecular targeting. The fact that >95% of the underlying molecular defects in FD occur at the same amino acid make it a candidate for compounds with specific activity at the protein harboring the R201 mutations. Drug discovery for such compounds can be accomplished through high throughput screening of molecular libraries with appropriate assays.
Summary
Fibrous dysplasia is a difficult disease to manage. The sporadic mosaic nature of the disease means that it is an uncommon disease with a variable expression. Each case is unique, and the approach that was appropriate in the previous case is likely not to be the best in this case. The intrinsic fibrous and osteomalacic nature of the tissue, and the propensity to involve the critical, weight-bearing structures of the proximal femur, make surgical intervention both necessary and challenging. These intrinsic problems are sometimes compounded by extrinsic endocrine disease, especially hypophosphatemia and hyperthyroidism, which tend to exacerbate the intrinsic problems.
In spite of all of these challenges, over the 70 or so years since the disease was first described, there has been an evolution in the approach to treatment. First, it is clear that most patients with FD warrant screening for accompanying endocrine dysfunctions, and screening and monitoring of scoliosis. Second, grafting materials usually fail and should not be a central aspect of the surgical approach. Third, intramedullary devices are in general superior to side plates and screws in most cases. Finally, there is some disease so severe that no current surgical approaches will affect final functional outcome, and in those patients doing less may be the best approach. In the future, we can look to new medical treatments that are on the horizon, and should work for the initiation of international collaborative studies to better define optimal surgical approaches to the treatment of this challenging disease.
Acknowledgment
This work was supported by the Division of Intramural Research, National Institute of Dental and Craniofacial Research of the Intramural Research Program, National Institutes of Health, Department of Health and Human Services.
References
- 1.McCune DJ. Osteitis fibrosa cystica: the case of a nine-year-old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child. 1936;52:743–744. [Google Scholar]
- 2.Albright F, Butler AM, Hampton AO, Smith P. Syndrome characterized by osteitis fibrosa disseminata, areas, of pigmentation, and endocrine dysfunction, with precocious puberty in females: report of 5 cases. N Engl J Med. 1937;216:727–746. doi: 10.1056/NEJM193704292161701. [DOI] [Google Scholar]
- 3.Lichtenstein L. Polyostotic fibrous dysplasia. Arch Surg. 1938;36:874–898. doi: 10.1001/archsurg.1938.01190230153012. [DOI] [Google Scholar]
- 4.Lichtenstein L, Jaffe HL. Fibrous dysplasia of bone: a condition affecting one, several or many bones, graver cases of which may present abnormal pigmentation of skin, premature sexual development, hyperthyroidism or still other extraskeletal abnormalities. Arch Pathol. 1942;33:777–816. [Google Scholar]
- 5.Dorfman HD, Czerniak B (1998) Fibroosseous lesions. In: Dorfman HD, Czerniak B (eds) Bone tumors. Mosby, St. Louis, pp 441–491
- 6.Collins MT, Bianco P (2003) Fibrous dysplasia. In: Favus MJ (ed) Primer on the metabolic bone diseases and disorders of mineral metabolism, 5th edn. American Society for Bone and Mineral Research, Washington D.C., pp 466–470
- 7.Bianco P, Robey PG, Wientroub S (2003) Fibrous dysplasia. In: Glorieux FH, Pettifor J, Juppner H (eds) Pediatric bone: biology and disease. Academic Press/Elsevier, New York, pp 509–539
- 8.Henschen F, Fallvon H. Osteitis fibosa with multiple tumors in the musculature. Verh Dtsch Ges Pathol. 1926;21:93–97. [Google Scholar]
- 9.Mazabraud A, Girard J. A peculiar case of fibrous dysplasia with osseous and tendinous localizations. Rev Rhum Mal Osteoartic. 1957;24:652–659. [PubMed] [Google Scholar]
- 10.Campanacci M (1999) Bone and soft tissue tumors: clinical features, imaging, pathology, and treatment, 2nd edn. Springer, Berlin Heidelberg New York
- 11.Bianco P, Kuznetsov SA, Riminucci M, Fisher LW, Spiegel AM, Robey PG. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gsalpha-mutated skeletal progenitor cells. J Clin Invest. 1998;101:1737–1744. doi: 10.1172/JCI2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bianco P, Robey P. Diseases of bone and the stromal cell lineage. J Bone Miner Res. 1999;14:336–41. doi: 10.1359/jbmr.1999.14.3.336. [DOI] [PubMed] [Google Scholar]
- 13.Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19:180–192. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 14.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune–Albright syndrome (see comments) N Engl J Med. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 15.Schwindinger WF, Francomano CA, Levine MA. Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune–Albright syndrome. Proc Natl Acad Sci USA. 1992;89:5152–5156. doi: 10.1073/pnas.89.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alman BA, Greel DA, Wolfe HJ. Activating mutations of Gs protein in monostotic fibrous lesions of bone. J Orthop Res. 1996;14:311–315. doi: 10.1002/jor.1100140221. [DOI] [PubMed] [Google Scholar]
- 17.Riminucci M, Fisher LW, Shenker A, Spiegel AM, Bianco P, Gehron Robey P. Fibrous dysplasia of bone in the McCune–Albright syndrome: abnormalities in bone formation (see comments) Am J Pathol. 1997;151:1587–1600. [PMC free article] [PubMed] [Google Scholar]
- 18.Riminucci M, Liu B, Corsi A, Shenker A, Spiegel AM, Robey PG, Bianco P. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol. 1999;187:249–258. doi: 10.1002/(SICI)1096-9896(199901)187:2<249::AID-PATH222>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 19.Corsi A, De Maio F, Ippolito E, Cherman N, Gehron Robey P, Riminucci M, Bianco P. Monostotic fibrous dysplasia of the proximal femur and liposclerosing myxofibrous tumor: which one is which? J Bone Miner Res. 2006;21:1955–1958. doi: 10.1359/jbmr.060818. [DOI] [PubMed] [Google Scholar]
- 20.Akintoye SO, Chebli C, Booher S, Feuillan P, Kushner H, Leroith D, Cherman N, Bianco P, Wientroub S, Robey PG, Collins MT. Characterization of gsp-mediated growth hormone excess in the context of McCune–Albright syndrome. J Clin Endocrinol Metab. 2002;87:5104–5112. doi: 10.1210/jc.2001-012022. [DOI] [PubMed] [Google Scholar]
- 21.Lee JS, FitzGibbon E, Butman JA, Dufresne CR, Kushner H, Wientroub S, Robey PG, Collins MT. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med. 2002;347:1670–1676. doi: 10.1056/NEJMoa020742. [DOI] [PubMed] [Google Scholar]
- 22.Cutler CM, Lee JS, Butman JA, FitzGibbon E, Kelly MH, Brillante B, Feuillan P, Robey P, Dufresne CR, Collins MT (2006) Long term outcome of optic nerve encasement and optic nerve decompression in patients with fibrous dysplasia: risk factors for blindness and safety of observation. Neurosurgery (in press) [DOI] [PubMed]
- 23.Leet AI, Magur E, Lee JS, Wientroub S, Robey PG, Collins MT. Fibrous dysplasia in the spine: prevalence of lesions and association with scoliosis. J Bone Joint Surg Am. 2004;86A:531–537. [PubMed] [Google Scholar]
- 24.Ruggieri P, Sim FH, Bond JR, Unni KK. Malignancies in fibrous dysplasia. Cancer. 1994;73:1411–1424. doi: 10.1002/1097-0142(19940301)73:5<1411::AID-CNCR2820730516>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz DT, Alpert M. The malignant transformation of fibrous dysplasia. Am J Med Sci. 1964;247:1–20. doi: 10.1097/00000441-196401000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Yabut SM, Jr, Kenan S, Sissons HA, Lewis MM. Malignant transformation of fibrous dysplasia: a case report and review of the literature. Clin Orthop. 1988;228:281–289. [PubMed] [Google Scholar]
- 27.Lopez-Ben R, Pitt MJ, Jaffe KA, Siegal GP. Osteosarcoma in a patient with McCune–Albright syndrome and Mazabraud’s syndrome. Skeletal Radiol. 1999;28:522–526. doi: 10.1007/s002560050556. [DOI] [PubMed] [Google Scholar]
- 28.Jhala DN, Eltoum I, Carroll AJ, Lopez-Ben R, Lopez-Terrada D, Rao PH, Pettenati MJ, Siegal GP. Osteosarcoma in a patient with McCune–Albright syndrome and Mazabraud’s syndrome: a case report emphasizing the cytological and cytogenetic findings. Hum Pathol. 2003;34:1354–1357. doi: 10.1016/j.humpath.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 29.Collins MT, Sarlis NJ, Merino MJ, Monroe J, Crawford SE, Krakoff JA, Guthrie LC, Bonat S, Robey PG, Shenker A. Thyroid carcinoma in the McCune–Albright syndrome: contributory role of activating Gs alpha mutations. J Clin Endocrinol Metab. 2003;88:4413–4417. doi: 10.1210/jc.2002-021642. [DOI] [PubMed] [Google Scholar]
- 30.Scanlon EF, Burkett FE, Sener SF, Green OC, Traisman HS, Marr TJ, Victor TA, Crist ML. Breast carcinoma in a 11-year-old girl with Albright’s syndrome. Breast. 1980;6:6–9. [Google Scholar]
- 31.Tanabeu Y, Nakahara S, Mitsuyama S, Ono M, Toyoshima S. Breast cancer in a patient with McCune–Albright syndrome. Breast Cancer. 1998;5:175–178. doi: 10.1007/BF02966691. [DOI] [PubMed] [Google Scholar]
- 32.Huston TL, Simmons RM. Ductal carcinoma in situ in a 27-year-old woman with McCune–Albright syndrome. Breast J. 2004;10:440–442. doi: 10.1111/j.1075-122X.2004.21490.x. [DOI] [PubMed] [Google Scholar]
- 33.Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leet AI, Chebli C, Kushner H, Chen CC, Kelly MH, Brillante BA, Robey PG, Bianco P, Wientroub S, Collins MT. Fracture incidence in polyostotic fibrous dysplasia and the McCune–Albright syndrome. J Bone Miner Res. 2004;19:571–577. doi: 10.1359/JBMR.0301262. [DOI] [PubMed] [Google Scholar]
- 35.Bianco P, Riminucci M, Majolagbe A, Kuznetsov SA, Collins MT, Mankani MH, Corsi A, Bone HG, Wientroub S, Spiegel AM, Fisher LW, Robey PG. Mutations of the GNAS1 gene, stromal cell dysfunction, and osteomalacic changes in non-McCune–Albright fibrous dysplasia of bone. J Bone Miner Res. 2000;15:120–128. doi: 10.1359/jbmr.2000.15.1.120. [DOI] [PubMed] [Google Scholar]
- 36.Corsi A, Collins MT, Riminucci M, Howell PG, Boyde A, Robey PG, Bianco P. Osteomalacic and hyperparathyroid changes in fibrous dysplasia of bone: core biopsy studies and clinical correlations. J Bone Miner Res. 2003;18:1235–1246. doi: 10.1359/jbmr.2003.18.7.1235. [DOI] [PubMed] [Google Scholar]
- 37.Ippolito E, Bray EW, Corsi A, De Maio F, Exner UG, Robey PG, Grill F, Lala R, Massobrio M, Pinggera O, Riminucci M, Snela S, Zambakidis C, Bianco P. Natural history and treatment of fibrous dysplasia of bone: a multicenter clinicopathologic study promoted by the European Pediatric Orthopaedic Society. J Pediatr Orthop B. 2003;12:155–177. doi: 10.1097/01.bpb.0000064021.41829.94. [DOI] [PubMed] [Google Scholar]
- 38.Hart ES, Kelly MH, Chen CC, Ziran N, Lee JS, Feuillan P, Kushner H, Robey P, Collins MT (2006) Onset and progression of fibrous dysplasia lesions, and relationship to functional outcome. J Bone Miner Res (in press) [DOI] [PubMed]
- 39.Collins MT, Chebli C, Jones J, Kushner H, Consugar M, Rinaldo P, Wientroub S, Bianco P, Robey PG. Renal phosphate wasting in fibrous dysplasia of bone is part of a generalized renal tubular dysfunction similar to that seen in tumor-induced osteomalacia. J Bone Miner Res. 2001;16:806–813. doi: 10.1359/jbmr.2001.16.5.806. [DOI] [PubMed] [Google Scholar]
- 40.Collins MT (2004) McCune–Albright syndrome. In: Filetti S (ed) Orphanet Encyclopedia, Paris. http://www.orpha.net/data/patho/GB/uk-McCune-Albright-Syndrome.pdf
- 41.Candeliere GA, Roughley PJ, Glorieux FH. Polymerase chain reaction-based technique for the selective enrichment and analysis of mosaic arg201 mutations in G alpha s from patients with fibrous dysplasia of bone. Bone. 1997;21:201–206. doi: 10.1016/S8756-3282(97)00107-5. [DOI] [PubMed] [Google Scholar]
- 42.Karadag A, Riminucci M, Bianco P, Cherman N, Kuznetsov SA, Nguyen N, Collins MT, Robey PG, Fisher LW. A novel technique based on a PNA hybridization probe and FRET principle for quantification of mutant genotype in fibrous dysplasia/McCune–Albright syndrome. Nucleic Acids Res. 2004;32(7):E63. doi: 10.1093/nar/gnh059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lumbroso S, Paris F, Sultan C. Activating Gsalpha mutations: analysis of 113 patients with signs of McCune–Albright syndrome—a European Collaborative Study. J Clin Endocrinol Metab. 2004;89:2107–2113. doi: 10.1210/jc.2003-031225. [DOI] [PubMed] [Google Scholar]
- 44.Hannon TS, Noonan K, Steinmetz R, Eugster EA, Levine MA, Pescovitz OH. Is McCune–Albright syndrome overlooked in subjects with fibrous dysplasia of bone? J Pediatr. 2003;142:532–538. doi: 10.1067/mpd.2003.153. [DOI] [PubMed] [Google Scholar]
- 45.Collins MT, Kushner H, Reynolds JC, Chebli C, Kelly MH, Gupta A, Brillante B, Leet AI, Riminucci M, Robey PG, Bianco P, Wientroub S, Chen CC. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res. 2005;20:219–226. doi: 10.1359/JBMR.041111. [DOI] [PubMed] [Google Scholar]
- 46.Kelly MH, Brillante B, Kushner H, Gehron Robey P, Collins MT. Physical function is impaired but quality of life preserved in patients with fibrous dysplasia of bone. Bone. 2005;37:388–394. doi: 10.1016/j.bone.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 47.Leet AI, Wientroub S, Kushner H, Brillante B, Kelly MH, Robey PG, Collins MT. The correlation of specific orthopaedic features of polyostotic fibrous dysplasia with functional outcome scores in children. J Bone Joint Surg Am. 2006;88:818–823. doi: 10.2106/JBJS.E.00259. [DOI] [PubMed] [Google Scholar]
- 48.O’Sullivan M, Zacharin M. Intramedullary rodding and bisphosphonate treatment of polyostotic fibrous dysplasia associated with the McCune–Albright syndrome. J Pediatr Orthop. 2002;22:255–260. [PubMed] [Google Scholar]
- 49.Ippolito E, Caterini R, Farsetti P, Potenza V. Surgical treatment of fibrous dysplasia of bone in McCune–Albright syndrome. J Pediatr Endocrinol Metab. 2002;15(Suppl 3):939–944. [PubMed] [Google Scholar]
- 50.Jung ST, Chung JY, Seo HY, Bae BH, Lim KY. Multiple osteotomies and intramedullary nailing with neck cross-pinning for shepherd’s crook deformity in polyostotic fibrous dysplasia: 7 femurs with a minimum of 2-years follow-up. Acta Orthop. 2006;77:469–473. doi: 10.1080/17453670610046415. [DOI] [PubMed] [Google Scholar]
- 51.Guille JT, Kumar SJ, MacEwen GD. Fibrous dysplasia of the proximal part of the femur. Long-term results of curettage and bone-grafting and mechanical realignment. J Bone Joint Surg Am. 1998;80:648–658. doi: 10.2106/00004623-199805000-00005. [DOI] [PubMed] [Google Scholar]
- 52.Enneking WF, Gearen PF. Fibrous dysplasia of the femoral neck. Treatment by cortical bone-grafting. J Bone Joint Surg Am. 1986;68:1415–1422. [PubMed] [Google Scholar]
- 53.Shih HN, Su JY, Hsu KY, Hsu RW. Allogeneic cortical strut for benign lesions of the humerus in adolescents. J Pediatr Orthop. 1997;17:433–436. [PubMed] [Google Scholar]
- 54.Segev E, Kollender Y, Bickels J, Flusser G, Issakov J, Wientroub S, Meller I. Cryosurgery in fibrous dysplasia: good result of a multimodality protocol in 16 patients. Acta Orthop Scand. 2002;73:483–486. doi: 10.1080/00016470216318. [DOI] [PubMed] [Google Scholar]
- 55.Stanton RP, Montgomery BE. Fibrous dysplasia. Orthopedics. 1996;19:679–685. doi: 10.3928/0147-7447-19960801-14. [DOI] [PubMed] [Google Scholar]
- 56.Di Figlia SE. Cortisone in polyostotic fibrous dysplasia. NY State J Med. 1951;51:2665. [PubMed] [Google Scholar]
- 57.Bell NH, Avery S, Johnston CC., Jr Effects of calcitonin in Paget’s disease and polyostotic fibrous dysplasia. J Clin Endocrinol Metab. 1970;31:283–290. doi: 10.1210/jcem-31-3-283. [DOI] [PubMed] [Google Scholar]
- 58.Tanner HC, Jr, Dahlin DC, Childs DS., Jr Sarcoma complicating fibrous dysplasia. Probable role of radiation therapy. Oral Surg Oral Med Oral Pathol. 1961;14:837–846. doi: 10.1016/S0030-4220(61)80014-5. [DOI] [PubMed] [Google Scholar]
- 59.Riminucci M, Kuznetsov SA, Cherman N, Corsi A, Bianco P, Gehron Robey P. Osteoclastogenesis in fibrous dysplasia of bone: in situ and in vitro analysis of IL-6 expression. Bone. 2003;33:434–442. doi: 10.1016/S8756-3282(03)00064-4. [DOI] [PubMed] [Google Scholar]
- 60.Liens D, Delmas PD, Meunier PJ. Long-term effects of intravenous pamidronate in fibrous dysplasia of bone. Lancet. 1994;343:953–954. doi: 10.1016/S0140-6736(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 61.Chapurlat RD, Delmas PD, Liens D, Meunier PJ. Long-term effects of intravenous pamidronate in fibrous dysplasia of bone. J Bone Miner Res. 1997;12:1746–1752. doi: 10.1359/jbmr.1997.12.10.1746. [DOI] [PubMed] [Google Scholar]
- 62.Chapurlat R, Delmas PD, Liens D, Meunier PJ. Long-term effects of intravenous pamidronate in fibrous dysplasia of bone. J Bone Miner Res. 2002;10:1746–1752. doi: 10.1359/jbmr.1997.12.10.1746. [DOI] [PubMed] [Google Scholar]
- 63.Pfeilschifter J, Ziegler R. Effect of pamidronate on clinical symptoms and bone metabolism in fibrous dysplasia and McCune–Albright syndrome. Med Klin (Munich) 1998;93:352–359. doi: 10.1007/BF03044679. [DOI] [PubMed] [Google Scholar]
- 64.Lala R, Matarazzo P, Bertelloni S, Buzi F, Rigon F, de Sanctis C. Pamidronate treatment of bone fibrous dysplasia in nine children with McCune–Albright syndrome. Acta Paediatr. 2000;89:188–193. doi: 10.1111/j.1651-2227.2000.tb01214.x. [DOI] [PubMed] [Google Scholar]
- 65.Zacharin M, O’Sullivan M. Intravenous pamidronate treatment of polyostotic fibrous dysplasia associated with the McCune Albright syndrome. J Pediatr. 2000;137:403–409. doi: 10.1067/mpd.2000.107836. [DOI] [PubMed] [Google Scholar]
- 66.Parisi MS, Oliveri MB, Gomez Acotto C, Mautalen C. Intravenous pamidronate increases bone mineral density and reduces bone remodeling markers in fibrous dysplasia. Bone. 2001;29:300–301. doi: 10.1016/S8756-3282(01)00531-2. [DOI] [Google Scholar]
- 67.Weinstein RS. Long-term aminobisphosphonate treatment of fibrous dysplasia: spectacular increase in bone density (see comments) J Bone Miner Res. 1997;12:1314–1315. doi: 10.1359/jbmr.1997.12.8.1314. [DOI] [PubMed] [Google Scholar]
- 68.Lane JM, Khan SN, O’Connor WJ, Nydick M, Hommen JP, Schneider R, Tomin E, Brand J, Curtin J. Bisphosphonate therapy in fibrous dysplasia. Clin Orthop. 2001;382:6–12. doi: 10.1097/00003086-200101000-00003. [DOI] [PubMed] [Google Scholar]
- 69.Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, Glorieux FH. Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab. 2003;88:4569–4575. doi: 10.1210/jc.2003-030050. [DOI] [PubMed] [Google Scholar]
- 70.Chapurlat R. Current pharmacological treatment for fibrous dysplasia and perspectives for the future. Joint Bone Spine. 2005;72:196–198. doi: 10.1016/j.jbspin.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 71.Woo SB, Hellstein JW, et al. Narrative [corrected] review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med. 2006;144:753–761. doi: 10.7326/0003-4819-144-10-200605160-00009. [DOI] [PubMed] [Google Scholar]
- 72.Bianco P, Riminucci M, Kuznetsov S, Robey PG. Multipotential cells in the bone marrow stroma: regulation in the context of organ physiology. Crit Rev Eukaryot Gene Expr. 1999;9:159–173. doi: 10.1615/CritRevEukarGeneExpr.v9.i2.30. [DOI] [PubMed] [Google Scholar]
- 73.Bianco P, Robey PG. Stem cells in tissue engineering. Nature. 2001;414:118–121. doi: 10.1038/35102181. [DOI] [PubMed] [Google Scholar]
- 74.Mankani MH, Kuznetsov SA, Shannon B, Nalla RK, Ritchie RO, Qin Y, Robey PG. Canine cranial reconstruction using autologous bone marrow stromal cells. Am J Pathol. 2006;168:542–550. doi: 10.2353/ajpath.2006.050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Devauchelle B, Badet L, Lengele B, Morelon E, Testelin S, Michallet M, D’Hauthuille C, Dubernard JM. First human face allograft: early report. Lancet. 2006;368:203–209. doi: 10.1016/S0140-6736(06)68935-6. [DOI] [PubMed] [Google Scholar]